
Abstract
Transmembrane protein 16A (TMEM16A), also called Ano1, is a Ca2+ activated Cl− channel expressed widely in mammalian epithelia, as well as in vascular smooth muscle and some tumors and electrically excitable cells. TMEM16A inhibitors have potential utility for treatment of disorders of epithelial fluid and mucus secretion, hypertension, some cancers and other diseases. 4-Aryl-2-amino thiazole T16Ainh-01 was previously identified by high-throughput screening. Here, a library of 47 compounds were prepared that explored the 5,6-disubstituted pyrimidine scaffold found in T16Ainh-01. TMEM16A inhibition activity was measured using fluorescence plate reader and short-circuit current assays. We found that very little structural variation of T16Ainh-01 was tolerated, with most compounds showing no activity at 10 μM. The most potent compound in the series, 9bo, which substitutes 4-methoxyphenyl in T16Ainh-01 with 2-thiophene, had IC50 ∼1 μM for inhibition of TMEM16A chloride conductance.
Introduction
Transmembrane protein 16A (TMEM16A) (also known as anoctamin1, ANO1, DOG1, ORAOV2, TAOS-2) is a Ca2+-activated Cl− channel (CaCC) that is expressed widely in mammalian tissues, including secretory epithelial cells, smooth muscle cells in the airways and reproductive tract, interstitial cells of Cajal and nociceptive neuronsCitation1,Citation2. TMEM16A is overexpressed in some human cancers and its expression has been correlated with tumor gradeCitation3,Citation4. Studies in TMEM16A knockout mice have implicated its involvement in tracheal developmentCitation5,Citation6 and mucociliary clearanceCitation7, with knockout mice showing mucus accumulation in the airwaysCitation8. TMEM16A knockout or knockdown is associated with diminished rhythmic contraction of gastric smooth muscle cellsCitation5, defective protein reabsorption in kidney proximal tubuleCitation9 and attenuated pain responseCitation10. TMEM16A knockout mice also manifest reduced blood pressure and decreased hypertensive response following vasoconstrictor treatmentCitation11.
TMEM16A contains eight putative transmembrane domains with intracellular NH2 and COOH termini and two calmodulin binding domainsCitation1,Citation2. Putative Ca2+ binding sites are located at E702 and E705Citation12. The TMEM16A protein appears to be structured as a homodimerCitation13,Citation14. TMEM16A is expressed in multiple splice variants that have variable sensitivity to cytosolic Ca2+Citation15,Citation16. An X-ray crystal structure (3.4 Å resolution) was recently solved of a fungal TMEM16 isoform with Ca2+-activated lipid scramblase activity (nhTMEM16), which has 39–42% homology to mammalian TMEM16ACitation17. nhTMEM16 contains 10 transmembrane segments per subunit and a region of six residues (including glutamate and aspartate), surrounding bound Ca2+ ions, providing a potential structural explanation for Ca2+-activation.
Pharmacological inhibition of TMEM16A has been proposed to be of utility for inflammatory and reactive airways diseases and hypertension, and perhaps for pain and cancerCitation1. TMEM16A activation has been considered as a therapeutic strategy to treat cystic fibrosis, gastrointestinal hypomotility and salivary gland hypofunctionCitation18–20. TMEM16A has recently been proposed as a target in chronic inflammatory diseaseCitation21. Non-selective CaCC inhibitors, which inhibit TMEM16A as well as non-TMEM16A (as yet unidentified) CaCCs have been identified by high-throughput screeningCitation22. TMEM16A-selective inhibitors have been identified from functional screens using TMEM16A-transfected cells, which include aminothiazole linked to a disubstituted pyrimidine (T16Ainh-A01; )Citation23. T16Ainh-A01 has been used in studies of TMEM16A function in vascular smooth muscle cells and mammalian blood vesselsCitation24, models of chronic hypoxic pulmonary hypertensionCitation25, epithelial fluid transportCitation26 and cancer cell proliferationCitation27.
Figure 1. The structure of lead inhibitor T16Ainh-A01.
