
Abstract
Fentanyl is a powerful opiate analgesic typically used for the treatment of severe and chronic pain, but its prescription is strongly limited by the well-documented side-effects. Different approaches have been applied to develop strong analgesic drugs with reduced pharmacologic side-effects. One of the most promising is the design of multitarget drugs. In this paper we report the synthesis, characterization and biological evaluation of twelve new 4-anilidopiperidine (fentanyl analogues). In vivo hot-Plate test, shows a moderate antinociceptive activity for compounds OMDM585 and OMDM586, despite the weak binding affinity on both μ and δ-opioid receptors. A strong inverse agonist activity in the GTP-binding assay was revealed suggesting the involvement of alternative systems in the brain. Fatty acid amide hydrolase inhibition was evaluated, together with binding assays of cannabinoid receptors. We can conclude that compounds OMDM585 and 586 are capable to elicit antinociception due to their multitarget activity on different systems involved in pain modulation.
Introduction
Opioids have been used for years in the control of pain from moderate to severe, and with controversial results especially in the management of chronic and neuropathic pain.
Addiction and tolerance, including nausea and vomiting, constipation and respiratory depression are well known adverse side effectsCitation1. Fentanyl is the prototype of the 4-anilidopiperidines opioid analgesics and is characterized by high potency and efficacy. Expansion of the structure-activity relationships (SAR) of fentanyl derivatives has led to the discovery of new morphinomimetics with diverse pharmacological profilesCitation2. Numerous research groups have studied the structure’s modifications of fentanyl, with the aim to reduce the pharmacologic side effects, acting on other opioid receptors such as the δ-receptorCitation3. It has been demonstrated that μ and δ-opioid receptors form heterodimers and their simultaneous stimulation can give a better antinociceptive activityCitation4. Fentanyl has been previously employed in the design of bivalent drugs by Hruby et al. Citation5 in light of the well-documented interaction between opioid and cannabinoid receptorsCitation6.
Stimulated by the previously described SAR on fentanyl and its bivalent analogues, which reported no general rules for the effect of the substitution on the propionyl amide moiety,Citation5 we synthesised 12 novel 4-anilidopiperidine derivatives replacing the tertiary propyl-amide group in fentanyl, with different aryl-ureas and aryl-carbamates found in several active FAAH and MAGL inhibitors with the intent to develop novel multitarget analgesics.Citation7,Citation8 FAAH and MAGL are hydrolase enzymes, responsible of the endocannabinoid cleveage, and their inhibitors have shown to possess nociceptive effects in different pain models, as a consequence of the increased level of endocannabinoidsCitation9,Citation10.
A first series of compounds bearing the 4-anilidopiperidine scaffold on fentanyl was synthesised, and several substituted O-arylcarbamates (compounds OMDM584–OMDM589) have been used in place of the propylamide function. A second series of N-arylureas was designed replacing the N-propyl moiety of fentanyl with several substituted N-aryl building blocks (compounds OMDM590–OMDM595). The synthesis of all compounds is depicted in Scheme 1, structural features are reported in .
Scheme 1. Reagents and conditions: (a) 1-phenethyl-N-phenylpiperidin-4-amine (0.5 mmol), Et3N (0.6 mmol), DCM, r.t. overnight.
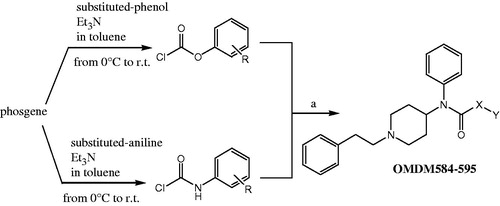
Table 1. Structures and yields of products OMDM584–595.
Results and discussion
Chemistry
Carbamates OMDM584–589 were synthesised starting from the corresponding chloroformate intermediate, which was prepared using phosgene in toluene with the appropriate substituted phenol and triethylamine (Scheme 1).
