
Abstract
Tyramine derivatives 3–27 were synthesized by using conventional and environmental friendly ultrasonic techniques. These derivatives were then evaluated for the first time for their α-glucosidase (Sources: Saccharomyces cerevisiae and mammalian rat-intestinal acetone powder) inhibitory activity by using in vitro mechanism-based biochemical assays. Compounds 7, 14, 20, 21 and 26 were found to be more active (IC50 = 49.7 ± 0.4, 318.8 ± 3.7, 23.5 ± 0.9, 302.0 ± 7.3 and 230.7 ± 4.0 μM, respectively) than the standard drug, acarbose (IC50 = 840.0 ± 1.73 μM (observed) and 780 ± 0.028 μM (reported)) against α-glucosidase obtained from Saccharomyces cerevisiae. Kinetic studies were carried out on the most active members of the series in order to determine their mode of inhibition and dissociation constants. Compounds 7, 20 and 26 were found to be the competitive inhibitors of α-glucosidase. These compounds were also screened for their protein antiglycation, and dipeptidyl peptidase-IV (DPP-IV) inhibitory activities. Only compounds 20, 22 and 27 showed weak antiglycation activity with IC50 values 505.27 ± 5.95, 581.87 ± 5.50 and 440.58 ± 2.74 μM, respectively. All the compounds were found to be inactive against DDP-IV enzyme. Inhibition of α-glucosidase, DPP-IV enzymes and glycation of proteins are valid targets for the discovery of antidiabetic drugs. Cytotoxicity of compounds 3–27 was also evaluated by using mouse fibroblast 3T3 cell lines. All the compounds were found to be noncytotoxic. The current study describes the synthesis α-glucosidase inhibitory activity of derivatives, based on a natural product tyramine template. The compounds reported here may serve as the starting point for the design and development of novel α-glucosidase inhibitors as antidiabetic agents.
Introduction
α-Glucosidase (EC 3.2.1.20) is an exo-type glycosidase enzyme that catalyzes the release of monosaccharides from the nonreducing end of the carbohydrates. α-Glucosidase inhibitors (AGIs) reduce the absorption of glucose from the small intestine. As a result, they can modulate the postprandial glucose levels in blood and serve as antidiabetic drugsCitation1,Citation2,Citation3. Acarbose, miglitol and voglibose are α-glucosidase inhibitors used for the treatment of type-2 diabetesCitation4. However, these drugs cause multiple side effects, such as diarrhea, abdominal discomfort, etc. A need of new, safe and effective inhibitors of α-glucosidase is therefore greatly felt. Similarly, dipeptidyl peptidase-IV (DPP-IV) is an important enzyme involve in the cleavage of X-proline dipeptides from the N-terminus of polypeptides. The inhibition of DPP-IV is an important target for drug discovery against diabetes. In addition to this, glycation of proteins in hyperglycemia leads to late diabetic complications. The inhibition of protein glycation is vigorously studied to slow down the on-set of diabetic complications.
Tyramine (4-hydroxyphenethylamine) is a naturally occurring monoamine, derived from the amino acid tyrosine. It is reported to possess various biological properties, such as antioxidantCitation5, α1-adrenergic agonisticCitation6 and monoamine oxidaseCitation7. It is reported that administration of tyramine to diabetic rats reduces the hyperglycemia, even at doses that have low effects on cardiovascular variablesCitation8.
The synthesis of imines or Schiff’s bases has special significance in organic synthesis and medicinal chemistryCitation9. For example, they have been widely used in asymmetric synthesis of α-aminonitriles, hydrogenation and cycloaddition reactionsCitation10. They have been reported to possess various biological activities, such as antimalarial, antibacterial, antifungal, antiviral, etcCitation11. These compounds also have various applications in coordination chemistryCitation11. The current study describes the synthesis of new imine derivatives of tyramine (3–27) by using conventional and ultrasonic-assisted chemistry and evaluation of their antidiabetic activity by using α-glucosidase, DPP-IV inhibition and protein antiglycation assays.
Over the years, sonochemistry (ultrasonic-assisted chemistry) has emerged as an important tool in organic synthesis. It is a type of green chemistry with several advantages, including cost-effectiveness, environmental friendly nature and time and yield efficiencyCitation12.
During the current study, the Schiff’s bases or benzylidineamine derivatives of tyramine (3–27) were synthesized because these classes have been reported to have a wide range of biological activities, including enzyme inhibition activitiesCitation13. Moreover, compounds containing benzoyl moiety are also reported to have enzyme-inhibiting activitiesCitation14. All the compounds were found to be new, except 9, 17, 18, 19, 21, 22 and 25. These derivatives have not been reported yet for their α-glucosidase inhibitory activity. An in vitro mechanism-based biochemical assay was employed to assess their α-glucosidase (source: Saccharomyces cerevisiae) inhibitory activity. Compounds 7, 14, 20, 21 and 26 were found to be more active (IC50 = 49.7 ± 0.4, 318.8 ± 3.7, 23.5 ± 0.9, 302.0 ± 7.3 and 230.7 ± 4.0 μM, respectively) than the standard drug, acarbose (IC50 = 840.0 ± 1.73 μM (obtained) and 780 ± 0.028 μM (reportedCitation15)). The cytotoxicity of these compounds was also evaluated. In addition, this series of compounds was also screened for antiglycation and dipeptidyl peptidase-IV (DPP-IV) inhibitory activities.
Both yeast and human α-glucosidase share about 40% sequence homologyCitation16. As a result, inhibitors of yeast α-glucosidase may also inhibit α-glucosidase enzyme obtained from human and other sources. Based on this assumption, we checked compounds 3–27 for the inhibition of α-glucosidase obtained from mammalian source (rat-intestinal acetone powder). We observed that compounds 20 and 22 possess good inhibitory activity with the IC50 = 175.4 ± 3.17 and 173.9 ± 3.8 μM, respectively, if compared with standard acarbose (IC50 = 67.1 ± 4.96 μM (observed) and IC50 = 52.8 μM or 34.11 μg/mL (reportedCitation17)). However, the inhibitory activity of these compounds against human α-glucosidase remained to be studied.
Experimental
Chemistry
General experimental conditions
Electron impact mass spectra (EI-MS) were recorded on a JEOL JMS-600H mass spectrometer with a MASPEC data system. The 1H-NMR spectra were recorded in deuterated solvents on Bruker Avance 300, 400, 500 and 600 MHz NMR spectrometers. The chemical shifts (δ) and coupling constants (J) were measured in ppm and Hz, respectively, relative to SiMe4 as internal standard. Thin-layer chromatography (TLC) analyses were performed on precoated ALUGRAM, SIL G/UV254 aluminum plates (Kieselgel 60, 20 × 20, 0.5 mm thick, E. Merk, Germany). The reagents and solvents were obtained from Aldrich (St. Louis, Missouri (MO)), E. Merck Darmstadt, (Germany), and Fluka (Buchs, Switzerland). Reagents and solvents were used without purification. Chromatograms, developed on TLC plates, were visualized under ultraviolet light at 254 nm for fluorescence quenching spots and 365 nm for florescent spots.
Ultrasonic equipment: Ultrasonic-mediated reactions were conducted in Elma Ultrasonic Cleaner LC30H (Elma Schmidbauer GmbH, Germany). All experiments were performed with output power of 240 W.