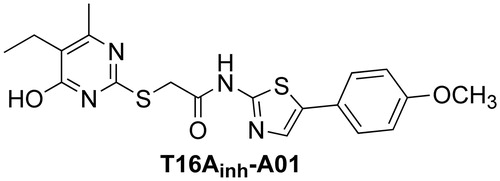
Herein, we present a systematic structural elaboration of the lead inhibitor T16Ainh-A01, including a variation of its pyrimidine (alkyl, small cycloalkyl and fluoroalkyl) and aminothiazole substituent (aromatic and heteroaromatic). On the pyrimidine, alkyl and cycloalkyl substituents were chosen to probe a possible hydrophobic pocket in the binding site, noting the presence of ethyl and methyl in the lead inhibitor. Fluoroalkyl substituents were considered given their resistance to oxidative metabolism, and ability to form electrostatic interactions. We hypothesized a more substantial interaction with the disubstituted pyrimidine and the binding site, while assuming the role of the aminothiazole substituent less important, and as a possible position for solubilizing groups. To probe this hypothesis, we designed inhibitors replacing 4-methoxyphenyl in T16Ainh-A01 with other aromatic rings, as well as a small selection of mono- and bicyclic heterocycles.
Materials and methods
Cell lines and culture
Fischer rat thyroid (FRT) cells were stably transfected with human TMEM16A (the abc isoform) and halide sensor YFP-H148Q/I152L/F46L. Cells were plated in 96-well black-walled microplates (Corning Inc., Corning, NY) at a density of 20 000 cell/well in Coon’s modified F-12 medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin. Assays were done 24 h after platingCitation20,Citation23.
TMEM16A functional assay
Each well of a 96-well plate containing the cultured cells was washed twice with phosphate-buffered saline (PBS) leaving 50 μl. Test compounds (0.5 μl in dimethyl sulfoxide (DMSO)) were added to each well at specified concentration. After 10 min, each well was assayed individually for TMEM16A-mediated I− influx by recording fluorescence continuously (400 ms/point) for 2 s (baseline), then 50 μl of 140 mM I− solution was added at 2 s, and then 50 μl of 70 mM I− solution containing 300 μM adenosine triphosphate (ATP) was added at 6.4 s. The 70 mM I− solution consisted of a 1:1 mixture of PBS and the 140 mM I− solution. The initial rate of I− influx following each of the solution additions was computed from fluorescence data by non-linear regressionCitation20,Citation23,Citation28.
Short-circuit current assay
FRT-TMEM16A cells were grown on Snapwell inserts as describedCitation20 and mounted in Ussing chambers (Physiologic Instruments, San Diego, CA). The basolateral membrane was permeabilized with amphotericin B (250 μg/ml) for 30 min, and a chloride gradient was applied in which the basolateral membrane was bathed with the HCO3-buffered solution, and in the apical solution 120 mM NaCl was replaced by sodium gluconate. Compounds were added to the apical solution. Cells were bathed for a 10-min stabilization period and aerated with 95% O2/5% CO2 at 37 °C before addition of 100 μM ATP. Short-circuit current was measured using an EVC4000 Multi-Channel V/I Clamp (World Precision Instruments, Sarasota, FL).
Chemistry: general
Unless otherwise indicated, all reaction solvents were anhydrous and obtained as such from commercial sources. All other reagents were used as supplied. Reverse-phase high-pressure liquid chromatography (RP-HPLC) analysis was performed using a Dionex Ultimate 3000 system, using a C18 column [3 × 150 mm]. Low-resolution electrospray ionization (ESI)-liquid chromatography mass spectrometry (LCMS) was carried out with an Agilent 1100 HPLC coupled to an Agilent 1956B mass spectrometry detector (MSD). RP-HPLC runs typically employed gradients of two solvents: [A] = H2O (0.05% trifluoroacetic acid (TFA)) and [B] CH3CN (0.05% TFA); RP-LCMS used the same solvent system with TFA replaced with formic acid (88% aq). The standard HPLC and LCMS gradients proceeded with [A:B] = 95:5 to [A:B] = 5:95 over 10 min. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on either a Bruker 300 or 500 MHz instrument. 1H NMR chemical shifts are relative to tetramethylsilane (TMS) (δ = 0.00 ppm), CDCl3 (δ 7.26), CD3OD (δ = 4.87 and 3.31), acetone-d6 (δ 2.05), or DMSO-d6 (δ 2.5). 13C NMR chemical shifts are relative to CD3OD (δ 49.2) or CDCl3 (δ 77.2). Microwave-assisted organic synthesis was performed using a Biotage Initiator instrument. Several compounds were prepared but also had a commercial supplier or were known: 2a–c (via general procedure 1); 7a, b, e and f (via general procedure 4); 8a–c, e, h–m, o and p (via general procedure 6); 9ag, ai, aj and ax (via general procedure 7).
