
Abstract
1,3,4-Thiadiazole was explored as a more polar, heterocyclic replacement for the phenyl ring in the 3-arylpropionic acid pharmacophore present in the majority of GPR40 agonists. Out of 13 compounds synthesized using a flexible, three-step protocol (involving no chromatographic purification), four compounds were confirmed to activate the target in micromolar concentration range. While the potency of the series should be subject of further optimization, the remarkable aqueous solubility and microsomal stability observed for the lead compound (8g) apparently attests to this new scaffold’s high promise in the GPR40 agonist field.
Introduction
Activation of free fatty acid receptor 1 (FFAR1, also known as GPR40) in pancreatic Islets of Langerhans, where it is highly expressed in hyperglycemic state, leads to a cascade of events that culminate in increased levels of insulin and eventual lowering of the glucose levelsCitation1. This mechanism of achieving the latter does not carry the danger of causing hypoglycemia (i.e. lowering of blood glucose concentration below physiologically acceptable levels). Therefore, agonists of GPR40 represent a therapeutically attractive alternative to the currently available treatments for type 2 diabetes mellitus (T2DM)Citation2. Unfortunately, the field of GPR40 agonist-based therapies was struck severely in late-2013 with discontinuation, due to idiosyncratic liver toxicity, of phase-III trial of Takeda’s frontrunner compound fasiglifam (TAK-875)Citation3. Currently, only one clinical investigation of a GPR40 agonist is underway, namely that of compound P11187 of undisclosed structure by PiramalCitation4. However, the proof of principle of the new approach to treat T2DM was, in principle, established in the course of TAK-875 investigationCitation5. Thus, current research effort worldwideCitation6 is aimed primarily at tackling the toxicity profile of GPR40 agonists.
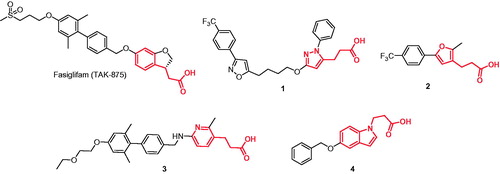
One of the ways to achieve the latter is to increase the total polar surface area (TPSA, Å2) of GPR40 agonists. Liver toxicity, of TAK-875 in particular, has been linked to the overly lipophilic character of the compoundCitation6. Indeed, TAK-875 and many other advanced compounds of this class are based on 3-phenylpropanoic scaffold and aim to mimic the endogenous ligands of GPR40 which are fatty acids. A much exploited strategy pursued in the design on higher-TPSA GPR40 agonists is to use more polar moieties (such as various heterocycles) to “decorate” compounds’ peripheryCitation7. The other, less popular approach is to replace 3-phenylpropanoic acid core with heterocyclic congeners. Compounds of this sort are a lot less abundant in the literature and can be exemplified by Takeda’s compounds 1Citation8, 2Citation9 and 3Citation10 as well as Amgen’s indole-based compound 4Citation11 (). In this article, we present our results related to the design, the synthesis of a series of hitherto unreported compounds containing 3–(1,3,4-thiadiazol-2-yl)propionic acid moiety (cLogP = −0.09) in lieu of 3-phenylpropanoic acid moiety (cLogP = 1.84)Citation12. Herein, we also report the results of characterization of the series as agonists of GPR40 receptor and determination of key ADMET properties of the most promising compounds.
Figure 1. Structures of TAK-875 and of the heterocyclic analogs of 3-phenylpropanoic acid-based GPR40 agonists 1–4.
Materials and methods
Chemical syntheses – general
All reactions were conducted in oven-dried glassware in atmosphere of nitrogen. Melting points were measured with a Buchi В-520 melting point apparatus and were not corrected. Analytical thin-layer chromatography (TLC) was carried out on Silufol UV-254 silica gel plates using appropriate mixtures of ethyl acetate and hexane. Compounds were visualized with short-wavelength UV light. 1H NMR and 13C NMR spectra were recorded on Bruker MSL-300 spectrometers in DMSO-d6 using TMS as an internal standard. Mass spectra were recorded using Shimadzu LCMS-2020 system with electron impact (EI) ionization. All and reagents and solvents were obtained from commercial sources and used without purification.