General procedure for the synthesis of compounds 3–27
Conventional method: About 1.0 mmol of aldehyde and ethanol (15 mL) were mixed together, along with the 0.8 mmol of acetic acid (0.05 mL). The reaction mixture was heated at reflux for 10–15 min, followed by the addition of tyramine (1 mmol or 0.137 g). The reaction mixture was allowed to reflux, and progress of the reaction was monitored by TLC. Upon completion (3–4 h), the solvent was evaporated under vacuo. The reaction mixture was washed with a mixture of hexanes-ethyl acetate (8:2), followed by washing with hexane. The solid product obtained was pure enough to be analyzed by spectroscopic techniques, such as 1H- and 13C-NMR, IR and mass spectroscopy. In some cases, the compounds were purified by re-crystallization or column chromatography.
Ultrasonication method: A reaction flask containing aldehyde (1.0 mmol), 15 mL ethanol and 0.05 mL acetic acid (0.8 mmol), was immersed in an ultrasonic bath containing water at room temperature. The mixture was exposed to ultrasound irradiation for varying time durations (). The reaction progress was monitored by TLC. After completion, the solvent was evaporated and washed with a mixture of hexanes and ethyl acetate to afford the desired pure products, similar reaction was performed with heating (80 °C) as well.
Table 1. Comparison between sonication and conventional synthesis of selected tyramine derivatives.
4-(2-(2-Nitrobenzylideneamino)ethyl)phenol (3)
1H-NMR (300 MHz, DMSO-d6): δ 9.11 (s, 1H, OH), 8.47 (s, 1H, CH), 8.01 (d, J = 7.8 Hz, 1H), 7.94 (d, J = 6.9 Hz, 1H), 7.80 (t, J = 7.2 Hz, 1H), 7.78 (q, J = 1.2 Hz, 1H), 7.04 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.1 Hz, 2H), 3.81 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 156.9 (C-2), 155.5 (C-4′), 148.8 (C-2″), 133.4 (C-5″), 131.1 (C-1″), 130.0 (C-1′), 129.7 (C-2′, C-6′), 129.6 (C-6″), 129.3 (C-4″), 124. 1 (C-3″), 115.0 (C-3′, 5′), 62.3 (C-8′), 35.7 (C-7′); MS (direct probe, positive EI) m/z 270.1 (M+), 163.1, 107.1; HREI-MS: Calculated for C15H14N2O3; 270.1004, Observed; 270.1003; νmax; 3421.5, 3080.1, 2927.7, 1608, 1517.9, 1338.5, 1253.6, 1105.1, 825.5.
4-(2-(4-Nitrobenzylideneamino)ethyl)phenol (4)
1H-NMR (300 MHz, CDCl3): δ 8.25 (d, J = 8.7 Hz, 2H), 8.16 (s, 1H), 7.84 (d, J = 8.7 Hz, 2H), 7.06 (d, J = 8.1 Hz, 2H), 6.74 (d, J = 8.4 Hz, 2H), 3.88 (t, J = 6.3 Hz, 2H), 2.98 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 159.5 (C-2), 155.5 (C-4′), 148.4 (C-4″), 141.6 (C-1″), 129.6 (C-2″,6″), 129.6 (C-1′), 128.7 (C-2′, 6′), 123.8 (C-3″, 5″), 115.0 (C-3′, 5′), 62.4 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 270.1 (M+), 163.0, 133.1, 107.0; HREI-MS: Calculated for C15H14N2O3; 270.1004, Observed; 270.1011; νmax; 3222.8, 3047.3, 1508.2, 1244.0, 981.7, 827.4.
4-(2-(3-Nitrobenzylideneamino)ethyl)phenol (5)
1H-NMR (600 MHz, DMSO-d6): δ 9.14 (s, 1H), 8.55 (t, J = 1.2 Hz, 1H), 8.39 (s, 1H), 8.29 (dd, J = 1.2 Hz, 1H), 8.14 (d, J = 7.8 Hz, 1H), 7.74 (t, J = 7.8 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 3.80 (t, J = 7.2 Hz, 2H), 2.84 (t, J = 7.8 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 159.2 (C-2), 155.5 (C-4′), 148.1 (C-3″), 137.6 (C-1′), 133.9 (C-6″), 130.8 (C-1′), 129.7 (C-2′, 6′), 124.9 (C-5″), 121.8 (C-4″), 115.0 (C-3′, 5′), 62.2 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 270.3 (M+), 163.1, 135.1, 107.1; HREI-MS: Calculated for C15H14N2O3; 270.1004, Observed; 270.0979; νmax; 3085.9, 2931.6, 1641.3, 1517.9, 1348.1, 1240.1, 1099.3, 821.6.
4-(2-(2-Bromo-4-methylbenzylideneamino)ethyl)phenol (6)
1H-NMR (300 MHz, DMSO-d6): δ 9.12 (s, 1H), 8.4 (s, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.48 (s, 1H), 7.25 (d, J = 8.1 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 3.79 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H), 2.31 (s, 3H) MS (direct probe, positive EI) m/z 319.0 ([M + 2]+), 317.0 (M+), 209.9, 184.9, 107.0; HREI-MS: Calculated for C16H16BrNO; 317.0415, Observed 317.0406; νmax; 3334.7, 2927.7, 1596.9, 1508.2, 1261.4, 823.5.
4-(2-(4-Chlorobenzylideneamino)ethyl)phenol (7)
1H-NMR (600 MHz, DMSO-d6): δ 9.13 (s, 1H), 8.23 (s, 1H), 7.72 (d, J = 9.0 Hz, 2H), 7.01 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 3.74 (t, J = 7.2 Hz, 2H), 2.79 (t, J = 7.8 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 159.7 (C-2), 155.5 (C-4′), 135.1 (C-4″), 135.0 (C-1″), 129.9 (C-1′), 129.7 (C-2′, 6′), 129.4 (C-2′, 6′), 128.8 (C-3″, 5″), 115.0 (C-3′, 5′), 62.3 (C-8′), 36.0 (C-7′); MS (direct probe, positive EI) m/z 261.1 ([M + 2]+), 259.1 (M+), 152.1, 125.0, 107.0; HREI-MS: Calculated for C15H14ClNO; 259.0764, Observed; 259.0763; νmax; 3417.6, 1643.2, 1506.3, 1238.2, 1091.6, 819.7.
4-(2-(2-Chlorobenzylideneamino)ethyl)phenol (8)
1H-NMR (600 MHz, DMSO-d6): δ 9.13 (s, 1H), 8.53 (s, 1H), 7.94 (dd, J = 1.8 Hz, 1H), 7.49 (m, J = 1.2 Hz, 2H), 7.40 (m, 1H), 7.02 (d, J = 7.8 Hz, 2H), 6.65 (t, J = 3.0 Hz, 2H), 3.81 (q, J = 6.6 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 157.0 (C-8′), 155.5 (C-4′), 133.8 (C-1′), 132.7 (C-2″), 132.0 (C-4″), 129.8 (C-1′), 129.7 (C-2′, 6′), 128.0 (C-6″), 127.4 (C-5″), 115.0 (C-3′, 5′), 62.5 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 261.1 ([M + 2]+), 259.1 (M+), 156.0, 139.0, 108.0; HREI-MS: Calculated for C15H14ClNO; 259.0764, Observed; 259.0744; νmax; 2929.7, 1616.2, 1510.2, 1448.4, 1247.9, 1029.9, 812.0.
