
Abstract
We designed and synthesized new series of diverse triazoles, isoxazoles, isoxazolines, and aziridines linked 4-methylumbelliferone 1 using intermolecular 1,3-dipolar cycloaddition reactions. Structures of these compounds were established on the basis of 1H NMR, 13C NMR, and ESI-HRMS. All prepared compounds were evaluated for their antimicrobial, anticoagulant, and anticholinesterase activities. Interestingly, among the tested molecules, some of the analogs displayed better activities than the parent 4-methylumbelliferone 1 such as 6a and 6d for their antifungal properties. Moreover, compounds 4, 5, 6, and 7 showed the importance of the added fragments to 4-methylumbelliferone 1 via the linker methylene to have good activity.
Introduction
The natural and synthetic coumarins have occupied an important role in the drug discovery process and have considerable pharmacological and chemical significanceCitation1. Literature survey shows that a large number of coumarin derivatives are found to be associated with diverse types of biological activities, such as anticancerCitation2, antioxidantCitation3, antimicrobialCitation4, anticoagulantCitation2 and they have proved to be promising anti-acetylcholinesteraseCitation5 agents. The coumarins are extremely variable in structure, due to various types of substitutions in their basic structure, for this reason we can found many studies focus on the modification of coumarins such as hydroxylated, alkoxylated, and alkylated to give more bioactive moleculesCitation6.
Different types of 1,2,3-triazole derivatives have enabled diverse applications in the fields of pharmaceuticalsCitation7, agrochemicalsCitation8, medicinal chemistryCitation9, and material sciencesCitation10. Because of their utility and numerous activities such as antitumoralCitation11, antifungalCitation12, antiproliferativeCitation13, antioxidant and anti-inflammatoryCitation14, several syntheses of 1,2,3-triazoles have been reported. One of the synthetic methods to assemble 1,2,3-triazoles is the Huisgen 1,3-dipolar cycloaddition of azides and alkynesCitation15. This cycloaddition reaction usually affords mixtures of 1,4 and 1,5-disubstituted 1,2,3-triazoles. Recently, Shakeel-u-Rehman and collaboratorsCitation16 reported, that 1,4-disubstituted 1,2,3-triazoles linked with 6-hydroxycoumarin are specifically prepared from azides and terminal alkynes under sharpless click chemistry conditions (CuSO4 5H2O and sodium ascorbate). Also, the regioisomeric 1,5-disubstituted triazoles are available from azides and terminal alkynes by the use of either magnesium acetylides or ruthenium catalystsCitation17.
On the other hand, heterocyclic compounds substituted with isoxazoles moieties have found increasing attention in organic syntheses, biochemistry and medicinal chemistry research due to their cytotoxicCitation18, antimycobacterialCitation19, antiviralCitation20, and anticonvulsantCitation21 activities.
In addition, heterocyclic compounds carrying isoxazolines units have also a pharmaceutical importance and some of them posse’s anticancerCitation22, antimicrobialCitation23, anticoagulantCitation2, and anti inflammatoryCitation24 activity.
It was revealed that aziridine ring is an element of a large number of natural products type alkaloidsCitation25 and various biologically active compounds such as antibioticsCitation26 and antitumor agentsCitation27. Because of their broad spectrum of biological activities and pharmacological propertiesCitation28, many efficient and more general synthetic strategies have been developed for the preparation of aziridinesCitation29.
Taking into account the various biological activities of triazoles, isoxazolines, isoxazoles, aziridines, and coumarins cited, respectively, in the literatureCitation30–33, it appeared to us interesting to think about the combination of these moieties hoping to access to more biologically effective compounds. In this context and as a continuation of our previous work on the synthesis of new fused 4-methylumbelliferone scaffoldsCitation34, we report here the synthesis of a diverse series of novel triazoles, isoxazoles, isoxazolines, and aziridines linked to 4-methylumbelliferone through [3 + 2] cycloaddition in the side chain. The synthetic compounds were then screened for their antimicrobial (antibacterial + antifungal), anticoagulant and anticholinesterase activities and the structure–activity relationship (SAR) has been discussed.
Materials and methods
Chemistry
All reactions were monitored by TLC using aluminium sheets of Merck silica gel 60 F254, 0.2 mm. Melting temperatures were determined on an electrothermal 9002 apparatus and were reported uncorrected. NMR spectra were recorded on a Bruker AC-300 spectrometer at 300 MHz (1H) and 75 MHz (13C). All chemical shifts were reported as δ values (ppm) relative to residual non deuterated solvent. ESI-HRMS were obtained with ESI-TOF (LCT, Waters) using the reflectron mode in the positive ion mode.
General procedure for O-allylation and O-propargylation of 4-methylumbelliferone (1)
The 4-methylumbelliferone 1 (2.6 g) was added in portions to a stirred suspension of NaH (1.21 g) in dry DMF (15 mL) at 20 °C. After the addition was over, the mixture was stirred at 20 °C for 30 min. A solution of propargyl bromide/allyl bromide (2 mL) in DMF (5 mL) was added dropwise to the mixture at 20 °C and the stirring was continued for 30 minCitation35,Citation36. The whole reaction mixture was poured into ice water and the solid separated was collected with filtration, dried and recrystallized from ethanol affording compounds 2 and 3, respectively.
4-Methyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (2)
Brown solid; 1.95 g; yield: 75%; mp.: 153–155 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.40 (s, 3H, H9), 2.58 (t, 1H, H4′, J = 2.4 Hz), 4.77 (d, 2H, H2′, J = 2.4 Hz), 6.15 (s, 1H, H3), 6.91–6.95 (m, 2H, H8 + H6), 7.52 (d, 1H, H5, J = 9.3 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 55.7, 75.9, 77.4, 101.9, 111.9, 112.1, 113.7, 125.1, 151.8, 154.5, 159.8, 160.5. ESI-HRMS [M + H]+ calcd. For (C13H11O3)+: 215.0708, found: 215.0711.
7-(Allyloxy)-4-methyl-2H-chromen-2-one (3)
Yellow solid; 1,69 g; yield: 65%; mp.: 149–151 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.37 (s, 3H, H9), 4.51 (m, 2H, H2′a,b), 5.25 (dd, 1H, H4a′, J = 10.5 Hz, J = 1.2 Hz), 5.36 (dd, 1H, H4b′, J = 17.4 Hz, J = 1.2 Hz), 5.91–6.00 (m, 1H, H3′), 6.03 (s, 1H, H3), 6.72 (d, 1H, H8, J = 2.4 Hz), 6.80 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 7.41 (d, 1H, H5, J = 8.7 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.6, 68.7, 101.2, 111.5, 112.2, 113.2, 118.4, 125.5, 131.7, 151.9, 154.7, 160.7, 161.0. ESI-HRMS [M + H]+ calcd. For (C13H13O3)+: 217.0865, found: 217.0870.
General procedure for the synthesis of triazolyl derivatives (4a–e)
To a mixture of dipolarophile 2 (3.0 mmol) dissolved in toluene (20 mL), the corresponding azides (3 eq), triethylamine (3 eq, 0.3 mol%) and copper (I) iodide catalyzed (0.5 eq) were added. This mixture was stirred at reflux for 12 h. The solvent was then removed under reduced pressure and the residue was loaded into a silica gel column and eluted with a mixture of chloroform/ethyl acetate (9/1) to give pure1,4-triazoles 4a–e.
4-Methyl-7-((1-phenyl-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (4a)
Yellow solid; 85 mg; Yield: 57%; m.p.: 170–172 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.40 (s, 3H, H9), 5.36 (s, 2H, H6′), 6.15 (s, 1H, H3), 6.95 (dd, 1H, H6, J = 2.4 Hz, J = 7.2 Hz), 6.97 (d, 1H, H8, J = 2.4 Hz), 7.46 (d, 1H, H5, J = 7.2 Hz), 7.44–7.57 (m, 3H, H9′ + H10′ + H11′), 7.76 (2d, 2H, H8′ + H12′, J = 7.8 Hz), 8.13 (s, 1H, H5′); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 61.7, 101.6, 111.8, 111.9, 113.6, 120.1 (C-8′), 120.1 (C-12′), 120.8, 125.2, 128.5, 129.3, 129.3, 136.3, 143.2, 151.9, 154.6, 160.5, 160.6. ESI-HRMS [M + H]+ calcd. For (C19H16N3O3)+: 334.1192, found: 334.1196.