General procedure for the preparation of compounds 7a--m12
To a suspension of elementary sulfur (1024 mg, 32.0 mmol) in dry dimethylformamide (DMF) (40 mL) was sequentially added (in dropwise fashion) triethylamine (4.45 mL, 32.0 mmol) and morpholine (2.12 mmol) and the resulting mixture was stirred for 30 min. Then, it was treated with a solution of respective 2-chloroacetamides 5 (1.0 mmol), all of which are known compounds. The mixture was stirred overnight, poured into water (100 mL) and the resulting precipitate was separated by filtration and air-dried. It was then suspended in acetone (100 mL) and the insoluble residue of excess of unreacted sulfur was filtered off and discarded. The filtrate was evaporated to dryness and the dry residue (predominantly consisting of 6) was dissolved in dry DMF (30 mL), treated with hydrazine hydrate (5.0 mL) and stirred for 12 h. The reaction mixture was poured into water, and the pH of the aqueous medium was adjusted to 5.0 with 2 M aqueous HCl. The resulting precipitate was filtered off, washed with water, air-dried and crystallized from ethanol to deliver analytically pure compounds seven in yields indicated.
N(1)–(4-methylphenyl)-2-hydrazino-2-thiooxacetamide (7a)
Yield 55%, yellow solid, m.p. 155–158°C; 1H NMR (300 MHz, DMSO-d6) δ 10.13 (s, 1H), 7.62 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 2.27 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 167.73, 158.15, 135.41, 134.16, 129.70, 120.49, 20.98.
N(1)– (2,4-difluorophenyl)-2-hydrazino-2-thiooxacetamide (7b)
Yield 71%, yellow solid, m. p. 174–178°C; 1H NMR (300 MHz, DMSO-d6) δ 10.19 (s, 1H), 7.91 (tt, J = 19.2, 9.6 Hz, 1H), 7.41 (ddd, J = 11.5, 9.0, 2.8 Hz, 1H), 7.28–7.04 (m, 1H), 7.19–7.08 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 165.81 (s), 159.51 (dd, J = 244.7, 11.7 Hz), 157.88 (s), 154.63 (dd, J = 248.4, 12.7 Hz), 125.01 (dd, J = 9.7, 2.3 Hz), 122.18 (dd, J = 11.5, 3.7 Hz), 111.93 (dd, J = 22.1, 3.7 Hz), 104.85 (dd, J = 27.1, 23.8 Hz).
N(1)-Phenyl-2-hydrazino-2-thiooxacetamide (7c)
Yield 55%, yellow solid, m. p. 152–156°C; 1H NMR (300 MHz, DMSO-d6) δ 10.21 (s, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.36 (t, J = 7.9 Hz, 5H), 7.14 (t, J = 7.4 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 167.73, 158.39, 137.91, 129.31, 125.03, 120.54.
N(1)– (3-chlorophenyl)-2-hydrazino-2-thiooxacetamide (7d)
Yield 64%, yellow solid, m. p. 162–166°C; 1H NMR (300 MHz, DMSO-d6) δ 10.38 (s, 1H), 7.94 (t, J = 2.0 Hz, 1H), 7.73–7.68 (m, 1H), 7.38 (t, J = 8.1 Hz, 1H), 7.20 (dd, J = 7.8, 1.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 167.62, 158.95, 139.49, 133.54, 130.93, 124.71, 120.17, 119.24.
N(1)– (4-methoxyphenyl)-2-hydrazino-2-thiooxacetamide (7e)
Yield 77%, yellow solid, m. p. 168–172°C; 1H NMR (300 MHz, DMSO-d6) δ 10.10 (s, 1H), 7.66 (t, J = 6.2 Hz, 2H), 6.92 (t, J = 6.1 Hz, 1H), 3.73 (s, 1H); 13C NMR (75 MHz, DMSO-d6) δ 167.79, 157.96, 156.59, 131.03, 122.15, 114.39, 55.68
N(1)– (2-fluorophenyl)-2-hydrazino-2-thiooxacetamide (7f)
Yield 70%, yellow solid, m. p. 172–176°C; 1H NMR (300 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.05 (ddd, J = 7.8, 5.6, 2.9 Hz, 1H), 7.40–7.29 (m, 1H), 7.28–7.19 (m, 2H); 13C NMR (75 MHz, DMSO-d6) δ 165.68 (s), 157.48 (s), 153.95 (d, J = 244.9 Hz), 126.59 (d, J = 7.8 Hz), 125.52 (d, J = 10.9 Hz), 125.25 (d, J = 3.6 Hz), 122.77 (s), 116.02 (d, J = 19.0 Hz).