4-(2-(Benzylideneamino)ethyl)phenol (9)
1H-NMR (400 MHz, DMSO-d6): δ 8.13 (s, 1H), 7.68 (q, J6″, 5″ = 1.5 Hz, J = 6.0 Hz, 2H), 7.39 (dd, J = 1.5 Hz, 3H), 7.08 (d, J = 8.0 Hz, 2H), 6.73 (d, J = 8.0 Hz, 2H), 3.82 (t, J = 7.5 Hz, 2H), 2.94 (t, J = 7.5 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 160.8 (C-2), 155.5 (C-4′), 136.1 (C-1″), 130.5 (C-1′), 130.0 (C-4″), 129.7 (C-2′, 6′), 128.6 (C-2″, 6″), 127.7 (C-3″, 5″), 115.0 (C-3′, 5′), 62.4 (C-8′), 36.1 (C-7′); MS (direct probe, positive EI) m/z 225.1 (M+), 118.0, 107.0, 91.0; HREI-MS: Calculated for C15H15NO; 225.1154, Observed; 225.1136; νmax; 3417.6, 2927.7, 1645.2, 1512.1, 1450.4, 1380.9, 1243.1, 823.5.
4-(2-(3-Chlorobenzylideneamino)ethyl)phenol (10)
1H-NMR (300 MHz, DMSO-d6): δ 9.13 (s, 1H), 8.23 (s, 1H), 7.73 (s, 1H), 7.66 (d, J = 7.2 Hz, 1H), 7.52 (t, J = 8.1 Hz, 2H), 7.46 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 8.1 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 3.76 (t, J = 7.2 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 160.0 (C-2), 155.5 (C-4′), 138.2 (C-1″), 133.5 (C-3″), 130.6 (C-1′), 130.2 (C-4″), 129.7 (C-5′), 129.6 (C-2′, 6′), 127.0 (C-6″), 126.5 (C-5′), 115.0 (C-3′, 5′), 62.2 (C-8′), 35.9 (C-7′); MS (direct probe, positive EI) m/z 261.2 ([M + 2]+), 259.2 (M+), 152.1, 125.1, 107.0; HREI-MS: Calculated for C15H14ClNO; 259.0764, Observed; 259.0764; νmax; 3334.7, 2923.9, 1602.7, 1514.0, 1382.9, 1261.4, 821.6.
4-(2-(3-Methylbenzylideneamino)ethyl)phenol (11)
1H-NMR (300 MHz, DMSO-d6): δ 9.12 (s, 1H, OH), 8.18 (s, 1H), 7.52 (s, 1H), 7.48 (d, J '= 7.5 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 7.26 (d, J = 7.5 Hz, 1H), 7.02 (t, J = 8.4 Hz, 2H), 6.67 (t, J = 4.5 Hz, 2H), 3.72 (t, J = 7.2 Hz, 2H), 2.81 (t, J = 7.2 Hz, 2H), 2.32 (s, 3H); 13C-NMR (75 MHz, DMSO-d6): δ 160.8 (C-2), 155.4 (C-4′), 137.8 (C-1″), 136.1 (C-3″), 131.1 (C-1′), 129.8 (C-4″), 129.6 (C-2′, 6′), 128.5 (C-2″), 128.0 (C-5″), 125.1 (C-6″), 114.9 (C-3′, 5′), 62.4 (C-8′), 36.0 (C-7′), 20.8 (CH3); MS (direct probe, positive EI) m/z 239.2 (M+), 132.1, 105.1; HREI-MS: Calculated for C16H17NO; 239.1310, Observed; 239.1294; νmax; 2916.2, 1639.4, 1514.0, 1450.4, 1244.0, 1043.4, 815.8.
4-(2-(2-Methylbenzylideneamino)ethyl)phenol (12)
1H-NMR (300 MHz, DMSO-d6): δ 9.12 (s, 1H), 8.44 (s, 1H), 7.75 (d, J = 7.5 Hz, 1H), 7.32 (t, J = 6.6 Hz, 1H), 7.24 (t, J = 9.9 Hz, 2H), 7.03 (d, J = 8.1 Hz, 2H), 6.66 (d, 2H, J = 8.1 Hz, 2H), 3.78 (t, J' = 7.2 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H), 2.38 (s, 3H); 13C-NMR (75 MHz, DMSO-d6): δ 159.6 (C-2), 155.4 (C-4′), 137.2 (C-2″), 133.9 (C-1″), 130.7 (C-1′), 129.9 (C-4″), 129.9 (C-6″), 129.7 (C-2′, 6′), 127.1 (C-3″), 125.9 (C-5″), 114.9 (C-3′, 5′), 62.8 (C-8′), 36.1 (C-7′), 18.8 (C-CH3); MS (direct probe, positive EI) m/z 239.2 (M+), 132.1, 105.1; HREI-MS: Calculated for C16H17NO; 239.1310, Observed; 239.1327; νmax; 2916.2, 2873.7, 1602.7, 1510.2, 1456.2, 1247.9, 817.8.
4-(2-(4-Methoxybenzylideneamino)ethyl)phenol (13)
1H-NMR (300 MHz, DMSO-d6): δ 9.11 (s, 1H), 8.15 (s, 1H), 7.65 (d, J = 8.7 Hz, 2H), 7.02 (q, J = 8.4 Hz, 4H), 6.65 (d, J = 8.4 Hz, 2H), 3.78 (s, 3H), 3.70 (t, J = 7.2 Hz, 2H), 2.79 (t, J = 7.2 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 161.0 (C-4″), 160.0 (C-2), 155.4 (C-4′), 130.0 (C-1′), 129.6 (C-2″, 6″), 129.3 (C-2′, 6′), 129.0 (C-1″), 114.9 (C-3′, 5′), 114.0 (C-3″, 5″), 62.3 (C-8′), 55.2 (C-OCH3), 36.2 (C-7′); MS (direct probe, positive EI) m/z 255.0 (M+), 148.1, 121.1; HREI-MS: Calculated for C16H17NO2; 255.1259, Observed; 255.1241; νmax; 2923.9, 1606.6, 1512.1, 1259.4, 1031.8, 821.6.
4-(2-(4-(Benzyloxy)benzylideneamino)ethyl)phenol (14)
1H-NMR (300 MHz, DMSO-d6): δ 9.13 (s, 1H), 8.14 (s, 1H), 7.64 (d, J = 8.7 Hz, 2H), 7.46 (t, J = 6.9 Hz, 2H), 7.41 (t, J = 6.6 Hz, 2H), 7.36 (t, J = 5.4 Hz, 1H), 7.06 (q, 4H), 6.64 (d, J = 8.1 Hz, 2H), 5.14 (s, 2H), 3.70 (t, J = 7.2 Hz, 2H), 2.78 (t, J = 7.2 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 160.1 (C-4″), 159.9 (C-2), 155.4 (C-4′), 136.7 (C-1″), 129.9 (C-1′), 129.6 (C-2′, 6′), 129.3 (C-2″, 6″), 129.1 (C-1″), 128.4 (C-3″, 5″), 127.8 (C-4″), 127.7 (C-2″, 6″), 114.9 (C-3′, 5′), 114.8 (C-3″, 5″), 69.3 (C-2a), 62.4 (C-8′), 36.2 (C-7′); MS (direct probe, positive EI) m/z 331.1 (M+), 224.1, 107.0, 91.1; HREI-MS: Calculated for C22H21NO2; 331.1572, Observed; 331.1563; νmax; 3429.2, 2860.2, 1647.1, 1606.6, 1255.6, 1037.6, 825.5.