General procedure 1: 4-aryl-2-aminothiazole bromoacetamides (2a–c) prepared from 4-aryl-2-aminothiazoles (1a–c)
Substituted 4-aryl-2-aminothiazole (1.0 eq, 2.5 mmol) (1a–c) was dissolved in anhydrous methylene chloride (0.3 M), followed by treatment with triethylamine (1.2 eq) and placed into an ice bath. The reaction mixture was stirred under argon until internal temp was about 0 °C and bromoacetyl bromide (1.05 eq) dissolved in dichloromethane (DCM) was added dropwise. Next, the reaction mixture was stirred under argon for 1 h at room temperature (RT). LCMS indicated consumption of starting material and formation of a product. The crude product was treated with HCl (0.1 M aq; 50 ml), transferred to a separatory funnel and extracted with 1:1 mixture of ethyl acetate and diethyl ether (50 ml). Then, the organic phase was washed with additional HCl (0.1 M aq), brine and was then dried over Na2SO4 and concentrated in vacuo.
General procedure 2: 2-amino heteroaryl thiazoles (4b–d) prepared from heteroaryl methyl ketones (3b–d)
Heteroaryl methyl ketone (1.0 eq; 8 mmol) (3b–d) was dissolved in EtOAc (0.1 M), followed by the addition of CuBr2 (2.0 eq). This reaction mixture was refluxed at 100 °C for 1 h. LCMS indicated consumption of starting material and the formation of the desired bromoketone intermediate. The reaction mixture was then left to cool to RT. Upon reaching RT, the reaction mixture was filtered by using a Buchner funnel, to remove excess precipitated CuBr2, and the filtrate was then added to a fresh round bottom flask (RBF). Thiourea (2.0 eq) was then added into the reaction mixture, which was then heated again for 1 h at 100 °C. Reaction mixtures typically changed from green to orange during the course of the reaction, with the formation of a precipitate. The mixture was then allowed to cool to RT. After reaching RT, the mixture was filtered with a Buchner funnel. The precipitate was then rinsed with ethyl acetate, in order to remove excess thiourea, which generated a crude product. The identity and purity of the product was confirmed by LCMS.
General procedure 3: 4-heteroaryl-2-aminothiazole chloroacetamide (5a–d) prepared from 4-heteroaryl-2-aminothiazoles (5a–d)
4-Heteroaryl 2-aminothiazole (4a–d) (1.0 eq; 0.4 mmol) was dissolved in 1,2-dichloroethane (DCE):dimethylformamide (DMF) (4:1 mixture, 0.1 M). Bromoacetic acid (7.0 eq), 4-dimethylaminopyridine (4-DMAP) (0.10 eq) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI HCl) (7.0 eq) were added sequentially. The reaction mixture was then refluxed at 100 °C for 1 h, and LCMS confirmed consumption of starting material (SM) and formation of the product. The reaction mixture was cooled to RT, and then taken up into Et2O:EtOAc (1:1; 50 ml), and washed with HCl (0.1 M aq.; 3 × 50 ml), then sat. aq. NaCl (50 ml), dried over Na2SO4 and concentrated in vacuo. Bromoacetamide intermediates were converted to chloroacetamide (3a–d) through the course of the reaction, presumably from chloride present in EDCI HCl. Identity as chloroacetamide and purity was confirmed by LCMS. The products were generally pushed to the next step without additional purification or characterization.