7-((1–(4-Methoxyphenyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4b)
Yellow solid; 96 mg; Yield: 64%; m.p.: 169–171 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.41 (s, 3H, H9), 3.88 (s, 3H, Ph-OCH3), 5.35 (s, 2H, H6′), 6.16 (s, 1H, H3), 6.98–7.05 (m, 2H, H8 + H6), 7.05 (2d, 2H, H9′ + H11′, J = 8.4 Hz), 7.54 (2d, 2H, H8′ + H12′, J = 8.4 Hz), 7.65 (d, 1H, H5, J = 7.5 Hz), 8.02 (s, 1H, H5′); 13C NMR (CDCl3, 75 MHz) δC: 18.6, 55.6, 62.3, 77.2, 102.2, 112.3, 112.4, 114.1, 114.1, 114.8, 121.3, 122.3, 122.3, 125.7, 130.3, 143.5, 152.4, 155.1, 160.0, 161.1. ESI-HRMS [M + H]+ calcd. For (C20H18N3O4)+: 364.1294, found: 363.1297.
7-((1–(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)-4-methyl-2H-chromen-2-one (4c)
Yellow solid; Yield: 100 mg; 67%; m.p.: 176–178 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.42 (s, 3H, H9), 5.37 (s, 2H, H6′), 6.17 (s, 1H, H3), 6.97–7.01 (m, 2H, H8 + H6), 7.51–7.56 (m, 4H, H8′+H9′+H11′+H12′), 7.73 (d, 1H, H5, J = 8.4 Hz), 8.10 (s, 1H, H5′); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 61.7, 101.7, 111.8, 111.9, 113.7, 120.6, 121.2, 121.2, 125.2, 129.5, 129.5, 134.3, 134.8, 143.6, 151.8, 154.6, 160.4, 160.5. ESI-HRMS [M + H]+ calcd. For (C19H15ClN3O3)+: 368.0802, found: 368.0808.
4-Methyl-7-((1-(m-tolyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (4d)
Yellow solid; 69 mg; Yield: 46%; m.p.: 168–170 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.42 (s, 3H, Ph-CH3), 2.47 (s, 3H, H9), 5.37 (s, 2H, H6′), 6.17 (s, 1H, H3), 6.99–7.02 (m, 2H, H8 + H6), 7.36–7.60 (m, 5H, H8′ +H10′ +H11′ +H12′ +H5), 8.08 (s, 1H, H5′); 13C NMR (CDCl3, 75 MHz) δC: 18.6, 21.3, 62.3, 101.8, 112.3, 112.5, 112.7, 121.2, 125.5, 125.9, 129.5, 129.6, 129.8, 130.7, 136.8, 143.6, 152.3, 155.1, 160.2, 161.1. ESI-HRMS [M + H]+ calcd. For (C20H18N3O3)+: 348.1343, found: 347.1348.
4-Methyl-7-((1-(naphthalen-1-yl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one (4e)
Yellow solid; 79 mg; Yield: 53%; m.p.: 173–175 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.41 (s, 3H, H9), 5.42 (s, 2H, H2′), 6.16 (s, 1H, H3), 7.00–7.05 (m, 2H, H8 + H6), 7.53–7.60 (m, 7H, Hnaph), 7.96 (d, 1H, H5, J = 7.5 Hz), 8.07 (s, 1H, H5′); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 61.8, 101.7, 111.8, 111.9, 113.6, 121.6, 123.1, 124.4, 125.2, 125.3, 126.6, 127.5, 127.8, 127.9, 130.1, 130.6, 132.9, 133.6, 142.5, 151.9, 154.6, 160.6. ESI-HRMS [M + H]+ calcd. (C23H18N3O3)+: 384.1348, found: 384.1349.
General procedure for the synthesis of isoxazoles (5a–e) and isoxazolines (6a–e)
A mixture of dipolarophile 2 or 3 (1.15 mmol) and arylnitrile oxides (3 eq) was dissolved in toluene (15 mL) in the presence of triethylamine (3 eq, 0.3 mol%), the solution was then refluxed for 12 h. The reaction was monitored with TLC after the completion of the reaction, we stopped it and we noted that there was a good amount of salt, so the salt was filtered off and the filtrate was evaporated. The product formed in each case was purified using precipitation with cyclohexane and ethyl acetate to yield compounds 5 or 6.
4-Methyl-7-((3-phenylisoxazol-5-yl) methoxy)-2H-chromen-2-one (5a)
Yellow solid; 52 mg; Yield: 35%; m.p.: 150–152 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.41 (s, 3H, H9), 5.30 (s, 2H, H2′), 6.18 (s, 1H, H3), 6.71 (s, 1H, H4′), 6.95 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 6.98 (d, 1H, H8, J = 2.4 Hz), 7.46–7.48 (m, 3H, H8′ + H9′ + H10′), 7.55 (d, 1H, H5, J = 8.7 Hz), 7.80–7.83 (m, 2H, H7′ + H11′); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 60.9, 101.4, 101.6, 111.8, 112.1, 114.0, 125.4, 126.3, 126.3, 128.0, 128.4, 128.4, 129.7, 151.8, 154.6, 160.0, 160.4, 162.1, 166.6. ESI-HRMS [M + H]+ calcd. For (C20H16NO4)+: 334.1070, found: 334.1079.
7-((3–(4-Methoxyphenyl) isoxazol-5-yl) methoxy)-4-methyl-2H-chromen-2-one (5b)
Brown solid; 67 mg; Yield: 45%; m.p.: 148–150 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.41 (s, 3H, H9), 3.86 (s, 3H, Ph-CH3), 5.26 (s, 2H, H2′), 6.18 (s, 1H, H3), 6.64 (s, 1H, H4′), 6.93 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 6.97 (d, 1H, H8, J = 2.4 Hz), 6.99 (d, 2H, H8′ + H10′, J = 8.1 Hz), 7.55 (d, 1H, H5, J = 8.7 Hz), 7.75 (d, 2H, H7′ + H11′, J = 8.1 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 54.8, 60.9, 101.2, 101.6, 111.8, 112.1, 113.8, 114.0, 114.0, 120.5, 125.3, 127.7, 127.7, 151.8, 154.6, 160.0, 160.4, 160.7, 161.7, 166.3. ESI-HRMS [M + H]+ calcd. (C21H18NO5)+: 364.1176, found: 364.1185.
7-((3–(4-Chlorophenyl) isoxazol-5-yl) methoxy)-4-methyl-2H-chromen-2-one (5c)
Brown solid; 60 mg; Yield: 40%; m.p.: 150–152 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.41 (s, 3H, H9), 5.28 (s, 2H, H2′), 6.18 (s, 1H, H3), 6.68 (s, 1H, H4′), 6.93 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 6.97 (d, 1H, H8, J = 2.4 Hz), 7.44 (d, 2H, H8′ + H10′, J = 8.4 Hz), 7.55 (d, 1H, H5, J = 8.7 Hz), 7.76 (d, 2H, H7′ + H11′, J = 8.4 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 60.9, 101.3, 101.5, 111.8, 112.1, 114.1, 125.4, 126.5, 127.6, 127.6, 128.7, 128.7, 135.8, 151.7, 154.6, 159.9, 160.4, 161.1, 167.0. ESI-HRMS [M + H]+ calcd. For (C20H15ClNO4)+: 368.0798, found: 368.0802.
7-((3–(4-Ethylphenyl) isoxazol-5-yl) methoxy)-4-methyl-2H-chromen-2-one (5d)
Yellow solid; 55 mg; Yield: 37%; m.p.: 155–157 °C; 1H NMR (CDCl3, 300 MHz) δH: 1.28 (t, 3H, Ph-CH2-CH3, J = 7.5 Hz), 2.42 (s, 3H, H9), 2.73 (q, 2H, Ph-CH2-CH3, J = 7.5 Hz), 5.30 (s, 2H, H2′), 6.19 (s, 1H, H3), 6.68 (s, 1H, H4′), 6.95 (dd, 1H, H6, J = 2.7 Hz, J = 8.4 Hz), 6.99 (d, 1H, H8, J = 2.7 Hz), 7.29 (d, 2H, H8′+ H10′, J = 8.1 Hz), 7.56 (d, 1H, H5, J = 8.4 Hz), 7.74 (d, 2H, H7′ + H11′, J = 8.1 Hz); 13C NMR (CDCl3, 75 MHz) δC: 15.3, 18.6, 28.8, 61.5, 101.9, 102.1, 112.4, 112.6, 112.7, 114.5, 125.6, 126.8, 126.8, 128.4, 128.4, 146.7, 152.2, 155.1, 160.5, 160.9, 162.5, 166.9. ESI-HRMS [M + H]+ calcd. For (C22H20NO4)+: 362.1386, found: 362.1392.