4-(2-(2-Methoxybenzylideneamino)ethyl)phenol (15)
1H-NMR (300 MHz, DMSO-d6): δ 9.11 (s, 1H), 8.53 (s, 1H), 7.81 (dd, J = 1.2 Hz, 1H), 7.44 (m, J = 1.5 Hz, 1H), 7.07 (m, 4H), 6.69 (t, J = 13.2 Hz, 2H), 3.74 (t, J = 7.2 Hz, 2H), 2.79 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 158.3 (C-2″), 155.9 (C-2), 155.5 (C-4′), 132.0 (C-4″), 130.0 (C-1′), 130.0 (C-2′, 6′), 126.5 (C-6″), 124.0 (C-1′), 120.5 (C-5″), 115.0 (3′, 5′), 111.8 (C-3″), 63.0 (C-8′), 55.6 (C-CH3), 36.5 (C-7′); MS (direct probe, positive EI) m/z 255.0 (M+), 148.0, 121.0; HREI-MS: Calculated for C16H17NO2; 255.1259, Observed; 255.1243; νmax; 3446.6, 2902.7, 1602.7, 1456.2, 1380.9, 1249.8, 1026.1, 825.5.
4-(2-(3-Methoxybenzylideneamino)ethyl)phenol (16)
1H-NMR (300 MHz, DMSO-d6): δ 9.14 (s, 1H, OH), 8.20 (s, 1H, CH), 7.36 (t, J = 7.5 Hz, 1H), 7.26 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.4 Hz, 4H), 6.66 (d, J = 8.4 Hz, 3H), 3.77 (s, 3H), 3.74 (t, J8′,7′ = 7.5 Hz, 2H), 2.81 (t, J7′,8′ = 7.2 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 160.7 (C-2), 159.4 (C-3″), 155.4 (C-4′), 137.6 (C-1′), 129.8 (C-1′), 129.7 (C-5′), 129.6 (C-2′, 6′), 120.6 (C-6″), 116.6 (C-4″), 114.9 (C-3′, 5′), 111.9 (C-2″), 62.3 (C-8′), 55.1 (C-OCH3), 36.0 (C-7′); MS (direct probe, positive EI) m/z 255.1 (M+), 148.0, 121.0, 107.0; HREI-MS: Calculated for C16H17NO2; 255.1259, Observed; 255.1247; νmax; 3421.5, 2935.5, 1639.4, 1585.4, 1510.2, 1271.0,1242.1, 1039.6, 819.7.
4-(2-(4-Methylbenzylideneamino)ethyl)phenol (17)
1H-NMR (300 MHz, DMSO-d6): δ 9.13 (s, 1H, OH), 8.18 (s, 1H, CH), 7.59 (d, J = 7.8 Hz, 2H), 7.24 (d, J = 7.8 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.4 Hz, 2H), 3.73 (t, J = 7.2 Hz, 2H), 2.80 (t, J = 7.2 Hz, 2H), 2.32 (s, 3H, CH3); 13C-NMR (75 MHz, DMSO-d6): δ 160.6 (C-2), 155.5 (C-4′), 140.2 (C-4″), 133.6 (C-1″), 129.9 (C-1′), 1129.7 (2′, 6′), 129.2 (2″, 6″), 127.7 (C-3″, 5″), 114.9 (C-3′, 5′), 62.4 (C-8′), 36.1 (C-7′), 21.0 (CH3); MS (direct probe, positive EI) m/z 239.1 (M+), 132.0, 105.0; HREI-MS: Calculated for C16H17NO; 239.1310, Observed; 239.1305; νmax; 3421.6, 2937.3, 2852.5, 1645.2, 1510.2, 1452.3, 1380.9, 1238.2, 815.8.
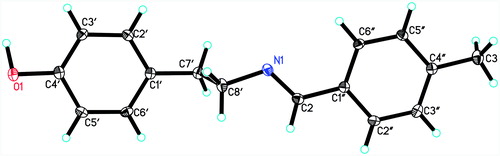
X-ray crystallographic analysis of compound 17: C16H17N1O1, Mr = 239.31, Orthorhomibic, space group P212121, a = 5.6936(10) Å, b = 14.614(3) Å, c = 15.597(3) Å, α3 = β = γ = 90°, V = 1297.8(4) Å3, Z = 4, ρcalc = 1.225 mg/m3, F(000) = 512, Mo Kαo = 0.71073 Å, max/min transmission 0.9909/0.9768, crystal dimensions 0.31 × 0.23 × 0.12, 1.91° < θ< 25.48°, 7105 reflections were collected, of which 2360 reflections were observed (Rint = 0.0258). The R values were: R1 = 0.0431, wR2 = 0.1224 for I > 2σ(I) and R1 = 0.0499, wR2 = 0.1649 for all data; max/min residual electron density: 0.670/−0.449e A°−3. Crystallographic data for compound 17 has been deposited in the Cambridge Crystallographic Data Center. The crystallographic information can be directly from CCDC Data Center (CCDC 1422584).
4-(2-(4-Cyanobenzylideneamino)ethyl)phenol (18)
1H-NMR (300 MHz, DMSO-d6): δ 9.12 (s, 1H), 8.32 (s, 1H), 7.92 (q, J = 8.7 Hz, J = 11.1 Hz, 4H), 7.02 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 3.81 (t, J = 6.9 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 159.8 (C-2), 155.5 (C-4′), 140.0 (C-1′), 132.6 (C-1′, 3″, 5″), 129.6 (C-2′, 6′), 128.3 (C-2″, 6″), 118.5 (C-CN), 114.9 (C-3′, 5′), 112.6 (C-4″), 62.4 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 251.0 (M+), 143.0, 116.0, 106.9; HREI-MS: Calculated for C16H14N2O; 250.1106, Observed; 250.1100; νmax; 3435.0, 2925.8, 1641.3, 1508.2, 1448.4, 1377.1, 1238.2, 1109.0, 829.3.
4-(2-(Naphthalen-1-ylmethyleneamino)ethyl)phenol (19)
1H-NMR (300 MHz, DMSO-d6): δ 9.11 (s, 1H), 8.99 (t, J = 2.4 Hz, 1H), 8.84 (s, 1H), 8.02 (q, J = 8.1 Hz, 2H), 7.87 (d, J = 6.9 Hz, 1H), 7.59 (m, 3H), 7.09 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 6.9 Hz, 1H), 3.89 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 7.2 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 161.1 (C-2), 155.5 (C-4′), 133.4 (C-1″), 131.3 (C-10″), 130.7 (C-1′), 130.6 (C-4″), 129.9 (C-9″), 129.8 (C-2′, 6′), 129.0 (C-5″), 128.5 (C-3″), 127.1 (C-6″), 126.1 (C-7″), 125.4 (C-2″), 124.6 (C-8″), 115.0 (C-3′, 5′), 63.3 (C-8′), 36.2 (C-7′); MS (direct probe, positive EI) m/z 275.2 (M+), 168.0, 141.1; HREI-MS: Calculated for C19H17NO; 275.1310, Observed; 275.1331; νmax: 3431.1, 2918.1, 1639.4, 1508.2, 1444.6, 1338.5, 1232.4, 1103.2, 813.9.
4-(2-(4-(Dimethylamino)benzylideneamino)ethyl)phenol (20)
1H-NMR (300 MHz, DMSO-d6): δ 9.09 (s, 1H), 8.05 (s, 1H), 7.50 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 6.71 (q, J = 8.7 Hz, 4H), 3.66 (t, J = 7.5 Hz, 2H), 2.77 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 160.2 (C-2), 155.4 (C-4′), 151.7 (C-2″, 4″), 130.1 (C-1′), 129.6 (C-2′, 6′), 129.0 (C-2″, 6″), 124.0 (C-1″), 114.9 (C-3′, 5′), 111.5 (C-3″, 5″), 62.5 (C-8′), 40.2 (N(CH3)2), 36.4 (C-7′); MS (direct probe, positive EI) m/z 268.2 (M+), 161.0, 146.1, 134.1, 117.0; HREI-MS: Calculated for C17H20N2O; 268.1576, Observed; 268.1596; νmax; 2898.8, 2852.5, 1610.5, 1514.0, 1452.3, 1373.2, 1242.1, 1178.4, 812.0.