General procedure 4: α-substituted β keto esters (7) using potassium carbonate
To a mixture of an unsubstituted β-keto ester (6) (1 eq) and iodo- or bromoalkane (1.05 eq) in DMF (0.1 M) was added K2CO3 (1.5 eq) and the mixture was allowed to briefly stir at RT under argon. Then, the reaction mixture was heated to 60 °C for 30 min. Some products (7f and g) were formed more effectively with the use of microwave irradiation (110 °C, 10 min). HPLC showed consumption of a starting material and formation of the product. The reaction mixture was taken up in H2O, extracted with DCM, washed with brine, dried over Na2SO4 and concentrated in vacuo. Crude products were subjected to the subsequent cyclization reactions without additional purification.
General procedure 5: α-substituted β keto isobutyl esters (7) using sodium tert-butoxide
To a mixture of an unsubstituted β-keto ester (6) (1 eq) and bromoalkane (1.05 eq) in tert-butanol (0.5 M) was added sodium tert-butoxide (1.2 eq) and the mixture was allowed to briefly stir at RT under argon, and was then heated to 90 °C for 24 h. HPLC showed consumption of a starting material and formation of the product. The reaction mixture was taken up in water, extracted with DCM, washed with brine, dried over Na2SO4 and concentrated in vacuo. Crude products were subjected to the subsequent cyclization reactions without additional purification.
General procedure 6: thiouracils (8a–p) generated by cyclization of unsubstituted (6) or substituted (7) β keto esters
A freshly prepared solution of sodium ethoxide was obtained by dissolving Na (10 eq) in EtOH (0.1 M), which was treated with a substituted or unsubstituted β-keto ester (6 or 7) (1 eq) followed by thiourea (2 eq). The reaction mixture was stirred under argon, heated to 100 °C in an oil bath, and allowed to reflux overnight. LCMS indicated consumption of starting material and formation of a product. The solvent was removed and the crude reaction mixture was acidified with 1 M HCl to pH = 3, extracted with DCM, dried over Na2SO4 and concentrated in vacuo to give crude thiouracil (8a–p) products, which were subjected to the coupling reaction without additional purification. Alternatively, the reactions could be affected by microwave irradiation (15 min at 150 °C).
General procedure 7: substituted thiopyrimidine aryl aminothiazoles (9aa–bu) from conjugation of thiouracils (8a–p) with 2-aminothiazole haloacetamides (2a–c or 5a–d)
To a 20 ml scintillation vial was added 4-aryl or 4-heteroaryl 2-aminothiazole haloacetamide (1.0 eq, typically 10–50 mg) (2 or 5), in DMF (0.1 M) followed by the addition of a substituted thiouracil (8) (1.0–1.2 eq). The reaction mixture was placed in an oil bath pre-heated to 60 °C. In the case of less reactive chloroacetamide (5a–d), NaI was added to facilitate the reaction (1 eq). Then, K3PO4 monohydrate (3 eq) was added and the vial was heated for 1 h. LCMS indicated consumption of starting materials and formation of product. The crude reaction mixture was diluted with EtOAc (20 ml) and washed five times with brine (20 ml), dried over Na2SO4 and concentrated in vacuo. Next, the crude reaction mixtures were purified by trituration with Et2O to give final products (9aa–bu). As specified individually, some compounds needed additional purification by preparative HPLC.
2-Chloro-N-(4-thiophen-2-yl-thiazol-2-yl)-acetamide (5b)
Utilizing general procedure 2, 1-thiophen-2-yl-ethanone (3b) (1000 mg, 7.92 mmol) was converted to 4-thiophen-2-yl-thiazol-2-ylamine (4b) which was isolated as a white solid (1980 mg, 95%). This was utilized in the next step, utilizing general procedure 3, to generate the title compound (5b) as a pink solid (28.4 mg, 25%). 1H NMR (500 MHz, DMSO-d6) δ 4.39 (s, 2H), 7.10 (t, J = 2 Hz, 1H), 7.49 (d, J = 5, 1H), 7.51 (s, 1H), 7.52 (d, J = 5 Hz, 1H).13C NMR (125 MHz, DMSO-d6) δ 42.7, 107.4, 124.3, 126.1, 128.5, 138.7, 144.4, 157.9, 165.6. ESI-LCMS (low resolution) m/z calculated for C9H7ClN2OS2 [M + H] 259.7, found [M + H] 259.3.