N(1)–(3-methylphenyl)-2-hydrazino-2-thiooxacetamide (7 g)
Yield 59%, yellow solid, m. p. 115–119°C; 1H NMR (300 MHz, DMSO-d6) δ 10.12 (s, 1H), 7.56 (s, 2H), 7.25 (dd, J = 11.4, 4.8 Hz, 1H), 6.96 (d, J = 7.3 Hz, 1H), 2.30 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 167.76, 158.20, 138.64, 137.78, 129.18, 125.75, 120.95, 117.57, 21.59.
N(1)– (4-fluorophenyl)-2-hydrazino-2-thiooxacetamide (7 h)
Yield 62%, yellow solid, m. p. 179–183°C; 1H NMR (300 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.78 (ddd, J = 8.5, 5.2, 2.9 Hz, 1H), 7.23–7.16 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 167.88 (s), 159.24 (d, J = 241.5 Hz), 158.53 (s), 134.35 (d, J = 2.6 Hz), 122.70 (d, J = 8.0 Hz), 115.88 (d, J = 22.4 Hz).
N(1)–(2-methylphenyl)-2-hydrazino-2-thiooxacetamide (7i)
Yield 73%, yellow solid, m. p. 151–156°C; 1H NMR (300 MHz, DMSO-d6) δ 10.09 (s, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.25 (dd, J = 13.5, 7.5 Hz, 2H), 7.11 (t, J = 7.4 Hz, 1H), 2.28 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 166.77, 157.44, 135.79, 130.94, 130.04, 126.94, 125.75, 122.08, 17.84.
N(1)-{[(5-Methyl-1,3,4-thiadiazol-2-yl)amino]-carbonyl}-2-hydrazino-2-thiooxacetamide (7j)
Yield 45%, yellow solid, m. p. 240–243°C; 1H NMR (300 MHz, CDCl3) δ 3.75–3.07 (m, 4H), 2.64 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 161.02, 160.63, 159.87, 158.34, 15.37.
N(1)– (3-methoxyphenyl)-2-hydrazino-2-thiooxacetamide (7k)
Yield 54%, yellow solid, m. p. 165–168°C; 1H NMR (300 MHz, CDCl3) δ 10.17 (s, 1H), 7.42 (t, J = 2.0 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.26 (t, J = 8.1 Hz, 1H), 6.72 (dd, J = 7.9, 2.1 Hz, 1H), 3.74 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 167.85, 159.98, 158.37, 139.02, 130.16, 112.67, 110.65, 106.20, 55.56.
N(1)– (3,4-difluorophenyl)-2-hydrazino-2-thiooxacetamide (7 l)
Yield 76%, yellow solid, m. p. 164–169°C; 1H NMR (300 MHz, CDCl3) δ 10.41 (s, 1H), 7.91 (ddd, J = 13.0, 7.4, 2.4 Hz, 1H), 7.65–7.55 (m, 1H), 7.43 (dd, J = 19.5, 9.2 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 167.77 (s), 158.94 (s), 149.53 (dd, J = 210.8, 12.9 Hz), 146.30 (dd, J = 210.4, 13.0 Hz), 135.06 (dd, J = 9.1, 3.0 Hz), 117.93 (d, J = 17.9 Hz), 117.43 (dd, J = 6.1, 3.4 Hz), 109.97 (d, J = 21.7 Hz).
N(1)- {[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]-carbonyl}-2-hydrazino-2-thiooxacetamide (7m)
Yield 40%, yellow solid, m. p. 183–189°C; 1H NMR (300 MHz, CDCl3) δ 3.65–3.19 (m, 3H), 3.07–2.95 (m, 1H), 1.29 (t, J = 7.5 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 167.13, 166.67, 159.90, 158.02, 23.21, 14.25.