2-((4-Hydroxyphenethylimino)methyl)phenol (21)
1H-NMR (300 MHz, DMSO-d6): δ 13.52 (s, 1H), 9.15 (s, 1H), 8.44 (s, 1H), 7.37 (d, J = 6.6 Hz, 1H), 7.32 (m, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.87 (t, J = 6.9 Hz, 2H), 6.67 (d, J = 8.4 Hz, 2H), 3.79 (t, J = 6.9 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 165.8 (C-2″), 160.8 (C-2), 155.6 (C-4′), 132.2 (C-6″), 131.5 (C-1′), 129.6 (C2′, 6′), 129.3 (C-4″), 118.5 (C-1′), 118.3 (C-5″), 116.4 (C-3″), 115.1 (C-3′, 5′), 60.0 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 241.2 (M+), 134.1, 107.1; HREI-MS: Calculated for C15H15NO2; 241.1103, Observed; 241.1093; νmax; 3377.1, 2925.8, 1635.5, 1448.4, 1380.9, 1230.5, 1159.1, 1004.8, 839.0.
5-((4-Hydroxyphenethylimino)methyl)-2-methoxyphenol (22)
1H-NMR (300 MHz, DMSO-d6): δ 9.10 (s, 1H), 8.09 (s, 1H), 7.29 (s, 1H), 7.08 (m, 3H), 6.80 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 8.4 Hz, 2H), 3.78 (s, 3H), 3.68 (t, J = 7.2 Hz, 2H), 2.78 (t, J = 7.5 Hz, 2H); 271.0, 164.0; 13C-NMR (100 MHz, DMSO-d6): δ 160.3 (C-2), 155.4 (C-4′), 149.1 (C-4″), 147.8 (C-5″), 130.0 (C-1″), 129.6 (C-2′, 6′), 127.9 (C-1′), 122.4 (C-2″), 115.1 (C-3″), 114.9 (C-3′, 5′), 109.8 (C-6″), 62.3 (C-8′), 55.4 (OCH3), 36.3 (C-7′); MS (direct probe, positive EI) m/z 271.0 (M+), 164.0, 132, 107, 82.9; HREI-MS: Calculated for C16H17NO3; 271.1208, Observed; 271.1214; νmax; 3440.8, 2956.7, 1662.5, 1585.4, 1515.9, 1431.1, 1323.1, 1232.4, 1128.3, 1020.3, 821.6.
4-(2-(2-Chloro-4-fluorobenzylideneamino)ethyl)phenol (23)
1H-NMR (300 MHz, CD3OD): δ 8.44 (s, 1H), 7.97 (d, J6″,5″ = 6.3 Hz, 1H), 7.26 (dd, J = 2.7 Hz, J = 2.4 Hz, 1H), 7.17 (m, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.69 (d, J = 8.7 Hz, 2H), 3.86 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 164.7 (C-4″), 161.4 (C-8′), 155.9 (C-4′), 155.5 (C-2″), 134.8, 134.6 (C-1′), 129.9 (C-6″), 129.8 (C-1′), 129.7 (C-2′, 6′), 117.1, 116.7 (C-3″), 115.3 (C-5″), 114.9 (C-3′, 5′), 62.4 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 279.1 ([M + 2]+), 277.1 (M+), 170.0, 143.0, 107.0; HREI-MS: Calculated for C15H13ClFNO; 277.0670, Observed; 277.0673; νmax; 3078.2, 2916.2, 1639.4, 1596.9, 1512.1, 1450.4, 1380.9, 1238.2, 1029.9, 908.4, 8, 821.6.
4-(2-(2,4-Dichlorobenzylideneamino)ethyl)phenol (24)
1H-NMR (400 MHz, DMSO-d6): δ 9.14 (s, 1H, OH), 8.48 (s, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.68 (d, J = 1.6 Hz, 1H), 7.49 (dd, J = 1.6 Hz, J = 1.2 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.65 (d, J = 8.0 Hz, 2H), 3.82 (t, J = 7.2 Hz, 2H), 2.82 (t, J = 7.2 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 156.1 (C-2), 155.5 (C-4′), 135.8 (C-1″), 134.6 (C-1′), 131.7 (C-2″), 129.7 (C-2′, 6′), 129.6 (C-3″), 129.3 (C-6″), 127.9 (C-4′), 115.0 (C-3′, 5′), 62.5 (C-8′), 35.8 (C-7′); MS (direct probe, positive EI) m/z 295.0 ([M + 2]+), 293.0 (M+), 186.0, 159.0, 107.0; HREI-MS: Calculated for C15H13Cl2NO; 293.0374, Observed; 293.0368, νmax; 2922.0, 1637.5, 1591.2, 1510.2, 1450.4, 1377.1, 1236.3, 1101.3, 1035.7, 817.8.
4-Chloro-2-(((4-hydroxyphenethyl)imino)methyl)phenol (25)
1H-NMR (300 MHz, DMSO-d6): δ 13.55 (s, 1H, OH), 9.18 (s, 1H, OH), 8.42 (s, 1H), 7.47 (d, J = 2.4 Hz, 1H), 7.32 (d, J = 2.4 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.8 Hz, 1H), 6.66 (d, J = 8.0 Hz, 2H), 3.78 (t, J = 7.2 Hz, 2H), 2.83 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 164.7 (C-2″), 160.0 (C-2), 155.7 (C-4′), 131.9 (C-4″), 130.5 (C-1′), 129.7 (C-2′, 6′), 129.2 (C-6″), 121.5 (C-1″), 119.5 (C-5″), 118.7 (C-3″), 115.1 (C-3′, 5′), 59.8 (C-8′), 35.7 (C-7′); MS (direct probe, positive EI) m/z 277.0 ([M + 2]+), 275.0 (M+), 168.0, 141.0, 107.1, 91.1, 77.1; HRESI-MS: Calculated for C15H14ClNO2; 275.0713, Observed; 275.0791; νmax; 2806.2, 2607.6, 1643.2, 1515.9, 1460.0, 1259.4, 1170.7, 1016.4, 829.3.
4-Fluoro-2-(((4-hydroxyphenethyl)imino)methyl)phenol (26)
1H-NMR (300 MHz, DMSO-d6): δ 13.25 (bs, 1H, OH), 9.17 (bs, 1H, OH), 8.42 (s, 1H), 7.28 (d, J = 3.0 Hz, 1H), 7.19 (m, 1H), 7.03 (d, J = 9.0 Hz, 2H), 6.88 (q, J = 3.0 Hz, 1H), 6.67 (d, J = 9.0 Hz, 2H), 3.79 (t, J = 6.0 Hz, 2H), 2.84 (t, J = 9.0 Hz, 2H); 13C-NMR (75 MHz, DMSO-d6): δ 164.7, 164.6 (C-2, J = 3.0 Hz), 156.9, 156.9 (C-2″, J = 0.7 Hz), 156.0, 155.6 (C-5″, J = 32.25 Hz), 152.9 (C-4′), 129.6 (C-2′, 6′), 129.2 (C-1′), 119.2, 118.9 (C-1″, J = 23.2 Hz), 118.6, 118.5 (C-4″, J = 6.7 Hz), 117.7, 117.6 (C-3″, J = 7.5 Hz), 116.6, 116.3 (C-6″, J = 23.2 Hz), 115.1 (C-3′, 5′), 60.1 (C-8′), 35.7 (C-7′); MS (direct probe, positive EI) m/z 259.2 (M+), 152.1, 125.0, 106.9, 94.9, 76.9; HRESI-MS: Calculated for C15H14NO2F: 259.1009, Observed: 259.1086; νmax; 2925.8, 1614.3, 1496.7, 1454.2, 1245.9, 1151.4, 1010.6, 835.1, 767.6.