2-(4-Hydroxy-5-methyl-6-trifluoromethyl-pyrimidin-2-ylsulfanyl)-N-(4-phenyl-thiazol-2-yl)-acetamide (9ao)
Utilizing general procedure 7 with thiouracil 8p (20 mg, 0.095 mmol) and aminothiazole bromoacetamide 2a (28 mg, 0.095 mmol), yellow solid was obtained (5 mg, 12%) after required preparative HPLC purification. 1H NMR (500 MHz, acetone-d6) δ 2.12 (s, 3H), 4.34 (s, 2H), 7.30 (t, J = 7 Hz, 1H), 7.40 (t, J = 7 Hz, 2H), 7.48 (s, 1H), 7.93 (d, J = 7 Hz, 2H). ESI-LCMS (low resolution) m/z calculated for C17H13F3N4O2S2 [M + H] 427.0, found [M + H] 427.2.
2-(4-Hydroxy-5,6-dimethyl-pyrimidin-2-ylsulfanyl)-N-(4-thiophen-2-yl-thiazol-2-yl)-acetamide (9bs)
Utilizing general procedure 7 with thiouracil 8l (15.0 mg, 0.097 mmol) and aminothiazole chloroacetamide 5b (25.00 mg, 0.097 mmol), brown solid was obtained (5.4 mg, 14.7%). 1H NMR (500 MHz, DMSO-d6) δ 1.84 (s, 3H), 2.12 (s, 3H), 4.06 (s, 2H), 7.10 (t, J = 3 Hz, 1H), 7.44 (s, 1H), 7.85 (d, J = 5 Hz, 1H), 7.95 (d, J = 4 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 10.4, 20.8, 33.6, 106.3, 114.0, 123.6, 125.4, 127.9, 136.9, 138.3, 143.6, 157.9, 167.2, 177.9, 221.9. ESI-LCMS (low resolution) m/z calculated for C15H14N4O2S3 [M + H] 379.5, found [M + H] 379.3.
2-(5-Ethyl-4-hydroxy-6-methyl-pyrimidin-2-ylsulfanyl)-N-(4-thiophen-2-yl-thiazol-2-yl)-acetamide (9bo)
Utilizing general procedure 7 with thiouracil 8h (16 mg, 0.097 mmol) and aminothiazole chloroacetamide 5b (25 mg, 0.097 mmol), light brown solid was obtained (4.3 mg, 11%). 1H NMR (500 MHz, acetone-d6) δ 1.06 (t, J = 7 Hz, 3H), 2.41 (s, 3H), 2.50 (q, J = 8 Hz, 2H), 4.17 (s, 2H), 7.07 (t, J = 7 Hz, 1H), 7.33 (s, 1H), 7.40 (d, J = 4, 1H), 7.49 (d, J = 3 Hz, 1H). 13C NMR (125 MHz, DMSO-d6) δ 13.0, 18.5, 32.9, 34.1, 107.0, 115.8, 124.3, 126.0, 128.5, 138.8, 144.28, 158.4, 162.0, 164.0 167.4, 174.4. ESI-LCMS (low resolution) m/z calculated for C16H16N4O2S3 [M + H] 393.5, found [M + H] 393.3.