The so obtained chloroformate was reacted with a solution of 1-phenethyl-N-phenylpiperidin-4-amine in DCM in presence of triethylamine at room temperature, to yield the desired crude product. In the same way, ureas OMDM590–595 were obtained from the corresponding urethane intermediates, which were prepared using phosgene in toluene with the appropriate substituted anilines and triethylamine (Scheme 1).
Then urethane intermediate was reacted with a solution of 1-phenethyl-N-phenylpiperidin-4-amine in DCM in presence of triethylamine at room temperature, to yield the desired crude product.
All crude compounds were purified on silica gel chromatography to give final products in good yields ().
In vitro assay
Biological in vitro tests were performed on all products, for μ and δ-opioid receptor affinity, on FAAH and MAGL ().
Table 2. In vitro assays on FAAH and MAGL inhibition.
Receptor binding assays
Competition binding experiments
In competition binding experiments, fentanyl reduced the [3H] DAMGO specific binding on μ-opioid receptor (MOR) with a very low 5.99 nM IC50 value, indicating a high affinity for MOR (Figure S1), as expected from previous reports, confirming the adequate performance of the radioligandCitation11.
The fentanyl analogues showed a wide range of affinity for MOR from mid nanomolar (IC50 > ∼500 nM) to micromolar (IC50 < ∼20 μM) (Figure S1) concentration. Compounds OMDM586, 591–593 showed the highest affinity for MOR with mid nanomolar IC50 values. Thus among the structural modifications of fentanyl, the combination of the amide group in position X with 4-, 3-chlorophenyl and 4-tert-butylphenyl groups in position Y (OMDM591–593) gives good affinity ().
Table 3. The affinity values (logIC50 ± SEM.) of OMDM584–595 compounds and fentanyl in [3H]DAMGO competition binding assays in rat brain membrane homogenates. The logIC50 values were calculated according to the competition binding curves (see Supplementary Figure 1S). for better comparison the two different basic structures are presented side by side.
Since compounds OMDM585 and 586 showed analgesic activities in pharmacological assays, we further characterized their binding for δ-opioid receptor (DOR), k-opioid receptor (KOR) and cannabinoid receptors (CBR), again in competition binding assays (Figure S2). Both compounds reduced total specific binding of the KOR selective [3H]U-69593 and DOR selective [3H]IleDelt II with an IC50 value in the micromolar range, showing poor affinity for these two opioid receptors (Fig S2A and B). In case of [3H]WIN55 212–2 binding experiments, the affinity of OMDM585 and 586 was weaker for CBR, since it decreased [3H]WIN55 212–2 specific binding by about 20–30% with an IC50 value in the micromolar range (Figure S2D, Table S1). For comparison the unlabelled forms of the tritiated ligands reported IC50 values in the nanomolar range, indicating that the radioligands performed adequately (Figure S2, Table S1).
[35S]GTPγS binding experiments
In G-protein activity stimulation measurements with [35S]GTPγS binding tests, fentanyl activated G-protein stimulation with a maximum efficacy of 159.8% (nearly 60% above basal activity) and with a potency of 110.4 nM (Table S2, Figure S3A and C), which corresponds with previous studiesCitation11.
Interestingly, all fentanyl analogues showed inverse agonist properties, since they reduced G-protein basal activity with very similar parameters: the maximum efficacy was between ∼80–100%, and their potency was between ∼3–5 μM (Figure S3A and C, Table S2). This effect however was not opioid receptor mediated, since it could not be reversed by the antagonist naloxone even in high, 10 μM concentrations, resulting in very similar maximum efficacy and potency values to those in the absence of naloxone (Figure S3B and D, Table S2). For comparison, the activity of fentanyl was markedly inhibited in the presence of naloxone and the G-protein activity reduced back to basal activity level (Figure S3B and D, Table S2). The inverse agonist properties of the fentanyl analogues are most probably due to the introduction of the benzene ring in position X, since this is shared by all analogues, but not by fentanyl (Scheme 1). In fact, the benzene ring together with the piperidine ring is also shared by rimonabant, which is an inverse agonist on CB1 receptorCitation12. However, the inverse agonist effect of rimonabant can be also mediated by a yet unidentified pertussis toxin sensitive GPCR.Citation13
Additionally, the fentanyl analogues described in this study also share some elements of the pharmacophore of other CB1 receptor inverse agonists, such as the two aromatic moieties, a hydrogen acceptor unit and a cyclic lipophilic part.Citation14
In vivo assays
Hot-Plate test was performed on all compounds to study the influence of different pharmacological properties on their analgesic profile. Results are shown in .