4-Methyl-7-((3–(4-nitrophenyl)isoxazol-5-yl)methoxy)-2H-chromen-2-one (5e)
Yellow solid; 90 mg; Yield: 60%; m.p.: 158–160 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.42 (s, 3H, H9), 5.32 (s, 2H, H2′), 6.19 (s, 1H, H3), 6.79 (s, 1H, H4′), 6.94 (dd, 1H, H6, J = 2.7 Hz, J = 8.7 Hz), 6.99 (d, 1H, H8, J = 2.7 Hz), 7.58 (d, 1H, H5, J = 8.7 Hz), 8.01 (d, 2H, H7′ + H11′, J = 8.7 Hz), 8.33 (d, 2H, H8′ + H10′, J = 8.7 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.6, 19.6, 61.4, 101.9, 102.0, 112.3, 112.8, 114.7, 124.2, 124.2, 125.9, 127.7, 127.7, 134.5, 148.8, 152.2, 155.1, 160.3, 160.8, 168.3. ESI-HRMS [M + H]+ calcd. For (C20H15N2O6)+: 379.0925, found: 379.0930.
4-Methyl-7-((3-phenyl-4,5-dihydroisoxazol-5-yl)methoxy)-2H-chromen-2-one (6a)
Yellow solid; 48 mg; Yield: 32%; m.p.: 145–147 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.38 (s, 3H, H9), 3.40 (dd, 1H, H4′a, J = 16.5 Hz, J = 7.5 Hz), 3.56 (dd, 1H, H4′b, J = 16.8, J = 10.8 Hz Hz), 4.15 (dd, 1H, H2′a, J = 14.1 Hz, J = 7.5 Hz), 4.23 (dd, 1H, H2′b, (J = 14.1 Hz, J = 5.1 Hz), 5.13–5.16 (m, 1H, H3′), 6.13 (s, 1H, H3), 6.81 (d, 1H, H8, J = 2.4 Hz), 6.86 (dd, 1H, H6, J = 2.4 Hz, J = 8.1 Hz), 7.27–7.43 (m, 3H, H8′ + H9′ + H10′), 7.47 (d, 1H, H5, J = 8.1 Hz), 7.69–7.72 (m, 2H, H7′ + H11′); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 36.9, 68.6, 76.9, 77.9, 101.3, 111.7, 111.9, 113.6, 125.1, 126.2, 126.2, 128.2, 128.2, 129.8, 131.0, 151.6, 155.9, 160.5, 160.8. ESI-HRMS [M + H]+ calcd. For (C20H18NO4)+: 336. 1231, found: 336.1236.
7-((3–(4-Methoxyphenyl)-4,5-dihydroisoxazol-5-yl)methoxy)-4-methyl-2H-chromen-2-one (6b)
Yellow solid; 67 mg; Yield: 45%; m.p.: 144–146 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.28 (s, 3H, H9), 3.35 (dd, 1H, H4′a, J = 16.5 Hz, J = 7.2 Hz), 3.43 (dd, 1H, H4′b, J = 16.5 Hz, J = 10.5 Hz), 3.75 (s, 3H, Hb), 4.03 (dd, 1H, H2′a, J = 14.2 Hz, J = 7.1 Hz), 4.12 (dd, 1H, H2′b, (J = 14.2 Hz, J = 5.2 Hz)), 5.00–5.04 (m, 1H, H3′), 6.03 (s, 1H, H3), 6.71 (d, 1H, H8, J = 2.7 Hz), 6.79 (dd, 1H, H6, J = 2.7 Hz, J = 9 Hz), 6.85 (d, 2H, H8′ + H10′, J = 9.6 Hz), 7.40 (d, 1H, H5, J = 9 Hz), 7.55 (d, 2H, H7′ + H11′, J = 9.6 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 37.6, 55.3, 56.2, 68.6, 77.6, 101.3, 111.7, 111.9, 113.7, 113.7, 121.2, 125.6, 128.3, 128.3, 151.9, 154.6, 155.4, 160.5, 160.7, 160.9. ESI-HRMS [M + H]+ calcd. For (C21H20NO5)+: 366.1336, found: 366.1341.
7-((3–(4-Chlorophenyl)-4,5-dihydroisoxazol-5-yl)methoxy)-4-methyl-2H-chromen-2-one (6c)
Brown solid; 87 mg; Yield: 58%; m.p.: 149–151 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.40 (s, 3H, H9), 3.36 (dd, 1H, H4′a, J = 16.8 Hz, J = 7.2 Hz), 3.55 (dd, 1H, H4′b, J = 16.8 Hz, J = 10.8 Hz), 4.18 (dd, 1H, H2′a, J = 14.5 Hz, J = 7.2 Hz), 4.21 (dd, 1H, H2′b, (J = 14.5 Hz, J = 5.1 Hz)), 5.14–5.19 (m, 1H, H3′), 6.16 (s, 1H, H3), 6.82 (d, 1H, H8, J = 2.4 Hz), 6.89 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 7.40 (d, 2H, H8′ + H10′, J = 8.4 Hz), 7.50 (d, 1H, H5, J = 8.7 Hz), 7.64 (d, 2H, H7′ + H11′, J = 8.4 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.6, 37.3, 69.2, 78.7, 101.7, 112.4, 112.8, 114.1, 118.5, 125.5, 128.2, 128.2, 128.8, 128.8, 136.3, 152.3, 155.1, 155.5, 161.0, 161.2. ESI-HRMS [M + H]+ calcd. For (C20H17NO4)+: 370.0846, found: 370. 0847.
7-((3–(4-Ethylphenyl)-4,5-dihydroisoxazol-5-yl)methoxy)-4-methyl-2H-chromen-2-one (6d)
Brown solid; 30 mg; Yield: 20%; m.p.: 145–147 °C; 1H NMR (CDCl3, 300 MHz) δH: 1.27 (t, 3H, Ph-CH2-CH3, J = 7.3 Hz), 2.40 (s, 3H, H9), 2.70 (q, 2H, Ph-CH2-CH3, J = 7.3 Hz), 3.35 (dd, 1H, H4′a, J = 16.5 Hz, J = 6.6 Hz), 3.50 (dd, 1H, H4′b, J = 16.8 Hz, J = 10.8 Hz), 4.17–4.20 (m, 2H, H2′a+ H2′b), 5.13–5.16 (m, 1H, H3′), 6.14 (s, 1H, H3), 6.83 (d, 1H, H8, J = 2.7 Hz), 6.90 (dd, 1H, H6, J = 2.7 Hz, J = 9 Hz), 7.27 (d, 2H, H8′ + H10′, J = 8.1 Hz), 7.50 (d, 1H, H5, J = 9 Hz), 7.63 (d, 2H, H7′ + H11′, J = 8.1 Hz); 13C NMR (CDCl3, 75 MHz) δC: 14.8, 18.1, 28.2, 37.1, 68.6, 77.7, 78.2, 101.8, 112.2, 113.6, 125.1, 126.0, 126.3, 126.3, 127.8, 127.8, 146.4, 151.9, 154.6, 155.9, 160.7, 160.9. ESI-HRMS [M + H]+ calcd. For (C22H22NO4)+: 364. 1545, found: 363.1549.
4-Methyl-7-((3–(4-nitrophenyl)-4,5-dihydroisoxazol-5-yl)methoxy)-2H-chromen-2-one (6e)
Yellow solid; 82 mg; Yield: 55%; m.p.: 159–161 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.47 (s, 3H, H9), 3.39 (dd, 1H, H4′a, J = 16.8 Hz, J = 7.2 Hz), 3.65 (dd, 1H, H4′b, J = 16.8 Hz, J = 10.8 Hz), 4.24–4.33 (m, 2H, H2′a + H2′b), 5.14–5.20 (m, 1H, H3′), 6.16 (s, 1H, H3), 6.92 (d, 1H, H8, J = 2.4 Hz), 6.96 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 7.65 (d, 1H, H5, J = 8.7 Hz), 7.93 (d, 2H, H7′+H11′, J = 9 Hz), 8.26 (d, 2H, H8′ + H10′, J = 9 Hz); 13C NMR (CDCl3, 75 MHz) δC: 17.9, 36.1, 69.5, 79.7, 101.6, 111.3, 112.4, 113.5, 123.8, 123.8, 126.3, 127.7, 127.7, 135.4, 143.1, 153.0, 154.6, 155.7, 159.9, 161.3. ESI-HRMS [M + H]+ calcd. For (C20H17N2O6)+: 381.1087, found: 381.1090.
General procedure for the synthesis of aziridine (7a–e)
A mixture of dipolarophile 3 (1 eq) and arylazides (3 eq) catalyzed (0.5 eq) was dissolved in toluene (15 mL) and the solution was then refluxed for 6 h. The reaction was monitored with TLC after the completion of the reaction; the reaction mixture was concentrated and cooled. The product formed in each case was purified by column chromatography using 9:1 mixture of chloroform and ethylacetate to yield compounds 7a–e.