2-Bromo-4-chloro-6-(((4-hydroxyphenethyl)imino)methyl)phenol (27)
1H-NMR (300 MHz, DMSO-d6): δ 14.55 (bs, 1H, OH), 9.23 (s, 1H, OH), 8.40 (s, 1H), 7.69 (d, J = 3.0 Hz, 1H), 7.39 (d, J = 3.0 Hz, 1H), 7.03 (d, J = 6.0 Hz, 2H), 6.68 (d, J = 6.0 Hz, 2H), 3.83 (t, J = 6.0 Hz, 2H), 2.88 (t, J = 9.0 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 165.2 (C-2″), 163.8 (C-2), 155.8 (C-4′), 135.6 (C-4″), 130.8 (C-1′), 129.6 (C-2′, 6′), 128.3 (C-6″), 117.5 (C-5″), 116.5 (C-1″), 115.2 (C-3′, 5′), 114.9 (C-3″), 55.4 (C-8′), 34.9 (C-7′); MS (direct probe, positive EI) m/z 357.2 ([M + 2]+), 355.2 (M+), 248.1, 221.1, 167.1, 107.1, 77.1, 44.0; HRESI-MS: Calculated for C15H13BrClNO2; 354.6262, Observed; 355.0052; νmax; 3008.7, 1637.5, 1492.8, 1369.4, 1228.6, 1139.9, 1043.4, 831.3.
α-Glucosidase inhibitory activity
α-Glucosidase (Saccharomyces cerevisiae, EC 3.2.1.20), (Sigma catalog # G0660) inhibitory activity was evaluated by using 0.1 M phosphate buffer (pH 6.8) at 37 °C. The enzyme (0.2 U/mL) in phosphate buffer saline was incubated with various concentrations of test compounds at 37 °C for 15 min. About 0.7 mM of the substrate, p-nitrophenyl-α-d-glucopyranoside, was added absorbance change at 400 nm was recorded up to 30 min by multiplate reader. DMSO (7.0% final) was used as control. Acarbose was used as positive control. Following formula was used to calculate the % inhibition:Citation20
DPP-IV inhibitory assay
Dipeptidyl peptidase (DPP-IV) (Human), (E.C. 3.4.1.4.5) inhibitory activity was evaluated by using glycine-proline-p-nitroaniline (Gly-Pro-PNA) as substrate, and sitagliptin as standard inhibitor. Test samples (10 μL) were pipetted onto a 96-well microplate along with DPP-IV (0.025 /mL) and Tris-HCl buffer pH 8.0 were preincubated for 20 min at 37 °C. Reaction was initiated with the addition of substrate (0.2 mM). The microplate was then incubated for 50 min at 37 °C. (Spectramax 384, Molecular Devices, Sunnyvale, CA,USA).
Absorbance was measured at 405 nm. The following formula was used to calculate the % inhibition:Citation21
Protein glycation inhibition assay
Bovine serum albumin (10 mg/mL), methyl glyoxal (14 mM), various concentrations of the test compounds (prepared in DMSO, 10% final concentration) and 0.1 M phosphate buffer (pH 7.4) containing sodium azide (30 mM) was incubated under aseptic conditions at 37 °C for 9 days. After 9 days, each sample was examined for the development of specific fluorescence (excitation, 330 nm; emission, 420 nm) against sample blank. Rutin was used as a positive control. All of the experiments were carried out in a 96-well microplate reader (SpectraMax M5, Molecular Devices, USA). The percent inhibition of AGEs formation in the test sample versus control was calculated for each inhibitor compound by using the following formula:Citation22
Cytotoxicity evaluation on 3T3 cells
3-[4, 5-Dimethylthiazole-2-yl]-2, 5-diphenyl-tetrazolium bromide (MTT) assay was used to determine the cytotoxicity of the compounds. Mouse fibroblast 3T3 cells were grown in Dulbecco’s modified Eagle medium (DMEM), along with 5% fetal bovine serum (FBS), 100 μg/mL of streptomycin and 100 IU/mL of penicillin in 75-cm2 flasks. The flasks were then incubated at 37 °C in 5% CO2. Cells were harvested, followed by counting with hemocytometer and dilution with a medium. Cell culture was prepared with the concentration of 5 × 104 cells/mL, followed by introduction (100 μL/well) into 96-well plates. Medium was removed after overnight incubation. Freshly prepared medium (200 μL) was added with various concentrations of test compounds (1–30 μM). After 48 h, 200 μL MTT dye (0.5 mg/mL) was added into all wells and again incubated for 4 h. DMSO (100 μL) was added to each well. The formation of formazan dye within the cells was calculated by measuring the absorbance at 540 nm, using a multiplate reader (SpectraMax plus, Molecular Devices). The cytotoxicity was estimated as the concentration causing 50% inhibition of 3T3 cells (IC50). Following formula was used to calculate the % inhibition:
The data were processed by using a software called Soft-Max Pro (Molecular Devices).Citation23
Statistical analysis
The EZ-Fit Enzyme Kinetics Program (Perrella Scientific Inc., Amherst, USA) was employed to calculate the IC50 values. All the experiments were performed in triplicate, and results are reported as the ± Standard Error of Mean values.
Results and discussion
Chemistry
Schiff base derivatives of tyramine 3–27 were synthesized in a single step by using various aldehydes. Both conventional and ultrasonic methods were used for the synthesis. In the conventional method, resulting compounds were purified by solvent–solvent extraction and obtained in good to excellent yields. In a few cases, the compounds were purified by using normal phase column chromatography. It was observed that ultrasonic procedure (with or without heating) not only provides excellent yields, but no major purification of resultant compounds was required. After completion of the reaction, the solvent was evaporated, and the washing yielded the resultant products. shows comparison of reaction time and yields between the conventional and sonication (with or without heating) techniques for selected compounds.
The stereochemistry of the products was deduced by NOESY-NMR and single-crystal X-ray diffraction analyses. shows the key NOESY interactions in the compounds 21 and 17. In compound 21, strong NOE correlations were observed between the C-2 olefinic proton and the C-6″ aromatic proton. NOE between C-2 olefinic proton and methylenic protons at C-8′ was also observed. These correlations indicated an E configuration of the C=N double bond. The NOE correlation between the two hydroxyls at C-4′ and C-2″ may well because of the intramolecular chemical exchange rather than NOECitation18.
Figure 1. Key NOESY correlations of compounds 17 and 21.
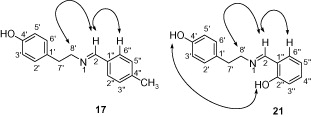
To further study the stereochemisty of the double bond, NOESY experiment was performed on another member of the series (compound 17). A similar NOESY correlation was observed between C-2 olefinic proton and C-6″/C-2″ aromatic proton and C-8′ methylene protons. Single-crystal X-ray diffraction analysis of compound 17 unambiguously established the E configuration of the C=N double bond in compounds 3–27 ().