Figure 1. In vivo activity of compounds OMDM584–595 by hot-Plate test. Data from hot plate experiments. **** is for p < 0.001 versus Vehicle; ** is for p < 0.01 versus Vehicle; * is for p < 0.05 versus Vehicle. N = 10.
Hot-plate data confirm that compounds OMDM585, 586 have a good analgesic activity only at high doses. However, this cannot be explained by the opioid activity, as the binding to μ and δ-opioid receptors is very weak, furthermore the FAAH and MAGL inhibition activity was negligible.
The inverse agonist activity on the G-Protein stimulation leaves us with the hypothesis that products OMDM585 and 586 are able to elicit their antinociceptive activity either by interaction to block one or more excitatory circuits in the brain rather than by their affinity to μ opioid and cannabinoid receptors at micromolar range.
Molecular docking
According to the experimental results, the inactive state of μ-opioid receptor was used for docking (PDB code: 4dkl). The lowest docking energy poses of the analogues only partly matched with the docked pose of fentanylCitation15, as shown in Table S3. Furthermore, because the analogues reported weak binding affinity and the agonist character of the parent molecule fentanyl changed, it is feasible to assume that their binding mode differs from that of fentanyl. To check this assumption, a distance based interaction model was matched against the docking energies. The interaction model was constructed as follows: the distances between selected atoms of the ligands and the receptor were measured, and the square root of the sum of the squares of the Pairwise Distances was used as a measure of binding distance, as shown in EquationEquation 1(1) .
(1)
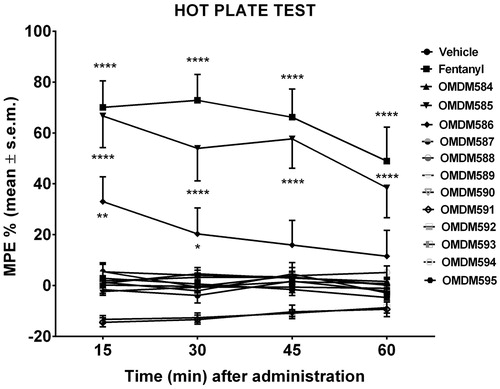
Where, is the distance between the ith selected atom of the ligand (li) and the receptor (ri), respectively. The corresponding atom pairs are shown in Table S4. The docked pose, possessing the minimum binding distance by the above formula, was selected for each ligand and the corresponding ligand efficiency and distance values were used to fit a linear regression model ().
Figure 2. Linear regression for the best interaction model of compounds OMDM584–595.
The Spearman rank correlation coefficients were calculated too. All atom pair combinations were checked to qualify the different interaction (binding) modes. The interaction models based on the previously published docking pose (Table S4, column A for ligand atoms), in which the anilide ring points toward the extracellular direction,Citation15 resulted in very weak regression models and mainly with negative slope. The negative slope is contradictory to the expectations that the deeper binding energy is a consequence of the shorter binding distance. This gave rise the possibility that the orientation of the bound analogues differ from that of fentanyl which can also be seen in Table S3.
To check this assumption, interaction models with the opposite orientation were tested (Table S4, column B for ligand atoms). This type of interaction models resulted in higher correlation between the ligand efficiencies and the calculated distances. The best model is shown in Figure S4.
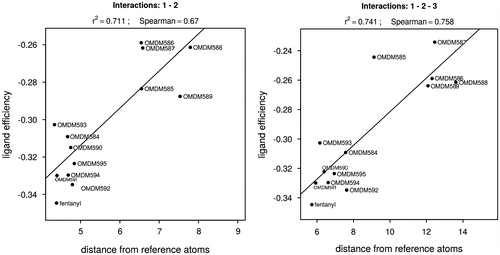
The key amino acids interactions are (i) Asp147-proton on piperidine N, (ii) Tyr326 side chain-anilide aromatic ring, (iii) Ile144 side chain-phenethyl aromatic ring ().