4-Methyl-7-((1-phenylaziridin-2-yl)methoxy)-2H-chromen-2-one (7a)
Yellow solid; 97 mg; Yield: 65%; m.p.: 142–144 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.15 (d, 1H, H4′a, J = 6.3 Hz), 2.25 (d, 1H, H4′b, J = 3.3 Hz), 2.31 (s, 3H, H9), 2.48–2.51 (m, 1H, H3′), 3.94 (dd, 1H, H2′a, J = 7.5 Hz, J = 10.5 Hz), 4.25 (dd, 1H, H2′b, J = 3.9 Hz, J = 10.5 Hz), 6.05 (s, 1H, H3), 6.80 (d, 1H, H8, J = 2.4 Hz), 6.91 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 6.93–7.19 (m, 5H, H7′+H8′+H9′+H10′+H11′), 7.44 (d, 1H, H5, J = 8.7 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 30.7, 37.3, 70.1, 101.3, 111.6, 112.1, 113.3, 120.1, 120.1, 122.2, 125.1, 128.5, 128.5, 151.9, 153.0, 154.7, 160.6, 161.2. ESI-HRMS [M + H]+ calcd. For (C19H18NO3)+: 308.1287, found: 307.1290.
7-((1–(4-Methoxyphenyl) aziridin-2-yl)methoxy)-4-methyl-2H-chromen-2-one (7b)
Yellow solid; 82 mg; Yield: 55%; m.p.: 137–139 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.15 (d, 1H, H4′a, J = 6.6 Hz), 2.26 (d, 1H, H4′b, J = 3.3 Hz,), 2.38 (s, 3H, H9), 2.46–2.49 (m, 1H, H3′), 3.74 (s, 3H, Hb), 3.99 (dd, 1H, H2′a, J = 7.5 Hz, J = 10.5 Hz), 4.29 (dd, 1H, H2′b, J = 3.9 Hz, J = 10.5 Hz), 6.12 (s, 1H, H3), 6.78 (2d, 2H, H7′ + H11′, J = 9 Hz), 6.87 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 6.93 (d, 1H, H8, J = 2.4 Hz), 6.97 (2d, 2H, H8′ + H10′, J = 9 Hz), 7.49 (d, 1H, H5, J = 8.7 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 30.9, 37.5, 55.0, 70.1, 101.3, 111.6, 112.1, 113.3, 113.8, 113.8, 120.9, 120.9, 125.1, 148.1, 151.9, 154.7, 154.9, 160.7, 161.2. ESI-HRMS [M + H]+ calcd. For (C20H20NO4)+: 337.1390, found: 338.1392.
7-((1–(4-Chlorophenyl) aziridin-2-yl)methoxy)-4-methyl-2H-chromen-2-one (7c)
Yellow solid; 112 mg; Yield: 75%; m.p.: 146–148 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.13 (d, 1H, H4′a, J = 6.6 Hz), 2.27 (d, 1H, H4′b, J = 3.3 Hz), 2.33 (s, 3H, H9), 2.45–2.49 (m, 1H, H3′), 3.90 (dd, 1H, H2′a, J = 7.5 Hz, J = 10.5 Hz), 4.25 (dd, 1H, H2′b, J = 3.9 Hz, J = 10.5 Hz), 6.07 (s, 1H, H3), 6.80 (d, 1H, H8, J = 2.4 Hz), 6.89 (2d, 2H, H7′ + H11′, J = 8.7 Hz), 6.90 (dd, 1H, H6, J = 2.4 Hz, J = 8.7 Hz), 7.13 (2d, 2H, H8′ + H10′, J = 8.7 Hz), 7.44 (d, 1H, H5, J = 8.7 Hz); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 30.8, 37.6, 69.8, 101.2, 111.6, 112.1, 112.6, 121.4, 121.4, 125.1, 127.3, 128.5, 128.5, 151.6, 151.9, 154.7, 160.7, 161.1. ESI-HRMS [M + H]+ calcd. For (C19H16ClNO3)+: 342.0896, found: 341.0897.
4-Methyl-7-((1-(m-tolyl) aziridin-2-yl)methoxy)-2H-chromen-2-one (7d)
Yellow solid; 72 mg; Yield: 48%; m.p.: 141–143 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.15 (d, 1H, H4′a, J = 6.3 Hz), 2.21 (s, 3H, Hb), 2.25 (d, 1H, H4′b, J = 3.3 Hz), 2.38 (s, 3H, H9), 2.48–2.51 (m, 1H, H3′), 4.08 (dd, 1H, H2′a, J = 7.5 Hz, J = 10.8 Hz), 4.35 (dd, 1H, H2′b, J = 3.9 Hz, J = 10.8 Hz), 6.16 (s, 1H, H3), 6.71–6.78 (m, 3H, H7′ + H9′+H11′), 7.01–7.09 (m, 3H, H8 + H6 + H10′), 7.65 (d, 1H, H5, J = 9.3 Hz); 13C NMR (CDCl3, 75 MHz) δC: 17.9, 20.9, 30.6, 37.5, 70.3, 101.6, 111.2, 112.5, 113.3, 117.5, 121.0, 122.8, 126.3, 128.6, 138.1, 153.1, 153.8, 154.7, 159.9, 161.5. ESI-HRMS [M + H]+ calcd. For (C20H20NO3)+: 322. 1439, found: 321.1443.
4-Methyl-7-((1-(naphthalen-1-yl) aziridin-2-yl)methoxy)-2H-chromen-2-one (7e)
Yellow solid; 67 mg; Yield: 45%; m.p.: 143–145 °C; 1H NMR (CDCl3, 300 MHz) δH: 2.38 (d, 1H, H4′a, J = 6.3 Hz), 2.42 (s, 3H, H9), 2.58 (d, 1H, H4′b, J = 3.3 Hz), 2.81–2.85 (m, 1H, H3′), 4.27 (dd, 1H, H2′a, J = 6.9 Hz, J = 10.2 Hz), 4.47 (dd, 1H, H2′b, J = 3.9 Hz, J = 10.2 Hz), 6.17 (s, 1H, H3), 6.95 (d, 1H, H8, J = 2.4 Hz), 6.99–7.03 (m, 2H, H6 + H7′), 7.40 (d, 1H, H5, J = 8.1 Hz), 7.55–8.50 (m, 6H, H8′+H9′+H10′+H11′+H12′+H13′); 13C NMR (CDCl3, 75 MHz) δC: 18.1, 31.9, 38.0, 70.6, 101.3, 111.7, 112.0, 113.4, 114.2, 122.2, 122.9, 125.0, 125.1, 125.2, 125.6, 127.5, 127.8, 133.7, 148.1, 151.9, 154.7, 160.6, 161.1. ESI-HRMS [M + H]+ calcd. For (C23H20NO3)+: 358. 1443, found: 358. 1448.
Biological evaluation
Antibacterial activityCitation37
The purified products were screened for their antibacterial activity by using the agar disc diffusion method toward Pseudomonas syringae pv, Syringae, Pseudomonas savatanoi, and Agrobacterium tumefasciens. Nutrient agar (NA) medium cooled at 45 °C was supplemented with a bacterial suspension (106 CFU/mL) and poured into Petri plates. After solidification, sterile Whatman paper discs (diameter 6 mm) were placed at the surface of the culture medium and 20 μL (1000 μg/mL) of the product dissolved in dimethyl sulfoxide (DMSO) was dropped onto each disc. The negative control plates had no product added to the filter paper whereas in the positive control plates, discs were impregnated with the same volume of ampicillin solution (5 mg/mL). The treated Petri dishes were incubated at 25 °C for 48 h. The antibacterial activity was evaluated by measuring the diameter of the inhibitory zones formed around the discs. The experiment was replicated twice.
Antifungal activityCitation38
Aspergillus flavus, Aspergillus niger, and Penicillium italicum were used for the screening of antifungal activity of the products tested by using the disc diffusion method. A conidial suspension of the tested fungi was prepared (104–105 CFU/mL) and added to potato dextrose agar (PDA) medium cooled at 45 °C and poured uniformly into Petri plates (diameter 90 mm). Sterilized paper discs (6 mm, Whatman No. 1 filter paper) were impregnated with 20 μL (1000 μg/mL) of the product dissolved in DMSO and placed on the culture plates whereas the negative control plates had no product added to the filter paper. In the positive control plates, discs were imbibed with the same volume of a carbendazim suspension (0.5 mg/mL). The diameter of the inhibition zone (mm) around the disc was measured after incubation at 25 °C for 4 d and compared with control. The test was performed in triplicate.