General procedure for the preparation of compounds 8a--mCitation12
Thiohydrazide 7 (0.1 mmol) was placed in a thick-walled crew-capped glass tube along with succinic anhydride (1.2 mmol) and glacial acetic acid (3.0 mL). The reaction mixture was heated at reflux temperature on vigorous stirring over 2 h, cooled down and poured into water (25 mL). The precipitate formed was filtered off and air-dried to deliver analytically pure compounds 8 in yields indicated.
3–(5-{[(4-Methylphenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8a)
Yield 73%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ ppm δ 12.44 (s, 3H), 11.02 (s, 3H), 7.70 (d, J = 8.4 Hz, 6H), 7.17 (d, J = 8.4 Hz, 6H), 3.37 (t, J = 7.0 Hz, 7H), 2.82 (t, J = 7.0 Hz, 6H), 2.27 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 174.03, 173.47, 166.40, 156.54, 135.59, 134.29, 129.62, 121.20, 33.14, 25.69, 20.98; MS m/z 292.3 (M + H+).
3–(5-{[(2,4-Difluorophenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8b)
Yield 87%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ ppm δ 12.44 (s, 1H), 10.90 (s, 1H), 7.58 (td, J = 8.8, 6.2 Hz, 1H), 7.43–7.35 (m, 1H), 7.18–7.10 (m, 1H), 3.38 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 174.29 (s), 173.43 (s), 165.25 (s), 160.70 (dd, J = 245.8, 11.7 Hz), 157.12 (s), 156.65 (dd, J = 250.8, 13.0 Hz), 129.13 (d, J = 10.0 Hz), 121.20 (dd, J = 12.6, 3.7 Hz), 111.92 (dd, J = 22.3, 3.6 Hz), 105.05 (dd, J = 26.9, 24.3 Hz), 33.15 (s), 25.71 (s); MS m/z 314.2 (M + H+).
3-[5-(Anilinocarbonyl)-1,3,4-thiadiazol-2-yl]propanoic acid (8c)
Yield 80%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ ppm 12.42 (s, 1H), 11.10 (s, 1H), 7.84 (t, J = 8.6 Hz, 2H), 7.38 (dd, J = 16.9, 9.2 Hz, 2H), 7.23–7.00 (m, 1H), 3.38 (t, J = 7.0 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ ppm δ 174.16, 173.49, 166.28, 156.76, 138.04, 129.25, 125.22, 121.29, 33.13, 25.68; MS m/z 278.2 (M + H+).
3–(5-{[(3-Chlorophenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8d)
Yield 76%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 12.44 (s, 3H), 11.30 (s, 3H), 7.99 (t, J = 1.9 Hz, 3H), 7.81 (dd, J = 15.5, 7.3 Hz, 3H), 7.40 (t, J = 8.1 Hz, 1H), 7.22 (dd, J = 7.9, 1.5 Hz, 3H), 3.38 (t, J = 7.0 Hz, 2H), 2.82 (t, J = 7.0 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 174.38, 173.44, 165.94, 157.05, 139.64, 133.48, 130.95, 124.87, 120.68, 119.66, 33.15, 25.73; MS m/z 312.8 (M + H+).
3–(5-{[(4-Methoxyphenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8e)
Yield 91%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 12.43 (s, 1H), 10.99 (s, 1H), 7.72 (d, J = 6.2 Hz, 1H), 6.94 (d, J = 6.2 Hz, 1H), 3.74 (s, 1H), 3.37 (t, J = 7.0 Hz, 1H), 2.82 (t, J = 7.0 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 173.93, 173.46, 166.47, 156.65, 156.32, 131.13, 122.79, 114.33, 55.66, 33.16, 25.68; MS m/z 308.2 (M + H+).
3–(5-{[(2-Fluorophenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8f)
Yield 81%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ ppm δ 12.45 (s, 1H), 10.82 (s, 1H), 7.61 (t, J = 7.8 Hz, 1H), 7.40–7.11 (m, 3H), 3.39 (t, J = 6.9 Hz, 2H), 2.83 (t, J = 6.9 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 174.28 (s), 173.45 (s), 165.40 (s), 156.22 (d, J = 247.9 Hz), 128.34 (d, J = 7.8 Hz), 127.50 (s), 124.79 (dd, J = 26.0, 7.9 Hz), 116.45 (d, J = 19.6 Hz), 33.14 (s), 25.72 (s). MS m/z 296.6 (M + H+).