Figure 3. Definition of interactions; on the left: orientation similar to that reported by Micovic et al.Citation15 on the right: opposite orientation.
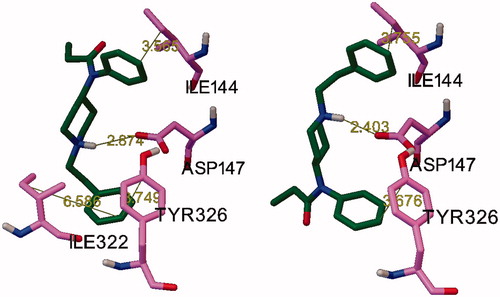
These results are in agreement with the finding that there are more sites to accommodate the analogues with the additional large substituent within the binding pocket in the orientation opposite to the literature pose of fentanyl. Comparing the different interaction models (atom pair combinations), the best linear fit was obtained with two atom pairs, 1 and 2 (Table S4). Involving any other interactions significantly decreased the linear fit, except in the case of the non-fentanyl-like orientation, for which the 1–2–3 interactions resulted in almost the same regression (, r2 = 0.741, Spearman’s rank correlation: 0.758).
The interaction modes of fentanyl analogues are shown in supplementary (Figure S4). The most important interaction between the synthesized analogues and MOR is a hydrophobic interaction. Compound OMDM584 interacts just with Tyr148 due to π-π stacking. OMDM585 can be stable in the binding site of MOR (Tyr128, Trp293 and Tyr326) via three π–π stacking interactions. However, the presence of some polar or charged residues in proximity of Fentanyl analogues may affect the MOR activation. The presence of Lys303 and Lys233, as the positive charged residues, in the vicinity of tert-butylphenyl moiety of OMDM587 can dramatically destabilize the protein:ligand complex. The same cases can be found for the other fentanyl analogues: Asp147 and Lys233 are located in the vicinity of phenyl and cholorophenyl moieties of OMDM585. Also, in the case of OMDM586, Lys233, Glu229 and Asn230 can have negative effects on MOR:ligand complex stability. In the active form of MOR, Tyr148 can participate in the hydrogen bonding network involving Lys233 and a water molecule. This study reported that the ionic interaction between ligand and Asp147 has been found in either active or inactive form of MOR. Based on our docking results, no similar interaction was found for fentanyl analogues.
Conclusions
Potential abuse, unwanted side effects and lack of analgesic efficacy are major problems in prescribing opioid analgesicsCitation16.
Bi- or poly-functional compounds can fight chronic and neuropathic pain at various levels, demonstrating some ability to cross the BBB, with reduced or none sign of tolerance and addiction, thus supporting the idea that a single drug with FAAH/MAGL inhibitor activity plus an opioid agonist activity, could be a very attractive design for the development of a “third generation” painkillersCitation6.
In this study, we investigated the potential of multi-target compounds combining opioid agonists and FAAH/MAGL inhibitors. The pharmacological profile of such compounds should join high efficacy in chronic pain states reducing the development of tolerance. Significant advantages over a multidrug approach could be: (i) oral administration, (ii) better pharmacokinetic properties, (iii) no drug–drug interactions and (iv) improved compartment targetingCitation17.
The data obtained so far, indicate that our products have weak affinity for μ-opioid receptors, and are not strong inhibitors of FAAH/MAGL. However, two products revealed a good antinociceptive activity but very weak potency when compared to fentanyl, therefore we concluded that the activity wasn’t mediated by the opioid system or endocannabinoids. In our studies, a strong inverse agonist activity in the stimulation of the G0/i protein was revealed, which can be related to several systems present in the brain, directly involved in pain modulation.
A direct binding to CBR1 receptor has been explored, but the so obtained data are in accord with the observed activity.