Anticoagulant activityCitation39
Activated partial thromboplastin time (aPTT) was performed using Platelin LS reagent (Trinity Biotech PLC, Cowicklow, Ireland) on a STAR analyzer (Diagnostica Stago, Asnières, France). Coagulation test was performed on 4-methylumbelliferone derivatives 2, 3, 4a–e, 5a–e, 6a–e, and 7a–e samples at various concentrations diluted in a pool of frozen normal plasma. 4-Methylumbelliferone 1 was used as reference.
BChE inhibitory activity assayCitation40
Butyrylcholinesterase (BChE) was used to hydrolyze butyrylthiocholine (colorless) to butyrate and thiocholine. Then, thiocholine reacts with 5,5′-dithiobis[2-nitrobenzoic acid] (DTNB) to form 5-mercapto-2-nitrobenzoic acid (5-MNBA), a yellow product. The rate of 5-MNBA formation, measured spectrophotometrically at 405 nm is proportional to the enzymatic activity of BChE in the sample. Human plasma (pool plasma from samples designated for biochemical analysis) was used as a source of BChE. 100 μL of compound (concentration of 1000 μg/mL) were added to 100 μL of plasma and the mixture was incubated at 37 °C for a determined time. After incubation, the enzyme activity was measured by Konelab 30® UV apparatus. The control (plasma and distilled water) was treated in the same conditions. All assays were carried out in duplicate. The anticholinesterase activity was calculated by the following formula:
The sample concentration providing 50% inhibition (IC50) was calculated by plotting inhibition percentages against concentrations of the sample.
Results and discussion
First, our key intermediate was the dipolarophiles 2 and 3, which can easily be obtained by O-propargylation and O-allylationCitation41,Citation42, respectively of 4-methylumbelliferone 1 in one step (Scheme 1). Indeed, in our investigation, dimethylformamide was found to be an excellent solvent for the reaction of allyl and propargylbromide with 4-methylumbelliferone 1, in the presence of NaH. The dipolarophiles 2 and 3 were obtained in 75% and 65% yield, respectively and which were identified by the spectroscopic means (1H, 13C, and ES-HRMS)Citation43,Citation35.
Scheme 1. Synthetic route of dipolarophiles 2 and 3.
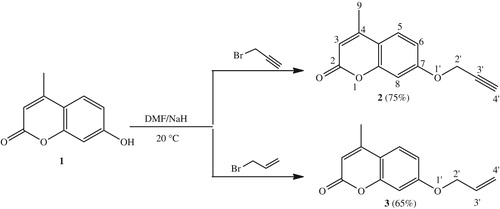
In recent years, the click chemistry is a powerful tool, allowing synthetic chemists to obtain selectivity, simple experimental set-up, tolerance to various functional groups and quantitative yieldsCitation36. The click reaction between azide and terminal alkyne catalyzed by Cu(I) has extensively facilitated the practical and efficient regiospecific formation of 1,4-disubstituted 1,2,3-triazolesCitation44. With this method, the reaction of the dipolarophile 2 with different arylazides generated in situ and copper (I) iodide catalyzed, in the presence of catalytic amount of triethylamine (3 eq) in refluxing toluene for 6–12 h, led to the formation of new triazoles linked 4-methylumbelliferone (Scheme 2). The isolated product yields were in the range of 50–55%. The structures of these compounds were confirmed according to their spectral data, where the 1H NMR spectra of compounds 4a–e indicated the existence of the triazolic moieties by showing essentially the resonance of the protons introduced by the arylazides. The 1H NMR spectrum for 4a as an example, showed a singlet at δH 5.36 relative to the methylenic protons H6′ and another singlet at δH 8.13 corresponding to the H5′ triazole proton. The 13C NMR spectrum showed a signal at δH 61.7 for the –O–CH2 (C6') carbon and two signals at δC 120.1 and 143.2 for the two characteristic triazole carbons C5′ and C4′, respectively. The mass spectrum recorded in ES-HRMS showed a pseudo-molecular ion peak [M + H]+ at m/z 334.1192 which is consistent with its molecular formula C19H16N3O3. The resulting 1,4-regioisomers were evidenced from the NOEs between H5′ (triazole)/H6′ (1.85 Å) and H5′ (triazole)/Harom (1.30 Å) and the absence of any NOE between H6′/Harom.
Scheme 2. Synthetic route of triazoles 4.
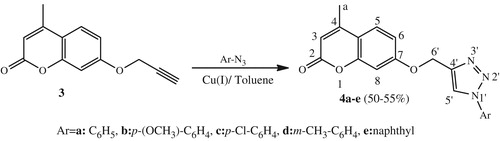
The 1,3-dipolar cycloaddition has became widely used as a highly efficient method for the synthesis of isoxazolesCitation45. Using this method, the reaction of the dipolarophile 2 with different aylnitrile oxides in refluxing toluene with a catalytic amount of triethylamine (3 eq) for 12 h led to the formation of new isoxazoles 5a–e (Scheme 3) in good to moderate yields (35–60%). The 1H NMR spectrum of compound 5a as an example showed in addition of the signals corresponding to the protons introduced by the dipole the appearance of a singlet at δH 5.30 relative to the proton H2′ and another singlet at δH 6.71 corresponding to the proton H4′. The 13C NMR spectrum of this cycloadduct 5a was also a good support for the proposed structure which exhibited characteristic signals at δC 60.9 corresponding to O–CH2 carbon, and at δC 101.4, 160.0, and 166.6 relative to isoxazoline carbons C4′, C3′, and C5′, respectively.
Scheme 3. Synthetic route of isoxazoles 5a–e and isoxazolines 6a–e.
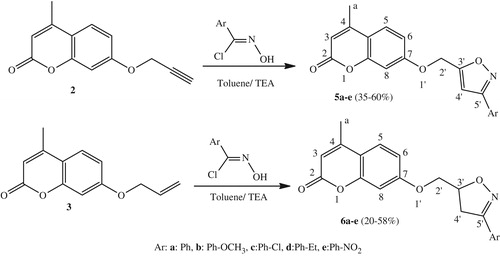
In another hand, we studied the behavior of the dipolarophile 3 with some arylnitrile oxides under conventional conditions by refluxing in toluene in the presence of a catalytic amount of triethylamine for 12 h. The reaction led to the formation of new cycloadducts 6a–e (20–58%) (Scheme 3).
Their structures were established on the basis of their spectral data (1H, 13C NMR, and ES-HRMS). In fact, the 1H NMR spectrum of compound 6a, as an example, showed the presence of two doublets of doublets at δH 3.40 (J = 16.8 Hz, J = 7.2 Hz) and at δH 3.56 (J = 16.8 Hz, J = 10.8 Hz) corresponding to the protons H4′a and H4′b, two other doublets of doublets at δH 4.15 (J = 14.1 Hz, J = 7.5 Hz) and δH 4.23 (J = 14.1 Hz, J = 5.1 Hz) attributable to protons H2′a and H2′b. The same spectrum showed a multiplet centered at δH 5.13 attributable to the H3′ of the stereogenic center. Further, the 13C NMR spectrum of this compound showed in addition to the carbons introduced by the dipole, the presence of a signal at δC 68.6 relative to –O–CH2 carbon and two signals at δC 36.9 and 77.9 relative to the isoxazoline carbons C4′ and C3′, respectively.
The resulting regiochemistry was evidenced from the NOEs between H4′ (isoxazoline)/H2′ (1.88 Å) and H4′ (isoxazoline)/Harom (1.32 Å) and the absence of any NOE between H2′/Harom.
The structures of cycloadducts 5a–e and 6a–e were further confirmed by mass spectrometry.
Following the same way, the dipolarophile 3 was treated according to the 1,3-dipolar cycloaddition with various arylazides as a dipole in refluxing toluene for 6 h, providing with complete regiosepecificity the awaited 1,2,3-triazoles I which lose a molecule of nitrogen to yield the corresponding aziridines 7a–e (Scheme 4) in excellent to moderate yields (65–75%).
Scheme 4. Synthetic route of aziridines 7a-e.
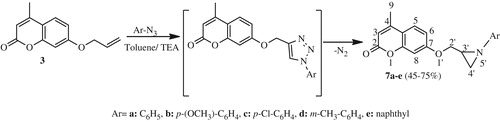
The structure of compounds 7a–e has been evidenced via their 1H and 13C NMR spectra. The 1H NMR spectrum of compound 7a as an example showed in addition to the protons introduced by the dipole the appearance of two doublets at δH 2.15 (J = 6.3 Hz) and at δH 2.25 (J = 3.3 Hz) attributable to the methylenic protons H4′a and H4′b of the aziridine ring, two doublets of doublets at δH 3.94 (J = 10.5 Hz, 7.5 Hz) and at δH 4.25 (J = 10.5 Hz, 3.9 Hz) corresponding to the diastereotopic methylenic protons H2′a and H2′b and a multiplet centered at δH 2.50 attributable to the proton H3′ of the stereogenic center. Further, the 13C NMR spectrum of the same compound showed the presence of signals at δC 30.7 and δC 37.3 relative to aziridine carbons C4′ and C3′, respectively and a signal at δC 70.1 corresponding to –O–CH2 carbon. The structures of compound 7a–e were further confirmed by their mass spectra.