Results and discussion
Chemistry
The targeted 5,6-disubstituted pyrimidine-linked aminothiazole scaffold was approached through the synthetic strategy outlined in Scheme 1. The synthesis commenced with the preparation of aminothiazole haloacetamide. Bromoacetylation of simple substituted 4-aryl-2-aminothiazoles (1a–c) was accomplished with bromoacetic bromide to generate the corresponding bromoacetamide (2a–c). Bromoketone 3a was commercially available and directly subjected to cyclization to aminothiazole 4a. Other 4-heteroaryl-2-aminothiazoles were not available, and were prepared in a one-pot two-step bromination/cyclization process from heteroaryl methyl ketones (3b–d) using CuBr2 followed by reaction with thiourea, generating aminothiazole products (4b–d) in good yields. Surprisingly, our attempts to form bromoacetamides of heteroaryl aminothiazoles 4a–d using highly reactive bromoacetyl bromide were not successful. Therefore, we coupled 4a–d with bromoacetic acid in the presence of EDCI HCl. Interestingly, transient bromoacetamides were converted to chloroacetamides (5a–d) through the course of the reaction presumably due to chloride present in EDCI HCl, as confirmed by LCMS. Fortunately, chloride was a sufficient leaving group in the subsequent alkylation reactions, albeit with the assistance of sodium iodide. The products of both routes are listed in , with the reactions generally occurring in good yield.
Scheme 1. Synthesis of 4-aryl/heteroaryl-2-aminothiazole inhibitor candidates. Reagents and conditions: (a) bromoacetyl bromide, Et3N, DCM, 0 °C; (b) for bromoketone 6a: thiourea, THF, 50 °C; (c) for methyl ketones 6b–d: CuBr2, EtOAc, 100 °C; then thiourea 100 °C; (d) bromoacetic acid, EDCI HCl, cat. 4-DMAP, DCE:DMF (1:1), 100 °C; (e) R-X, base, DMF, 60 °C or MW 110 °C (see experimental); (f) Na/EtOH, thiourea, 100 °C; (g) K3PO4-H2O, DMF. For chloroacetamides (X = Cl), NaI was added to facilitate substitution.
Table 1. Yields from preparation of 4-aryl 2-aminothiazole bromoacetamides (2a–c) and 4-heteroaryl 2-aminothiazole chloroacetamides (5a–d). The heteroaryl aminothiazole intermediates were prepared from either a heteroaryl bromoketone (3a) or methylketones (3b–d).
α-substituted β-keto esters were prepared for cyclization with thiourea to generate thiouracils, with the results of alkylation summarized in . Methyl acetoacetate (6a) and isobutyl acetoacetate (6b) were alkylated to β-keto esters (7a–d) by simple substitution. Isobutyl esters were used to decrease the trans-esterification during the reaction, and also to decrease the volatility of β-keto ester products, aiding in isolation. Methyl 3-cyclopropyl-3-oxopropionate (6c) was used to prepare a small homologous series (7e–g) of β-keto esters. Isolated yields were fair to quantitative.
Table 2. Yields from α-alkylation of β-keto esters (6 → 7).
Upon generation of a small library of α-substituted β-keto esters (7a–g), the compounds were cyclized to the corresponding thiouracils (8a–g) by treatment of with thiourea under basic conditions (). Additionally, a selection of commercially available α-substituted β-keto esters (7h–j) and α-non-substituted species (6c–h) were also cyclized to the corresponding thiouracils (8h–p).
Table 3. Yields from cyclization of β-keto esters (6 or 7) to mono- and di-substituted thiouracils (8a–p) using thiourea.
The final synthetic task was coupling of the mono- and disubstituted thiouracils (8a–p) with 4-aryl and 4-heteroaryl 2-aminothiazole haloacetamides (2a–c and 5a–d) to generate the inhibitor candidates (9aa–bu). Each of the thiouracils was coupled with one or more 2-aminothiazole haloacetamides in the presence of K3PO4 monohydrate in DMF at 60 °C, with the results summarized in . Poorly electrophilic 2-aminothiazole chloroacetamide (5a–d) required the addition of sodium iodide to facilitate the alkylation, by the Finkelstein mechanism. The reactions generally worked well, giving acceptable isolated yields of product, allowing construction of the 47-member library of inhibitor candidates. While the coupling reactions proceeded to completion, the slight impurity of a small number of products necessitated purification by preparative HPLC (see Supplementary material).
Table 4. Coupling yields and TMEM16A inhibition of a library of thiopyrimidine aryl aminothiazoles (9aa–bu). Yields (%) are of the isolated or purified products. IC50 (μM) for inhibition of TMEM16A anion conductance using a fluorescence plate reader assay. The purity of active compounds was >95% based on HPLC-LCMS analysis at 254 nm, combined with the absence of impurities observed by inspection of 1H NMR spectra.