Further, in silico molecular docking studies have been performed and definitely ruled out the involvement of the known systems in the antinociceptive activity. The comparison of the best regression model for the conventional, fentanyl-like orientation and the opposite one indicates that the latter is more feasible. It suggests that the analogues may recognise MOR in a different manner than fentanyl, due to the size of the new substituents.
Based on these results, the most important interactions between the ligands and the receptor are (i) Asp147-proton on piperidine N, (ii) Tyr326 side chain-anilide aromatic ring, (iii) Ile144 side chain- phenethyl aromatic ring. This is in agreement with the experimental results showing that the analogues lost the agonistic character.
We may conclude that the pharmacologic profile observed in the murine tests may be explained by the modulation of several receptors in the brain, related to the stimulation of the cerebral activity and usually responsible for enhancing the noxious stimuli, like adrenergic, glutamaergic, substance P and others. Although it has been previously reported that the fentanyl analgesic activity could be readily maintained into small multitarget ligands, the incorporation of potent endocannabinoid activity is more intriguing and challengingCitation18.
Further studies are currently underway in our laboratories to design multitarget molecules with potent inhibitory activity at FAAH enzyme and as strong opioid agonists.
Experimental section
Material and methods
All chemical reagents were commercially available unless otherwise indicated. TLC was performed by using Merck F254 silica plates (Kenilworth, NJ) and components were analyzed by using UV light and an iodine chamber. Melting points were determined on a Buchi apparatus and are uncorrected. Column chromatographies were carried out using Merck silica gel 60 (230–400 mesh). IR spectra were recorded on a Spectrum-One FT-ATR spectrometer (Perkin-Elmer, Waltham, MA). Band position and absorption ranges are given in csup>/sup>. 1H and 13C NMR spectra were obtained on a Bruker Avance 400 MHz (Pittsburgh, PA), using CDCl3 as solvent.
Chemistry
Synthetic procedure for the carbamates OMDM584–589
To a stirred 15% phosgene solution in toluene (3.3 mL, 5 mmol) a solution of the appropriately-substituted phenol (1.0 mmol) and Et3N (0.168 mL, 1.2 mmol) in dry toluene (5 mL) was added drop-wise at 0 °C. The reaction mixture was stirred for 3 h at room temperature and evaporated under vacuum. The crude chloroformate was dissolved in dry DCM (4 mL) and a solution of 1-phenethyl-N-phenylpiperidin-4-amine (0.140 g, 0.50 mmol) and Et3N (0.084 mL, 0.6 mmol) in dry DCM (2 mL) was added drop-wise at room temperature with stirring. The reaction mixture was stirred overnight at room temperature, diluted with water, and extracted with AcOEt. The organic phase was washed with NaHCO3 s.s. and twice with water, dried (Na2SO4), and evaporated under vacuum. The residue was purified by column chromatography.
Phenyl(1-phenethylpiperidin-4-yl)(phenyl)carbamate (584)
Yield 80% (hexane/AcOEt, 7:3 as chromatographic eluent); mp 138–139 °C (from hexane/CH2Cl2); IR 2952, 2763, 1716, 1595, 1494, 1320, 1199, 1032, 991 csup>/sup>; 1H NMR (400 MHz) δ 1.59 (2H, m), 1.94 (2H, m), 2.15 (2H, m), 2.55 (2H, m), 2.74 (2H, m), 3.04 (2H, d, J = 11.6 Hz), 4.33 (1H, m), 7.12–7.34 (15H, m); 13C NMR (100 MHz) δ 30.79, 33.84, 53.04, 56.45, 60.41, 121.61, 125.18, 126.06, 127.92, 128.39, 128.63, 128.94, 129.10, 129.99, 137.73, 140.17, 151.36, 154.07. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H29N2O2+: 401.2151, found: 401.2168.