Figure 2. ORTEP drawing of the single-crystal X-ray structure of compound 17.
Bioactivity
α-Glucosidase obtained from yeast has been widely used for the identification of in vitro inhibitors of α-glucosidase enzyme. However, mammalian α-glucosidase (rat intestinal acetone powder) activity may serve as better test model for the identification, design and development of antidiabetic agents, because of its close similarities with the mammalian systems. α-Glucosidase enzymes from bacterial, yeast and mammalian sources are known to have fairly conserved active sitesCitation19. Therefore, compounds 3–27 have been studied on both assays.
Compounds 3–27 were evaluated for their α-glucosidase (Saccharomyces cerevisiae) inhibitory activity in vitro (). Compounds 7, 14, 20, 21 and 26 were found to be more active (IC50 = 49.7 ± 0.4, 318.8 ± 3.7, 23.5 ± 0.9, 302.0 ± 7.3 and 230.7 ± 4.0 μM, respectively) than the standard drug, acarbose (IC50 = 840.0 ± 1.73 μM), while other members of the series exhibited less than 50% inhibition. Compounds 3–27 were also screened against α-glucosidase obtained from mammalian source (rat-intestinal acetone powder) also. We observed that compounds 20 and 22 possess good inhibitory activity with the IC50 = 175.4 ± 3.17 and 173.9 ± 3.8 μM, respectively, compared with standard drug, acarbose (IC50 = 67.1 ± 4.96 μM (Observed) and IC50 = 52.8 μM or 34.11 μg/mL (reported)). It is interesting to note that compound 20 was found to be active against both mammalian and yeast α-glucosidase, while compound 22 was found to be active against mammalian α-glucosidase only. Similarly, compound 7, which was just about 2-fold less active than compound 20, did not inhibit the mammalian α-glucosidase. The limited data presented here suggested that the structure–activity–relationship (SAR) for the yeast and mammalian (rat-intestinal acetone powder) α-glucosidases is different; however, certain correlations were also observed, for example, compound 20, the most active member of the series (IC50 = 23.5 ± 0.9 μM) against α-glucosidase from Saccharomyces cerevisiae was also found to be active against α-glucosidase obtained from mammalian rat-intestinal acetone powder as well (IC50 = 175.4 ± 3.17 μM).
Table 2. α-glucosidase inhibitory activity, and cytotoxicity of compounds 3–27.
Compound 20 was found to be the most active member of the series (IC50 = 23.5 ± 0.9 μM), which possess electron-donating dimethylamino group at the para or C-4″ position. The activity decreased to about half (IC50 = 49.7 ± 0.4 μM) when a chloro group was present at C-4″ position (as in compound 7). This may be due to negative inductive effect of the halogens. Change in the position of the chloro substituent from para to the ortho and meta positions, resulted in a complete loss of activity, as in compounds 8 and 10. The presence of the chloro groups at the C-4″ and C-2″ also resulted in a complete loss of activity (as in compound 24).
Compound 26 was found to be another active member of the series (IC50 = 230.7 ± 4.0 μM), with hydroxyl and fluoro groups at C-2″ and C-5″ positions, respectively. The activity decreased many folds when there was a substitution of a benzyloxy group at the C-4″ position (as in compound 14, IC50 = 318.8 ± 3.7 μM). Substitution of a methoxy group at the C-4″ and C-2″ positions (ortho and meta) resulted in a complete loss of activity, as in compounds 15 and 16. Although the methoxy group is also electron donating but the high electro-negativity of oxygen may be a reason for the lower activities of these compounds, in comparison to compounds 20 and 26, which also possess electron-donating groups.
Interestingly, among all the compounds with substitution at C-2″, only compound 21 showed enzyme inhibition activity (IC50 = 302.0 ± 7.3 μM).
In order to further study the antidiabetic potential of the compounds 3–27, they were evaluated for their protein glycation and DPP IV inhibition activities. Only compounds 20, 22 and 27 showed weak antiglycation activity with IC50 values 505.27 ± 5.95, 581.87 ± 5.50 and 440.58 ± 2.74 μM, respectively (). All other compounds were found to be inactive in both the assays.
Table 3. Antiglycation activityof compounds 3–27.
The cytotoxicity of compound 3–27 was evaluated against the mouse fibroblast 3T3 cell lines. All compounds were turned out to be noncytotoxic ().
Kinetic studies
In order to study the mechanism of inhibition, kinetics studies were performed on the most active compounds 7, 20 and 26. The study showed them to be competitive inhibitors with Ki values 60, 9 and 89 μM, respectively (). Lineweaver–Burk plots were used to determine the type of inhibition. The reciprocals of the rate of the reaction were plotted against the reciprocal of substrate concentrations to monitor the effect of the inhibitor on both Km and Vmax values. showed no effect on Vmax of α-glucosidase enzyme in the presence of different concentrations of compounds 7, 20 and 26, while the Km values increased. This showed a competitive behavior. The secondary replots of Lineweaver–Burk plots were plotted to determine the Ki values (). By plotting the slope of each line in the Lineweaver–Burk plots against different concentrations of compounds 7, 20 and 26, the Ki values were calculated. The Ki values were deduced from Dixon plots, by plotting the reciprocal of the rate of reaction against different concentrations of compounds 7, 20 and 26 ().
Figure 3. The inhibition of α-glucosidase enzyme by compound 7, (A) is the Lineweaver–Burk plot of reciprocal of rate of reaction (velocities) versus reciprocal of substrate (p-nitrophenyl α-d-glucopyranoside) in the absence of inhibitor and in the presence of 125.00 μM (○), 62.5 μM (•), 31.25 μM (□), 7.8 μM (Δ) and 3.9 μM (▴) of compound 7. (B) Is the secondary re-plot of Lineweaver–Burk plot between the slopes of each line on Lineweaver–Burk plot versus different concentrations of compound 7, (C) is the Dixon plot of reciprocal of rate of reaction (velocities) versus different concentrations of compound 7.
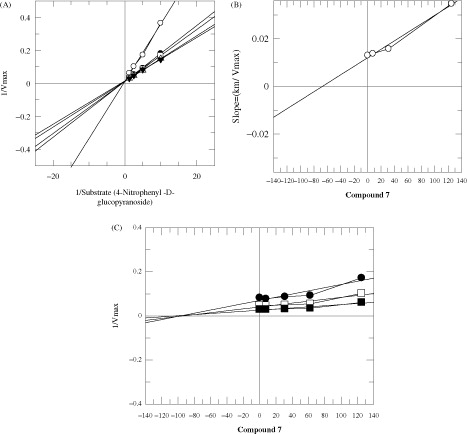
Figure 4. The inhibition of α-glucosidase enzyme by compound 20 (A) is the Lineweaver–Burk plot of reciprocal of rate of reaction (velocities) versus reciprocal of substrate (p-nitrophenyl α-d-glucopyranoside) in the absence of inhibitor and in the presence of 31.25 μM (○), 25 μM (•), 15 μM (□), 10 μM (▪) and 7.8 μM (Δ) of compound 20. (B) Is the secondary re-plot of Lineweaver–Burk plot between the slopes of each line on Lineweaver–Burk plot versus different concentrations of compound 20, (C) is the Dixon plot of reciprocal of rate of reaction (velocities) versus different concentrations of compound 20.