Biological characterization
Compounds 9aa–bu were evaluated for inhibition of TMEM16A anion channel function using a cell-based functional assay as described previouslyCitation20,Citation23. The compounds were added to FRT cells stably expressing human TMEM16A and the iodide-sensitive fluorescent protein YFP-H148Q/I152L/F46L and assayed from the kinetics of iodide uptake using a fluorescence plate reader. Initial testing was done at 10 μM. IC50 values for active compounds were determined from concentration-inhibition measurements, as summarized in . The fluorescence plate reader results were used to select candidates for the more definitive, albeit lower throughput, short-circuit (apical membrane) current assay.
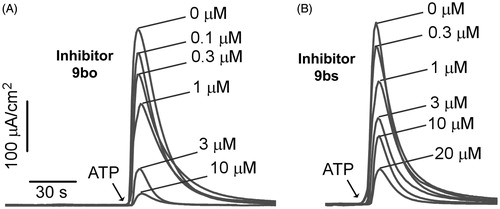
Surprisingly, most of the synthesized compounds were inactive at 10 μM showing little tolerance for variation of the thiouracil or aromatic ring in T16Ainh-A01. Cycloalkyl or fluoroalkyl substituents at R1, or alkyl or cycloalkyl substituents at R2, generally produced inactive compounds. An exception was 9ao, which incorporated a trifluoromethyl group at R1 and methyl at R2, but with reduced potency (IC50 = 6.2 μM) compared to T16Ainh-A01. Previously, it was shown that the R3 substituent could be varied as different substituted aromatic rings, with preservation of potencyCitation23. For the majority of compounds reported herein, R3 was Ph, 4-Cl-Ph, or 4-MeO-Ph, with nearly all compounds inactive at 10 μM. A small series of compounds explored replacement of R3 with heterocycles (9bn–bu), while keeping the thiouracil substitution found in T16Ainh-A01 (R1 = methyl, R2 = ethyl) or a homolog (R1 = R2 = methyl). Of these compounds, all with bicyclic heterocycles were inactive. Gratifyingly, two inhibitors with R3 = 2-thiophene (9bo, IC50 = 3.5 μM; 9bs, IC50 = 2.9 μM) were active.
Compounds 9bs and 9bo were evaluated by a short-circuit current electrophysiological assay of TMEM16A functionCitation23, with concentration-dependence shown in . IC50 values were ∼1 μM for 9bo and ∼3 μM for 9bs. The IC50 for T16Ainh-A01 is ∼1 μM as reported previouslyCitation23.
Figure 2. Short-circuit current measured in TMEM16A-expressing FRT cells. Inhibitors were added 5 min prior to TMEM16A activation by 100 μM ATP. Concentration-dependent inhibition by (A) 9bo (IC50 ∼ 1 μM); (B) 9bs (IC50 ∼ 3 μM).
Conclusion
In conclusion, a library of 47 5,6-disubstituted pyrimidine analogs (9aa–bu) of the lead 4-aryl-2-aminothiazole inhibitor (T16Ainh-A01) was synthesized in a modular strategy utilizing haloacetamide (2a–c or 5a–d) and thiouracil (8a–p) building blocks. This study currently represents the first systematic exploration of 4-aryl-2-aminothiazoles as inhibitors of TMEM16A. T16Ainh-A01 is a good starting point for optimization due to its applications in studying TMEM16A function in smooth muscle cells, hypertension and cancer, and because of its low micromolar potencyCitation23. Most of the compounds synthesized here were inactive at 10 μM, while three compounds showed measurable activity (9ao, 9bo and 9bs). The most potent compound, 9bo, with IC50 ∼1 μM, may serve as an alternative to lead compound T16Ainh-A01.
Supplementary material available online
IENZ_1135912_Supp.pdf
Download PDF (327.5 KB)Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Related Research Data
References
- Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev 2014;94:419–59.