4-Chlorophenyl(1-phenethylpiperidin-4-yl) (phenyl)carbamate (585)
Yield 63% (hexane/AcOEt, 7:3 as chromatographic eluent); mp 134–135 °C (from hexane/CH2Cl2); IR 2951, 2764, 1714, 1595, 1488, 1319, 1210, 1089, 996 csup>/sup>; 1H NMR (300 MHz) δ 1.58 (2H, m), 1.94 (2H, m), 2.14 (2H, m), 2.54 (2H, m), 2.73 (2H, m), 3.03 (2H, d, J = 11.7 Hz), 4.31 (1H, m), 6.96–7.41 (14H, m); 13C NMR (75 MHz) δ 30.82, 33.87, 53.02, 55.65, 60.39, 122.96, 126.04, 128.02, 128.38, 128.61, 128.98, 129.08, 129.89, 130.44, 137.56, 140.18, 149.88, 153.62. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H28ClN2O2+: 435.1761, found: 435.1765.
3-Chlorophenyl(1-phenethylpiperidin-4-yl)(phenyl) carbamate (586)
Yield 51% (hexane/AcOEt, 75:25 as chromatographic eluent); mp 96–97 °C (from hexane/CH2Cl2); IR 2949, 2760, 1717, 1589, 1493, 1299, 1203, 1020, 993 csup>/sup>; 1H NMR (300 MHz) δ 1.58 (2H, m), 1.95 (2H, m), 2.14 (2H, m), 2.55 (2H, m), 2.73 (2H, m), 3.04 (2H, d, J = 11.7 Hz), 4.30 (1H, m), 6.94–7.41 (14H, m); 13C NMR (75 MHz) δ 30.78, 33.86, 53.01, 55.70, 60.38, 109.97, 119.98, 122.23, 125.40, 126.04, 128.05, 128.37, 128.61, 128.99, 129.77, 129.86, 134.30, 140.17, 151.85, 153.42. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H28ClN2O2+: 435.1761, found: 435.1765.
4-(tert-Butyl)phenyl(1-phenethylpiperidin-4-yl)(phenyl) carbamate (587)
Yield 80% (hexane/AcOEt, 75:25 as chromatographic eluent); mp 152–154 °C (from hexane/CH2Cl2); IR 2944, 2795, 1710, 1597, 1494, 1292, 1223, 1022, 998 csup>/sup>; 1H NMR (300 MHz) δ 1.27 (9H, s), 1.60 (2H, m), 1.95 (2H, m), 2.14 (2H, m), 2.54 (2H, m), 2.73 (2H, m), 3.03 (2H, d, J = 11.1 Hz), 4.32 (1H, m), 6.95 (2H, d, J = 7.5 Hz), 7.14–7.39 (12H, m); 13C NMR (75 MHz) δ 30.85, 31.39, 33.87, 34.34, 53.07, 55.48, 60.41, 120.83, 125.97, 126.02, 127.79, 128.36, 128.61, 128.86, 129.98, 137.87, 140.22, 147.89, 149.02, 154.19. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C30H37N2O2+: 457.2777, found: 457.2781.
4-(Trifluoromethyl)phenyl(1-phenethylpiperidin-4-yl) (phenyl)carbamate (588)
Yield 26% (hexane/AcOEt, 7:3 as chromatographic eluent); mp 132–134 °C (from hexane/CH2Cl2); IR 2946, 2790, 1709, 1598, 1497, 1305, 1214, 1126, 1065, 995 csup>/sup>; 1H NMR (300 MHz) δ 1.60 (2H, m), 1.95 (2H, m), 2.15 (2H, m), 2.55 (2H, m), 2.74 (2H, m), 3.04 (2H, d, J = 11.4 Hz), 4.32 (1H, m), 7.14–7.42 (12H, m), 7.56 (2H, d, J = 8.1 Hz); 13C NMR (75 MHz) δ 30.77, 33.86, 53.00, 55.76, 60.38, 121.92, 126.06, 126.44 (q. J = 36.75 Hz), 128.14, 128.39, 128.62, 129.05, 129.84, 137.43, 140.16, 153.20, 153.91. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C27H28F3N2O2+: 469.2025, found: 469.2023.