Antibacterial activity
The antibacterial activity of the newly synthesized compounds 2, 3, 4, 5, 6, and 7 were evaluated against some bacteria, namely P. syringae pv, Syringae, P. savatanoi, and A. tumefasciens. The results mentioned in showed the inactivity of compound 1 against all the bacteria used. The antibacterial effect of some synthesized hybride molecules from the series 4, 5, 6, and 7 showed the importance of the added fragments to 4-methylumbelliferone 1 via the linker methylene to have activity. The same results showed that the propargylated compound 3 is slightly more active than the allylated derivative 2 against the three bacterial strains used. This slight difference in activity may be explained by the linearity of the propargyl moiety in comparison to the planar structure of the allyl group in compound 2.
Table 1. Antibacterial activity of 4-methylumbelliferone and its synthesized derivatives 2, 3, 4a–e, 5a–e, 6a–e, and 7a–e.
Most 1,4-triazoles 4 and isoxazoles 5 did not display any activity against the used bacteria. However, compound 4b bearing a p-MeO-C6H4 as a substituent at its triazole moiety which exhibited a moderate activity toward P. syringae pv, Syringae, and P. savatanoi (IZ = 8 mm), also compound 4e with a p-NO2-C6H4 in the trizaole ring displayed activity only against P. syringae pv, Syringae (IZ = 7 mm). In the series 5, only derivatives 5d and 5e bearing a m-CH3-C6H4 and p-NO2-C6H4, respectively, as substituents at the isoxazole moiety, were found to be active toward A. tumefasciens (IZ = 10 and 9 mm, respectively).
Most isoxazolines 6 displayed an acceptable antibacterial activity against A. tumefasciens (IZ = 8.5–10 mm) except compound 6a with a phenyl group in its isoxazoline moiety. On the other hand, only compounds 6a and 6d bearing a phenyl and m-CH3-C6H4, respectively, as substituents at their isoxazoline system, exhibited the same activity toward P. savatanoi (IZ = 10.5 mm), while against P. syringae pv, Syringae only compound 6a (C6H5) showed an activity (IZ = 11.5 mm). These finding showed that the noted activities of compounds 6a–e against the three bacteria used varies depending on the aryl group introduced by each aldehyde used in their preparation and the sensitivity against these compounds depends of the strain used.
The most significant activity of isoxazolines 6 compared to those of the isoxazoles 5 bearing the same aryl groups especially against A. tumefaciens allowed to conclude the importance of the spatial arrangement of the isoxazoline ring in compounds 6 with the creation of a stereocenter (C3′), which certainly influenced the activity in one direction or another. We cite as an example the case of compounds 6a and 6d which have been found active against P. savatanoi (IZ = 10.5 mm) while the corresponding isoxazoles 5a and 5d were found to be inactive against the same bacteria and unlike isoxazoles 5d (IZ = 10 mm) and 5e (IZ = 9 mm) are slightly more active than the corresponding iosoxazolines 6d (IZ = 8.5 mm) and 6e (IZ = 8.5 mm) against A. tumefaciens.
Most compounds 7a–e displayed an antibacterial activity against the microorganisms used.
Therefore, compound 7a bearing a phenyl in its aziridine moiety exhibited a good activity (IZ = 13 and 11 mm) against P. savatanoi and A. tumefasciens, respectively. This result compared to those of the other derivatives from the same series shows that the presence of the non substituted phenyl group introduced by the arylazide used seems at the origin of this relatively high activity.
Compounds 7c with a p-Cl-C6H4 (IZ = 11 mm) exhibited a good activity against P. syringae pv, Syringae compared to the rest of aziridines. This result can be explained by the presence of a chlorine atom in para position of the N-linked phenyl group in the aziridine fragment.
On the other hand, it has been noticed that the arylation of the nitrogen atom in the aziridine moiety in compound 7e by a naphthyl group resulted in the loss of activity toward A. tumefasciens whereas this substituent was found in favor of the activity of the same derivative against both P. syringae pv, Syringae, and P. savatanoi (IZ = 10.5 and 10 mm, respectively). This finding reinforces the independence of action of this compound against three strains used.
Overall, from all the results explained above and given in , we found that the aziridine system introduced with various substituents on its nitrogen atom in compounds 7 brings more antibacterial efficiency than isoxazoles (5) and isoxazolines (6) and even propargyl (2) and allyl (3) fragments and appears to act independently of the bacterial strain.
Antifungal activity
As it can be concluded from the data given in , only compounds 6a and 6d showed activity against P. italicum (IZ = 9.5 mm) and A. flavus (IZ = 9.5 mm), respectively. Despite the presence of the coumarin moiety in all tested molecules 2, 3, 4, 5, and 7, no significant activity was obtained. This finding let us to think that the linkage of proargyl, allyl, triazoles, isoxazoles, and aziridines systems to 4-methylumbelliferone 1, was not probably in favor of this activity but the antifungal potent of compounds 6 appears due to the nature of the aryl group attached to the isoxazoline ring.
Table 2. Antifungal activity of compounds 6a and 6d.
Anticoagulant activity
The in vitro anticoagulant activity of the 4-methylumbelliferone 1 and its synthesized derivatives 2, 3, 4a–e, 5a–e, 6a–e, and 7a–e were assessed by measuring the activated partial thromboplastin time at three different concentrations 200, 500, and 1000 μg/mL. The results given in showed that compounds tested at the concentration of 1000 μg/mL exhibited moderate to significant anticoagulant activities (aPTT = 41.7–74.3 s).
Table 3. Anticoagulant activity of 4-methylumbelliferone 1 and its synthesized derivatives 2, 3, 4a–e, 5a–e, 6a–e, and 7a–e.
As shown in , the aPTT assay indicated that the starting material 4-methylumbelliferone 1 (aPTT = 41 s) was found to be significantly less active than all the synthesized hybride molecules (aPTT = 54.6–74.3 s). This finding showed the contribution of all the fragments introduced (propargyl, allyl, triazole, isoxazole, isoaxazoline, and aziridine) each react in its own way to improve the anticoagulant activity of 4-methylumbelliferone 1.
As can be seen in , the O-allylated derivative 3 (aPTT = 72.7 s) was found to be more active than the O-propargylated one 2 (aPTT = 63.9 s). This finding may be interpreted by the planar structure of the allyl group in compound 3 in comparison to the linearity of the propargyl moiety in compound 2.
The results obtained show that compounds 4a–e displayed remarkable anticoagulant activity with aPTT values ranging from 55.6 s (compound 4a) to 60.8 s (compound 4c). The variation of this activity is easily explained by the nature of the aromatic system attached to the triazole ring. In fact, the slightly important activity of compounds 4b (p-OCH3-C4H6, aPTT = 58.6 s), 4c (p-Cl-C6H4, aPTT = 60.8 s), and 4d (m-CH3-C6H4, aPTT = 56.8 s) compared to that of 4a (C6H5, aPTT = 55.6 s) showed that the substitution of the phenyl system could be in favor of the anticoagulant activity. On the other hand, it was found that the atom of chlorine (in compound 4c) was the substituent which brought more activity compared to the others from the same series. Also we noticed that the naphthyl group in the compound 4e (aPTT = 59.7 s) has not remarkably improved the activity compared to that of 4a (aPTT = 55.6 s) where we have a phenyl group. The extension of the field of conjugation of the nitrogen doublet with the naphthyl group may be at the origin of this slight improvement of the noted activity.
Similarly, compounds 5a–e acted almost in the same way as compounds 4. Their activities ranged from 54.6 to 59.9 s and the compound 5a (aPTT = 54.6 s) with an unsubstituted phenyl group was found the least active in the series while compound 5d (aPTT = 59.9 s) bearing a m-CH3-C6H4 proved the most active.