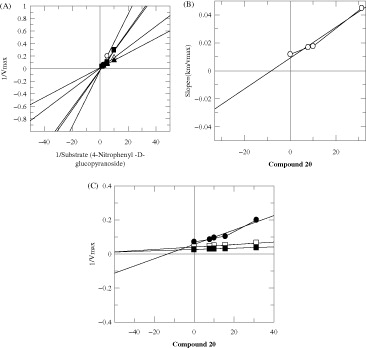
Figure 5. The inhibition of α-glucosidase enzyme by compound 26 (A) is the Lineweaver–Burk plot of reciprocal of rate of reaction (velocities) versus reciprocal of substrate (p-nitrophenyl α-d-glucopyranoside) in the absence of inhibitor and in the presence of 200.00 μM (○), 125 μM (•), 62.5 μM (□), 31.25 μM (Δ) and 15.625 μM (▴) of compound 26 (B) is the secondary replot of Lineweaver–Burk plot between the slopes of each line on Lineweaver–Burk plot versus different concentrations of compound 26, (C) is the Dixon replot of Ki.
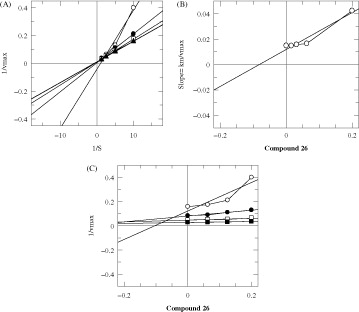
Table 4. The mode of inhibition of compounds 7, 20 and 26.
Conclusion
In conclusion, we have synthesized a class of novel α-glucosidase inhibitors, based on a natural product template, tyramine. The compounds reported here may serve as the starting points for the design and development of novel α-glucosidase inhibitors as anti-diabetic agents.
Declaration of interest
The authors have declared that there are no conflicts of interest.
Related Research Data
References
- van de Laar FA, Lucassen PL, Akkermans RP, et al. α-Glucosidase inhibitors for patients with type 2 diabetes results from a cochrane systematic review and meta-analysis. Diabetes Care 2005;28:154–63
- Chai T, Mohan M, Ong H, Wong F. Antioxidant, iron-chelating and anti-glucosidase activities of Typha domingensis Pers (Typhaceae). Trop J Pharm Res 2014;13:67–72
- Ajish KR, Antu KA, Riya MP, et al. Studies on α-glucosidase, aldose reductase and glycation inhibitory properties of sesquiterpenes and flavonoids of Zingiber zerumbet Smith. Nat Prod Res 2015;29:947–52
- (a) Hanefeld M, Cagatay M, Petrowitsch T, et al. Acarbose reduces the risk for myocardial infarction in type 2 diabetic patients: meta-analysis of seven long-term studies. Eur Heart J 2004;25:10–16 (b) Ihara Y, Toyokuni S, Uchida K, et al. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999;48:927–32 (c) Drent M, Tollefsen A, Van Heusden F, et al. Dose-dependent efficacy of miglitol, an alpha-glucosidase inhibitor, in type 2 diabetic patients on diet alone: results of a 24-week double-blind placebo-controlled study. Diabetes, Nutr Metab 2002;15:152–9 (d) Kim YM, Wang MH, Rhee HI. A novel α-glucosidase inhibitor from pine bark. Carbohydr Res 2004;339:715–17
- Qu YH, Yang JH, Chen SS. Synthesis of octopamine derivatives and investigation of their antioxidation activities. Chin J Mar Drugs 2012;3:006
- Bryar BA, Fregly MJ, Field FP. Changes in vascular responsiveness following chronic exposure to cold in the rat. J Appl Physiol 1983;55:823–9
- Kim J, Hirano M, Uchimura H, et al. Effects of thyroidectomy on monoamine oxidase activities toward tyramine and serotonin in the circumventricular nuclei of the rat. Experientia 1979;35:302–3
- Lino C, Sales T, Gomes P, et al. Anti-diabetic activity of a fraction from Cissus verticillata and tyramine, its main bioactive constituent, in alloxan-induced diabetic rats. Am J Pharmacol Toxicol 2007;2:178–88
- Patel MS, Patel DD, Patel VS, et al. Synthesis of Schiff bases of N-based methylene derivatives. Int J Sci Res 2014;2:580–5
- Ali E, Naimi-Jamal M, Dekamin M. Highly efficient and rapid synthesis of imines in the presence of nano-ordered MCM-41-SO 3 H heterogeneous catalyst. Scientia Iranica 2013;20:592–7
- da Silva CM, da Silva DL, Modolo LV, et al. Schiff bases: a short review of their antimicrobial activities. J Adv Res 2011;2:1–8
- (a) Bendale AR, Bhatt R, Nagar A, et al. Schiff base synthesis by unconventional route: an innovative green approach. Der Pharma Chemica 2011;3:34–8 (b) Nikpassand M, Fekri LZ, Sharafi S. An efficient and green synthesis of novel azo Schiff base and its complex under ultrasound irradiation. Orient J Chem 2013;29:1041–6
- (a) Markwardt F, Landmann H, Walsmann, et al. Comparative studies on the inhibition of trypsin, plasmin, and thrombin by derivatives of benzylamine and benzamidine. Eur J Biochem 1968;6:502–6 (b) Soman G, Narayanan J, Martin BL, Graves DJ. Use of substituted (benzylidineamino) guanidines in the study of guanidino group-specific ADP-ribosyltransferase. Biochemistry 1986;25:4113–19
- Sawada K, Okada S, Kuroda A, et al. 4-(Benzoylindolizinyl) butyric acids; novel nonsteroidal inhibitors of Steroid 5. ALPHA.-reductase. III. Chem Pharm Bull 2001;49:799–813
- Kouam SF, Khan SN, Krohn K, et al. α-Glucosidase inhibitory anthranols, kenganthranols AC, from the stem bark of Harungana m adagascariensis. J Nat Prod 2006;69:229–33
- Herscovics A. Importance of glycosidases in mammalian glycoprotein biosynthesis. Biochimica Et Biophysica Acta (BBA)-General Subjects 1999;1473:96–107
- Shihabudeen HMS, Priscilla DH, Thirumurugan K. Cinnamon extract inhibits α-glucosidase activity and dampens postprandial glucose excursion in diabetic rats. Nutr Metab 2011;8:46 (p11)
- Mori S, Berg JM, van Zijl PC. Separation of intramolecular NOE and exchange peaks in water exchange spectroscopy using spin-echo filters. J Biomolecul NMR 1996;7:77–82
- Yamamoto K, Nakayama A, Yamamoto Y, Tabata S. Val216 decides the substrate specificity of α‐glucosidase in Saccharomyces cerevisiae. Eur J Biochem 2004;271:3414–20
- Choudhary MI, Shah S, Khan SN, Khan MTH. Alpha-glucosidase and tyrosinase inhibitors from fungal hydroxylation of tibolone and hydroxytibolones. Steroids 2010;75:956–66
- Nongonierma AB, Mooney C, Shields DC, FitzGerald RJ. Inhibition of dipeptidyl peptidase IV and xanthine oxidase by amino acids and dipeptides. Food Chem 2013;141:644–53
- (a) Choudhary MI, Basha FZ, Abbas G, et al. Science at the interface of chemistry and biology: discoveries of α-glucosidase inhibitors and antiglycation agents. Pure ApplChem 2007;79:2263–8 (b) Cho SJ, Roman G, Yeboah F, Konishi Y, The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem 2007;14:1653–71
- Pauwels R, Balzarini J, Baba M, et al. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 1988;20:309–21