- Picollo A, Malvezzi M, Accardi A. TMEM16 proteins: unknown structure and confusing functions. J Mol Biol 2015;427:94–105.
- Qu Z, Yao W, Yao R, et al. The Ca(2+)-activated Cl(-) channel, ANO1 (TMEM16A), is a double-edged sword in cell proliferation and tumorigenesis. Cancer Med 2014;3:453–61.
- Wanitchakool P, Wolf L, Koehl GE, et al. Role of anoctamins in cancer and apoptosis. Philos Trans R Soc Lond B Biol Sci 2014;369:20130096
- Huang F, Rock JR, Harfe BD, et al. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci USA 2009;106:21413–18.
- Rock JR, Futtner CR, Harfe BD. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol 2008;321:141–9.
- Ousingsawat J, Martins JR, Schreiber R, et al. Loss of TMEM16A causes a defect in epithelial Ca2+-dependent chloride transport. J Biol Chem 2009;284:28698–703.
- Rock JR, O’Neal WK, Gabriel SE, et al. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl- secretory channel in mouse airways. J Biol Chem 2009;284:14875–80.
- Faria D, Rock JR, Romao AM, et al. The calcium-activated chloride channel Anoctamin 1 contributes to the regulation of renal function. Kidney Int 2014;85:1369–81.
- Lee B, Cho H, Jung J, et al. Anoctamin 1 contributes to inflammatory and nerve-injury induced hypersensitivity. Mol Pain 2014;10:5.
- Heinze C, Seniuk A, Sokolov MV, et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J Clin Invest 2014;124:675–86.
- Yu K, Duran C, Qu Z, et al. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res 2012;110:990–9.
- Fallah G, Römer T, Detro-Dassen S, et al. TMEM16A(a)/anoctamin-1 shares a homodimeric architecture with CLC chloride channels. Mol Cell Proteomics 2011;10:M110 004697.
- Sheridan JT, Worthington EN, Yu K, et al. Characterization of the oligomeric structure of the Ca(2+)-activated Cl- channel Ano1/TMEM16A. J Biol Chem 2011;286:1381–8.
- Tian Y, Kongsuphol P, Hug M, et al. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J 2011;25:1058–68.
- Tian Y, Schreiber R, Kunzelmann K. Anoctamins are a family of Ca2+-activated Cl- channels. J Cell Sci 2012;125:4991–8.
- Brunner JD, Lim NK, Schenck S, et al. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 2014;516:207–12.
- Mall MA, Galietta LJ. Targeting ion channels in cystic fibrosis. J Cyst Fibros 2015;14:561–70.
- Sondo E, Caci E, Galietta LJ. The TMEM16A chloride channel as an alternative therapeutic target in cystic fibrosis. Int J Biochem Cell Biol 2014;52:73–6.
- Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J 2011;25:4048–62.
- Sala-Rabanal M, Yurtsever Z, Berry KN, Brett TJ. Novel roles for chloride channels, exchangers, and regulators in chronic inflammatory airway diseases. Mediat Inflamm 2015;2015:497387
- De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol 2008;73:758–68.
- Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem 2011;286:2365–74.
- Davis AJ, Shi J, Pritchard HA, et al. Potent vasorelaxant activity of the TMEM16A inhibitor T16A(inh) A01. Br J Pharmacol 2013;168:773–84.
- Sun H, Xia Y, Paudel O, et al. Chronic hypoxia-induced upregulation of Ca2+-activated Cl- channel in pulmonary arterial myocytes: a mechanism contributing to enhanced vasoreactivity. J Physiol 2012;590:3507–21.
- Mroz MS, Keely SJ. Epidermal growth factor chronically upregulates Ca(2+)-dependent Cl(-) conductance and TMEM16A expression in intestinal epithelial cells. J Physiol 2012;590:1907–20.
- Duvvuri U, Shiwarski DJ, Xiao D, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res 2012;72:3270–81.
- Kumar S, Namkung W, Verkman AS, Sharma PK. Novel 5-substituted benzyloxy-2-arylbenzofuran-3-carboxylic acids as calcium activated chloride channel inhibitors. Bioorg Med Chem 2012;20:4237–44.