3–(5-{[(3-Methylphenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8 g)
Yield 84%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 12.43 (s, 1H), 11.01 (s, 1H), 7.67 (s, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.24 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 7.5 Hz, 1H), 3.37 (t, J = 7.0 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H), 2.30 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 174.08, 173.44, 166.36, 156.69, 138.45, 138.07, 129.06, 125.83, 121.73, 118.45, 33.16, 25.71, 21.67; MS m/z 292.3 (M + H+).
3–(5-{[(4-Fluorophenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8 h)
Yield 95%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ ppm δ 12.44 (s, 1H), 11.19 (s, 1H), 7.84 (dt, J = 10.4, 4.3 Hz, 2H), 7.20 (dd, J = 15.1, 6.1 Hz, 2H), 3.38 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 174.15 (s), 173.44 (s), 166.19 (s), 159.28 (d, J = 241.5 Hz), 156.70 (s), 134.53 (d, J = 2.7 Hz), 123.17 (d, J = 8.0 Hz), 115.86 (d, J = 22.4 Hz), 33.16 (s), 25.71 (s); MS m/z 296.4 (M + H+).
3–(5-{[(2-Methylphenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8i)
Yield 91%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 12.44 (s, 1H), 10.60 (s, 1H), 7.47–7.33 (m, 1H), 7.33–7.08 (m, 3H), 3.38 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.24 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 174.20, 173.61, 165.92, 156.93, 135.15, 133.97, 130.97, 127.25, 126.72, 126.68, 33.05, 25.60, 18.09; MS m/z 292.3 (M + H+).
3–(5-{[(5-Methyl-1,3,4-thiadiazol-2-yl)amino]-carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8j)
Yield 65%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 13.04 (s, 1H), 3.37 (t, J = 6.9 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.63 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 174.02, 173.44, 160.03, 33.05, 25.67, 15.67; MS m/z 300.2 (M + H+).
3–(5-{[(3-Methoxyphenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8k)
Yield 78%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 12.43 (s, 1H), 11.06 (s, 1H), 7.55–7.40 (m, 2H), 7.26 (t, J = 8.2 Hz, 1H), 6.73 (dd, J = 8.2, 2.3 Hz, 1H), 3.74 (s, 3H), 3.38 (t, J = 7.0 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 174.19, 173.48, 166.26, 159.86, 156.76, 139.25, 130.08, 113.40, 110.61, 106.99, 55.50, 33.12, 25.69; MS m/z 308.4 (M + H+).
3–(5-{[(3,4-Difluorophenyl)amino]carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8 l)
Yield 90%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ ppm δ 12.44 (s, 1H), 11.35 (s, 1H), 7.94 (ddd, J = 13.0, 7.5, 2.5 Hz, 1H), 7.67 (ddd, J = 6.0, 3.7, 1.9 Hz, 1H), 7.44 (dd, J = 19.7, 9.2 Hz, 1H), 3.38 (t, J = 7.0 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H); 13C NMR (75 MHz, DMSO-d6) δ 174.36 (s), 173.42 (s), 165.88 (s), 156.93 (s), 149.53 (dd, J = 203.2, 13.0 Hz), 146.30 (dd, J = 202.8, 12.9 Hz), 135.20 (dd, J = 9.0, 3.0 Hz), 117.96 (d, J = 17.7 Hz), 117.70 (dd, J = 6.1, 3.4 Hz), 110.29 (d, J = 21.7 Hz), 33.15 (s), 25.73 (s); MS m/z 314.2 (M + H+).
3–(5-{[(5-Ethyl-1,3,4-thiadiazol-2-yl)amino]-carbonyl}-1,3,4-thiadiazol-2-yl)propanoic acid (8m)
Yield 72%, white solid, m. p. 195–196°C; 1H NMR (300 MHz, DMSO-d6) δ 12.94 (s, 1H), 3.37 (t, J = 6.9 Hz, 2H), 3.00 (q, J = 7.5 Hz, 2H), 2.82 (t, J = 6.9 Hz, 2H), 2.41 (s, 1H), 1.30 (t, J = 7.5 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δ 174.09, 173.46, 33.03, 29.20, 25.66, 23.45, 13.85; MS m/z 314.4 (M + H+).