3-(Trifluoromethyl)phenyl(1-phenethylpiperidin-4-yl)(phenyl)carbamate (589)
Yield 13% (hexane/AcOEt, 7:3 as chromatographic eluent); mp 88–90 °C (from hexane/CH2Cl2); IR 2951, 2786, 1718, 1597, 1496, 1278, 1206, 1161, 1030, 995 csup>/sup>; 1H NMR (300 MHz) δ 1.60 (2H, m), 1.95 (2H, m), 2.15 (2H, m), 2.55 (2H, m), 2.74 (2H, m), 3.04 (2H, d, J = 11.7 Hz), 4.32 (1H, m), 7.15–7.41 (14H, m); 13C NMR (75 MHz) δ 30.77, 33.876, 53.01, 55.78, 60.39, 118.90, 121.95, 125.22, 126.06, 128.14, 128.39, 128.62, 129.06, 129.61, 129.86, 131.37, 131.80, 137.41, 140.17, 151.42, 153.38. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H28ClN2O2+: 435.1761, found: 435.1765.
General procedure for the synthesis of ureas OMDM590–595
To a stirred 15% phosgene solution in toluene (3.3 mL, 5 mmol) a solution of the appropriate aniline (1.0 mmol) and Et3N (0.35 mL, 2.5 mmol) in dry toluene (5 mL) was added drop-wise at 0 °C. The reaction mixture was stirred for 3 h at room temperature and evaporated under vacuum. The residue of the crude urethane was dissolved in dry toluene (4 mL) and a solution of 1-phenethyl-N-phenylpiperidin-4-amine (0.140 g, 0.50 mmol) in dry toluene (2 mL) was added drop-wise at room temperature with stirring. The reaction mixture was stirred overnight at room temperature, diluted with water, and extracted with AcOEt. The organic phase was washed with NaHCO3 s.s. and twice with water, dried (Na2SO4), and evaporated under vacuum. The residue was purified by column chromatography.
1-(1-Phenethylpiperidin-4-yl)-1,3-diphenylurea (590)
Yield 31% (hexane/AcOEt, 4:6 as chromatographic eluent); mp 152–155 °C (from hexane/CH2Cl2); IR 3426, 2922, 2758, 1674, 1519, 1490, 1437, 1313, 1282, 1232, 1079 csup>/sup>; 1H NMR (300 MHz) δ 1.48 (2H, m), 1.90 (2H, m), 2.18 (2H, m), 2.55 (2H, m), 2.73 (2H, m), 3.00 (2H, d, J = 11.4 Hz), 4.60 (1H, m), 5.86 (1H, s), 6.94–7.49 (15H, m); 13C NMR (75 MHz) δ 31.12, 33.85, 52.77, 53.16, 60.46, 119.19, 122.77, 125.99, 128.35, 128.60, 128.71, 128.99, 130.01, 131.27, 137.23, 138.88, 140.21, 154.10. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H29N3O+: 399.2232, found: 399.2251.
3-(4-Chlorophenyl)-1–(1-phenethylpiperidin-4-yl)-1-phenylurea (591)
Yield 61% (hexane/AcOEt, 6:4 as chromatographic eluent); mp 163–165 °C (from hexane/CH2Cl2); IR 3406, 2944, 2762, 1666, 1504, 1491, 1395, 1304, 1232, 1088 csup>/sup>; 1H NMR (300 MHz) δ 1.46 (2H, m), 1.89 (2H, m), 2.17 (2H, m), 2.54 (2H, m), 2.73 (2H, m), 3.02 (2H, d, J = 11.4 Hz), 4.58 (1H, m), 5.86 (1H, s), 7.14–7.51 (14H, m); 13C NMR (75 MHz) δ 31.10, 33.87, 52.92, 53.15, 60.46, 120.42, 126.02, 127.67, 128.37, 128.61, 128.66, 129.15, 130.10, 131.22, 137.02, 137.53, 140.20, 153.91. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H28ClN3O+: 433.1843, found: 433.1849.