For compounds 6a–e, the anticoagulant activity varies between aPTT =57.2 and 63.5 s. Compound 6e with a p-NO2-C6H4 showed the highest activity (aPTT = 63.5 s), while the less active compound in this series is 6d (aPTT = 57.2 s) whose phenyl borne by the isoxazoline ring is para-ethylated. On the other hand, the comparable activity of compounds 6b (p-OCH3-C6H4, aPTT = 63.2 s) and 6e (p-NO2-C6H4, aPTT = 63.5 s) shows that the donor or attractor electronic effect exerted by the substituent on the aromatic ring attached to the isoxazoline moiety does not appear to affect considerably the activity but the nature of the substituent seems behind the difference in activity between the various compounds. The relatively low activity of isoxazoles 5 compared to that of most of isoxazolines 6 can only be explained by the appearance of the new seterocenter C3′ in the structure of the latters.
Compounds 7a–e displayed anticoagulant activity (aPTT = 54.9–74.3 s) comparable to that of the other tested derivatives except compound 7d bearing a m-CH3-C4H6 substituent in its aziridine ring (aPTT = 74.3 s). The comparison of this activity to that of the rest of the compounds of the same series shows clearly the certain contribution of the m-CH3-C4H6 fragment in the noted activity. This derivative was found to be the most active among all the tested compounds.
As shown in , the most active compounds 3 and 7d at the concentration of 1000 mg/mL exhibited remarkable anticoagulant activity as indicated by the significant prolongation of aPTT in a dose-dependent manner.
Table 4. Anticoagulant activity of compounds 3 and 7d determined by measuring the activated partial thromboplastin time (s) at different concentrations.
Anticholinesterase activity
These results were expressed as IC50 values showing the activity of the synthesized of 4-methylumbelliferone 1 and most of its synthesized derivatives as shown in . The results showed that the dipolarophile 3 (IC50 = 0.3266 ± 0.01 mg/mL) was found to be more active than its analogous 2 (IC50 = 0.4131 ± 0.015 mg/mL). This finding could probably be due to the planar structure of the allyl group in compound 3 compared to the linearity of the propargyl moiety in compound 2. Moreover, the high activity of these alkylated derivatives 2 and 3 compared to that of the starting material 1 (IC50 = 0.692 ± 0.055 mg/mL) showed the contribution of the two alkyl fragments introduced to the improvement of the activity of the latter.
Table 5. Anticholinesterase activity of 4-methylumbelliferone 1 and most of its synthesized derivatives 2, 3, 4, 5, 6, and 7.
On the other hand, the obtained results showed the certain intervention of the triazole ring introduced in the compounds 4a (IC50 = 0.326 ± 0.010 mg/mL), 4b (IC50 = 0.360 ± 0.012 mg/mL), and 4d (IC50 = 0.334 ± 0.021 mg/mL) to practically double the activity of compound 1 (IC50 = 0.692 ± 0.055 mg/mL). The comparable inhibitory potency of compounds 4a (C6H5), 4b (p-OCH3-C6H4), and 4d (m-CH3-C6H4) allows to think that the nature of the aryl fragment attached to the corresponding triazole in these three cases seems without effect. The limited number of tested derivatives (only three) fails to consider unambiguously that the aromatic system attached at C2 of the triazole ring manages the behavior of these compounds against BChE. The diversification of fragments borne by the triazole ring in a larger series of derivatives could lead to a plausible conclusion.
All the isoxazoles derivatives 5a–e (IC50 = 0.634–0.390 mg/mL) proved to inhibit more the BChE than the starting compound 1 (IC50 = 0.692 ± 0.055 mg/mL). This finding showed the importance of the isoxazole moiety to improve the anti-BChE potential of compounds 5. The derivatives 5d and 5e bearing a p-Et-C6H4 and a p-NO2-C6H4 borne by their isoxazoline rings were found to be the most active (IC50 = 0.390 ± 0.012 and 0.495 ± 0.020 mg/mL, respectively). This result shows the role of the substitutent fixed in para position of the aromatic ring, indeed the ethyl group (electron-donating) in compound 5d seems to have more effect to enhance the contribution of the isoxazole to improve the activity of compound 1.
On the other hand, the electron-withdrawing group (NO2) in para position of the aryl system borne by the isoxazole in compound 5e may explain the relatively high activity of the latter.
Except the derivative 6b (p-OCH3-C6H4) (IC50 = 0.865 ±0.015 mg/mL), the cycloadducts 6a (IC50 = 0.338 ± 0.010 mg/mL), 6d (IC50 = 0.415 ± 0.018 mg/mL), and 6e (IC50 = 0.183 ± 0.020 mg/mL) have been shown to be more active than the starting material 1 (IC50 = 0.692 ± 0.055 mg/mL). This finding still shows the effect of the NO2 group observed in the case of 5e which is added that of the new stereocenter C3′. The same phenomena was noted with compound 6a (IC50 =0.338 ± 0.010 mg/mL) when compared to its isoxazole analogous 5a (IC50 = 0.556 ± 0.010 mg/mL). In the case of the ethyl group as substituent, our results did not show any remarkable effect of the newly formed stetreocenter to improve or to reduce the anti-BChE activity of compound 6d (IC50 = 0.415 ± 0.018 mg/mL) compared to that of its analogous 5d (IC50 = 0.390 ± 0.012 mg/mL). In the contrary, in the case of compound 6b (IC50 = 0.865 ± 0.015 mg/mL), the co-presence of the p-OCH3-C6H4 and the new stereocenter C3′ in the isoxazoline moiety reduce considerably the anti-BChE activity of 4-methylumbelliferone 1. The above results allowed concluding that the activity of compounds 6 depends along the new asymmetric center and the aryl group fixed at the isoxazoline moiety.
Compounds 7a–e were found considerably more active than 4-methylumbelliferone 1. It is clear that independently of the aryl group attached, the aziridine moiety has improved the anti-BChE activity of 4-methylumbelliferone 1. Our results showed that the activity was improved more than 4.5 times when we joined the 4-methylumbelliferone 1 to an aziridine moiety bearing a p-chlorophenyl ring through the spacer methylene (compound 7c, IC50 = 0.151 ± 0.010 mg/mL). The activity of this series varies depending on the aryl group attached to the aziridine, it decreases in the order 7c (p-Cl-C6H4, IC50 = 0.151 ± 0.010 mg/mL), 7a (C6H5, IC50 = 0.243 ± 0.020 mg/mL), 7e (p-Cl-C6H4, IC50 = 0.292 ± 0.020 mg/mL), 7d and 7b (p-OCH3-C6H4, IC50 = 0.427 ± 0.012 mg/mL). This finding shows clearly the effect of the aryl group attached to the aziridine ring on the anti-BChE activity.
Conclusion
The present work constitutes a contribution to the valorization of 4-methylumbelliferone 1 as a coumarin derivative to access to new biaocative hybride molecules. This coumarin has been transformed into two propargylated and allylated dipolarophiles 2 and 3, respectively to be used as precursors for synthesizing the target coumarin-containing triazole, isoxazoles, isoxazolines, and aziridines via intermolecular 1,3-dipolar cycloaddition reactions. The newly prepared cycloadducts have been tested for their possible antimicrobial, anticoagulant and anticholinesterase activities. The study of the SAR permitted to conclude in most cases the contribution of the added heterocyle (triazle, isoxazole, isoxazoline, and aziridine) to the starting coumarin via the methylene group as a linker to improve or even to bring activity. The nature of the aryl fragment attached to the added heterocyles and the created stereocenter in the case of the isoxazolines 6 have been the origin of some obtained results.
Declaration of interest
The authors declare no conflicts of interests. The authors alone are responsible for the content and writing of this article.