Determination of biological activity and ADMET characterization of compounds 8a--m
The calcium flux assay to determine agonistic activity of compounds 8a--m was conducted on Chinese hamster ovary (CHO) cells stably expressing GPR40 using the protocol earlier described by usCitation2. The experimental details of this assay and of the procedures used to determine aqueous solubility, metabolic stability in the presence of mouse liver microsomes and plasma protein binding are provided in Supplementary Material.
Results and discussion
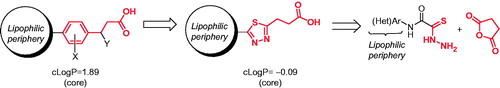
In order to mimic known GPR40 ligand with a 3-(1,3,4-thiadiazol-2-yl)propanoic acid, one should perhaps consider decorating the latter with lipophilic tail periphery and thus increasing affinity to the receptor (which has fatty acids like eicosatrienoic acid as endogenous ligands)Citation13. In this work, we selected anilide periphery for exploration of the SAR around this novel GPR40 scaffolds. We reasoned that a series of compounds like this would be conveniently attainable via cyclocondensation of the respective thiohydrazides with succininc anhydride ().
Figure 2. Design of new 1,3,4-thiadiazole-containing inhibitors explored in this work.
The synthesis of the compounds was based on the previously reported protocolCitation12 and was realized as shown in Scheme 1. Known 2-chloroacetamides 5 were reacted with excess elementary sulfur and morpholine (in the presence of triethylamine). Without purification, the intermediate N-aryl-2-morpholino-2-thiooxacetamides 6 was reacted with hydrazine hydrate to furnish good yields of the stable hydrazino products 7. The latter were condensed with succinic anhydride in glacial acetic acid to provide the target 3-(1,3,4-thiadiazol-2-yl)propanoic acids 8 in good yields (). The synthesis is highly flexible with respect to the anilde (or heteroaromatic) portion and is practical as it involves no chromatographic purification.
Scheme 1. Synthesis of 3-(1,3,4-thiadiazol-2-yl)propanoic acids 8a--m studied in this work.
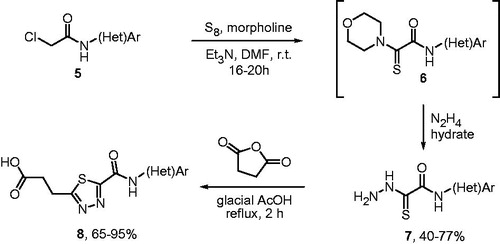
Table 1. Compounds 7(8)a--m synthesized in this work and GPR40 activation data obtained for 8a--m.
The 13 compounds 8a--m thus synthesized were evaluated for agonistic activity toward GPR40 receptor at 5 μM concentration using CHO cell line stably expressing GPR40 and calcium flux fluorescent imaging plate reader (FLIPR) platform (Supplementary Material). Four compounds that displayed >50% activation of the receptor (8d, 8 g, 8j and 8 l, ) at that concentration were selected for dose-response experiments to determine their EC50 values. The agonistic activity was confirmed and the compounds were identified as novel single-to-double-digit micomolar agonists of GPR40 ().
Table 2. EC50 values for compounds 8d, 8 g, 8j and 8 l.
One of these potent hits (8 g) was selected for further characterization based on its potency as well as the good EC50 curve fit (). The compounds displayed fair plasma protein binding (with >10% free fraction in plasma) and truly remarkable metabolic stability (determined on compound’s exposure to mouse liver microsome preparation) and aqueous solubility at physiological pH ().
Figure 3. Dose-response curves for compounds 8d, 8 g, 8j and 8l used to determine EC50 values.
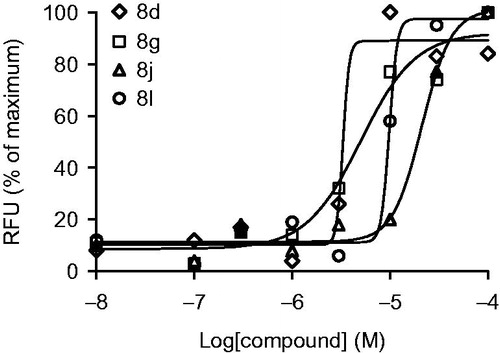
Table 3. Further in vitro characterization of lead compound 8g.
Conclusion
While the potency of the initial hits presented in this paper is lower than that of many advanced compounds,Citation2 they are full agonists of GPR40 receptor and can therefore serve as excellent starting points for further SAR-guided optimization. In particular, the sensitivity of the agonistic potency to the nature of the (hetero)anilide periphery observed in this work, provides a clue that this could be the part of the molecule that should be further manipulated and perhaps elaborated into a slightly more lipophilic one, in order to counterbalance the hydrophilicity introduced by 1,3,4-thiadiazole core and increase the affinity to the receptor and the resulting agonistic activity. The high promise of this new scaffold is significantly boosted by its remarkable ADMET profile, in particular, aquenous solubility and metabolic stability. These features (which we view as a positive corollary to the replacement of the benzene ring with 1,3,4-thiadiazole undertaken in this work) are indispensable from drug development perspective and certainly increase the value of the newly identified scaffold in the GPR40 agonist field.
Declaration of interest
The authors declare no conflict of interest. The authors are solely responsible for the content and results presented in this paper.
Supplementary material available at online
IENZ_1142984_Supplementary_Material.pdf
Download PDF (1.9 MB)Acknowledgements
This research was supported by the Russian Scientific Fund (project grant 1450-00069).
References
- Ito Y, Kawamata Y, Harada M, et al Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003;422:173–6
- Defossa E, Wagner M. Recent developments in the discovery of FFA1 receptor agonists as novel oral treatment for type 2 diabetes mellitus. Bioorg Med Chem Lett 2014;24:2991–3000
- Watterson KR, Hudson BD, Ulven T, Milligan G. Treatment of type 2 diabetes by free fatty acid receptor agonists. Front Endocrinol 2014;5:137
- Available from: https://clinicaltrials.gov
- Kaku K, Enya K, Nakaya R, et al Efficacy and safety of fasiglifam (TAK-875), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebo-controlled, phase III trial. Diabetes Obes Metab 2015;17:675–81
- Mancini AD, Poitout V. GPR40 agonists for the treatment of type 2 diabetes: life after 'TAKing' a hit. Diabetes Obes Metab 2015;17:622–9
- Zahanich I, Kondratov I, Naumchyk V, et al Phenoxymethyl 1,3-oxazoles and 1,2,4-oxadiazoles as potent and selective agonists of free fatty acid receptor 1 (GPR40). Bioorg Med Chem Lett 2015;25:3105–11
- Maekawa T, Hara R, Odaka H, et al Preparation of 1,2-azole derivatives with hypoglycemic and hypolipidemic activity. PCT Int Appl WO 2003099793A1, 564 Pp; Chem Abstr 2003;140:951003
- Hamamura K, Sasaki S, Amano Y, et al Preparation of furan derivatives for treatment of abnormal lipid metabolism, arteriosclerosis, and diabetes. PCT Int Appl WO 2004022551A1, 325 Pp; Chem Abstr 2004;140:220326
- Yasuma T, Negoro N, Sasaki S. Preparation of aminophenylpropanoic acid derivatives as antidiabetic agents. PCT Int Appl WO 2005087710 A1, 371 Pp; Chem Abstr 2005;143:1021733
- Sharma R, Akerman M, Cardozo MG, et al Preparation of coumarin and related carbocycle and heterocyclic analogs useful for treating metabolic disorders. PCT Int Appl WO 2007106469 A2, 194 Pp; Chem Abstr 2007;147:1064386
- Yarovenko VN, Shirokov AV, Krupinova ON, et al Synthesis of oxamic acids thiohydrazides and carbamoyl-1,3,4-thiadiazoles. Russ J Org Chem 2003;39:1133–9
- Ichimura A, Hirasawa A, Hara T, Tsujimoto G. Free fatty acid receptors act as nutrient sensors to regulate energy homeostasis. Prostaglandins Other Lipid Mediat 2009;89:82–8