References
- Vaarla K, Kesharwani RK, Santosh K, et al. Synthesis, biological activity evaluation and molecular docking studies of novel coumarin substituted thiazolyl-3-aryl-pyrazole-4-carbaldehydes. Bioorg Med Chem Lett 2015;25:5797–803
- Thakur A, Singla R, Jaitak V. Coumarins as anticancer agents: a review on synthetic strategies, mechanism of action and SAR studies. Eur J Med Chem 2015;101:476–95
- Melagraki G, Afantitis A, Igglessi-Markopoulou O, et al. Synthesis and evaluation of the antioxidant and anti-inflammatory activity of novel coumarin-3-aminoamides and their alpha-lipoic acid adducts. Eur J Med Chem 2009;44:3020–6
- Lad HB, Giri RR, Brahmbhatt DI. An efficient synthesis of some new 3-bipyridinyl substituted coumarins as potent antimicrobial agents. Chin Chem Lett 2013;24:227–9
- Razavi SF, Khoobi M, Nadri H, et al. Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem 2013;64:252–9
- Dubery IA. Effect of hydroxylated and methoxylated coumarins on the regulatory properties of phenylalanine ammonia-lyase from Citrus sinensis. Phytochemistry 1990;29:2107–8
- El MA, El Hadrami EM, Ben-Tama A, et al. Synthesis and characterization of new 1,4 and 1,5-disubstituted glucopyranosyl 1,2,3-triazole by 1,3-dipolar cycloaddition. J Mol Struct 2009;929:6–9
- Sarmiento-Sánchez JI, Ochoa-Terán A, Rivero IA. Conventional and microwave assisted synthesis of 1,4-disubstituted 1,2,3-triazoles from Huisgen cycloaddition. Arkivoc 2011;ix:177–88
- Ramprasad J, Nayak N, Dalimba U, et al. One-pot synthesis of new triazole-imidazo[2,1-b][1,3,4]thiadiazole hybrids via click chemistry and evaluation of their antitubercular activity. Bioorg Med Chem Lett 2015;25:4169–73
- Ali AA, Chetia M, Saikia B, et al. AgN(CN)2/DIPEA/H2O-EG: a highly efficient catalytic system for synthesis of 1,4-disubstituted-1,2,3 triazoles at room temperature. Tetrahedron Lett 2015;56:5892–5
- Avanzo RE, Anesini C, Fascio ML, et al. 1,2,4-Triazole D-ribose derivatives: design, synthesis and antitumoral evaluation. Eur J Med Chem 2012;47:104–10
- Jiang Z, Gu J, Wang C, et al. Design, synthesis and antifungal activity of novel triazole derivatives containing substituted 1,2,3-triazole-piperdine side chains. Eur J Med Chem 2014;82:490–7
- Mavrova AT, Wesselinova D, Tsenov JA, Lubenov LA. Synthesis and antiproliferative activity of some new thieno[2,3-d]pyrimidin-4(3H)-ones containing 1,2,4-triazole and 1,3,4-thiadiazole moiety. Eur J Med Chem 2014;86:676–83
- Bhalgat CM, Irfan Ali M, Ramesh B, Ramu G. Novel pyrimidine and its triazole fused derivatives: synthesis and investigation of antioxidant and anti-inflammatory activity. Arabian J Chem 2014;7:986–93
- Strakova I, Turks M, Strakovs A. Synthesis of triazole-functionalized tetrahydroindazolones by 1,3-dipolar cycloadditions between azides and alkynes. Tetrahedron Lett 2009;50:3046–9
- Shakeel-u-Rehman Masood-ur-Rahman Tripathi VK, et al. Synthesis and biological evaluation of novel isoxazoles and triazoles linked 6-hydroxycoumarin as potent cytotoxic agents. Bioorg Med Chem Lett 2014;24:4243–6
- Boz E, Tüzün NŞ. Reaction mechanism of ruthenium-catalyzed azidee-alkyne cycloaddition reaction: a DFT study. J Organomet Chem 2013;724:167–76
- Rao PS, Kurumurthy C, Veeraswamy B, et al. Synthesis of novel 5-(3-alkylquinolin-2-yl)-3-aryl isoxazole derivatives and their cytotoxic activity. Bioorg Med Chem Lett 2014;24:1349–51
- Changtam C, Hongmanee P, Suksamrarn A. Isoxazole analogs of curcuminoids with highly potent multidrug-resistant antimycobacterial activity. Eur J Med Chem 2010;45:4446–57
- Schmidtke M, Wutzler P, Zieger R, et al. New pleconaril and [(biphenyloxy)propyl]isoxazole derivatives with substitutions in the central ring exhibit antiviral activity against pleconaril-resistant coxsackievirus B3. Antiviral Res 2009;81:56–63
- Eddington ND, Cox DS, Roberts RR, et al. Synthesis and anticonvulsant activity of enaminones. 4. Investigations on isoxazole derivatives. Eur J Med Chem 2002;37:635–48
- Kaur K, Kumar V, Sharma AK, Gupta GK. Isoxazoline containing natural products as anticancer agents: a review. Eur J Med Chem 2014;77:121–33
- Bano S, Alam MS, Javed K, et al. Synthesis, biological evaluation and molecular docking of some substituted pyrazolines and isoxazolines as potential antimicrobial agents. Eur J Med Chem 2015;95:96–103
- Ghidini E, Capelli AM, Carnini C, et al. Discovery of a novel isoxazoline derivative of prednisolone endowed with a robust anti-inflammatory profile and suitable for topical pulmonary administration. Steroids 2015;95:88–95
- Ismail FMD, Levitsky DO, Dembitsky VM. Aziridine alkaloids as potential therapeuticagents. Eur J Med Chem 2009;44:3373–87
- Nonn M, Kiss L, Forró E, et al. Synthesis of densely functionalized cispentacin derivatives through selective aziridination and aziridine opening reactions: orthogonally protected di- and triaminocyclopentanecarboxylates. Tetrahedron 2014;70:8511–19
- Degennaro L, Trinchera P, Luisi R. Recent advances in the stereoselective synthesis of aziridines. Chem Rev 2014;114:7881–929
- D’hooghe M, Kenis S, Vervisch K, et al. Synthesis of 2-(aminomethyl)aziridines and their microwave-assisted ring opening to 1,2,3-triaminopropanes as novel antimalarial pharmacophores. Eur J Med Chem 2011;46:579–87
- Alcaide A, Llebaria A. Synthesis of 1-thio-phytosphingolipid analogs by microwave promoted reactions of thiols and aziridine derivatives. Tetrahedron Lett 2012;53:2137–9
- Cao L, Liu C, Tang X, et al. Highly selective synthesis of 1- polyfluoroaryl-1,2,3-triazoles via a one-pot three-component reaction. Tetrahedron Lett 2014;55:5033–7
- Kumar V, Kaur K. Fluorinated isoxazolines and isoxazoles: a synthetic perspective. J Fluorine Chem 2015;180:55–97
- Venugopala KN, Rashmi V, Odhav B. Review on natural coumarin lead compounds for their pharmacological activity. Biomed Res Int 2013;2013:963248
- Medjahed W, Tabet Zatla A, Kajima Mulengi J, et al. The synthesis of N-acyl-2-hydroxymethyl aziridines of biological interest. Tetrahedron Lett 2004;45:1211–13
- Zayane M, Romdhane A, Daami-Remadi M, Ben Jannet H. Access to new antimicrobial 4-methylumbelliferone derivatives. J Chem Sci 2015;127:1619–26
- Aziz-ur-Rehman IT, Abbasi MA, Nafeesa K, et al. Synthesis, spectral characterization and α-Chymotrypsin activity of 7-O-substituted derivatives of 7-hydroxy-4-methyl-1-benzopyran-2-one. Asian J Chem 2014;26:326–30
- Totobenazara J, Burke AJ. New click-chemistry methods for 1,2,3-triazoles synthesis: recent advances and applications. Tetrahedron Lett 2015;56:2853–9
- Marmonier A. Antibiotiques Technique de diffusion en gélose méthode des disques. Bactériologie Médicale Techniques Usuelles. Paris: SIMEP SA; 1987;4:237–43
- Barry AL, Thornsberry C. Susceptibility tests: diffusion test procedures. In: Balows Hausler WJ Jr, Heraman KL, Isenberg HD, Shadomy HJ, eds. Manual of clinical microbiology. 5th ed. Washington (DC): American Society for Microbiology; 1991:1117–25
- Ben Mansour M, Dhahri M, Hassine M, et al. Highly sulfated dermatan sulfate from the skin of the ray Raja montagui: anticoagulant activity and mechanism of action. Comp Biochem Physiol 2010;156B:206–15
- Ben Nejma A, Nguir A, Ben Jannet H, et al. New septanoside and 20-hydroxyecdysone septanoside derivative from Atriplex portulacoides roots with preliminary biological activities. Bioorg Med Chem Lett 2015;25:1665–70
- Fan G, Mar W, Park MK, et al. A novel class of inhibitors for steroid 5α-reductase: synthesis and evaluation of umbelliferone derivatives. Bioorg Med Chem Lett 2001;11:2361–3
- Oh JK, Stöeva V, Rademacher J, et al. Synthesis, characterization,and emulsion polymerization of polymerizable coumarin derivatives. J Polym Sci Part A Polym Chem 2004;42:3479–89
- Kosiova I, Kovackova S, Kois P. Synthesis of coumarin–nucleoside conjugates via Huisgen 1,3-dipolar cycloaddition. Tetrahedron 2007;63:312–20
- Goyard D, Praly JP, Vidal S. Synthesis of 5-halogenated 1,2,3-triazoles under stoichiometric Cu(I)-mediated azide–alkyne cycloaddition (CuAAC or ‘Click Chemistry’). Carbohydr Res 2012;362:79–83
- Mabrour M, Bougrin K, Benhida R, et al. An efficient one-step regiospecific synthesis of novel isoxazolines and isoxazoles of N-substituted saccharin derivatives through solvent-free microwave-assisted [3 + 2] cycloaddition. Tetrahedron Lett 2007;48:443–7