3-(3-Chlorophenyl)-1–(1-phenethylpiperidin-4-yl)-1-phenylurea (592)
Yield 48% (CHCl3/AcOEt, 8:2 as chromatographic eluent); mp 164–166 °C (from hexane/CH2Cl2); IR 3282, 2923, 2769, 1638, 1587, 1513, 1426, 1306, 1242, 1084 csup>/sup>; 1H NMR (300 MHz) δ 1.48 (2H, m), 1.90 (2H, m), 2.18 (2H, m), 2.55 (2H, m), 2.73 (2H, m), 3.03 (2H, d, J = 11.4 Hz), 4.56 (1H, m), 5.88 (1H, s), 6.92–7.49 (14H, m); 13C NMR (75 MHz) δ 31.06, 33.84, 52.97, 53.14, 60.44, 117.09, 119.15, 122.76, 126.04, 128.38, 128.62, 129.23, 129.67, 130.15, 131.17, 136.92, 140.11, 140.19, 153.76. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C26H28ClN3O+: 433.1843, found: 433.1849.
3-(4-(tert-Butyl)phenyl)-1–(1-phenethylpiperidin-4-yl)-1-phenylurea (593)
Yield 79% (CH2Cl2/AcOEt, 7:3 as chromatographic eluent); mp 168–170 °C (from hexane/CH2Cl2); IR 3415, 2958, 1670, 1588, 1511, 1405, 1322, 1283, 1236, 1083 csup>/sup>; 1H NMR (300 MHz) δ 1.26 (9H, s), 1.45 (2H, m), 1.88 (2H, m), 2.18 (2H, m), 2.55 (2H, m), 2.73 (2H, m), 3.02 (2H, d, J = 11.1 Hz), 4.59 (1H, m), 5.79 (1H, s), 7.13–7.49 (14H, m); 13C NMR (75 MHz) δ 31.15, 31.37, 33.88, 34.17, 52.74, 53.20, 60.50, 119.09, 125.54, 126.01, 128.36, 128.62, 128.93, 129.97, 131.32, 136.24, 137.38, 140.25, 145.74, 154.30. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C30H37ClN3O+: 455.2858, found: 455.2867.
1-(1-Phenethylpiperidin-4-yl)-1-phenyl-3–(4-(trifluoromethyl)phenyl)urea (594)
Yield 74% (CH2Cl2/AcOEt, 7:3 as chromatographic eluent); mp 182–184 °C (from hexane/CH2Cl2); IR 3400, 2954, 2803, 1671, 1594, 1509, 1408, 1317, 1235, 1116, 1076 csup>/sup>; 1H NMR (300 MHz) δ 1.50 (2H, m), 1.90 (2H, m), 2.18 (2H, m), 2.55 (2H, m), 2.74 (2H, m), 3.03 (2H, d, J = 11.4 Hz), 4.56 (1H, m), 6.03 (1H, s), 7.15–7.51 (14H, m); 13C NMR (75 MHz) δ31.07, 33.88, 53.05, 53.13, 60.46, 118.44, 125.92, 125.98, 126.05, 128.39, 128.62, 129.32, 130.21, 131.15, 136.84, 140.20, 142.07, 153.61. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C27H28F3N3O+: 467.2106, found: 467.2116.
1-(1-Phenethylpiperidin-4-yl)-1-phenyl-3–(3-(trifluoromethyl)phenyl)urea (595)
Yield 79% (CH2Cl2/AcOEt, 7:3 as chromatographic eluent); mp 111–112 °C (from hexane/CH2Cl2); IR 3419, 2949, 2805, 1668, 1526, 1491, 1336, 1277, 1116, 1097 csup>/sup>; 1H NMR (300 MHz) δ 1.46 (2H, m), 1.90 (2H, m), 2.18 (2H, m), 2.55 (2H, m), 2.74 (2H, m), 3.03 (2H, d, J = 11.4 Hz), 4.59 (1H, m), 5.99 (1H, s), 7.14–7.59 (14H, m); 13C NMR (75 MHz) δ31.09, 33.89, 53.06, 53.16, 60.47, 115.82, 119.27, 122.20, 126.046, 128.39, 128.63, 129.16, 129.31, 130.21, 131.18, 131.36, 136.86, 139.47, 140.21, 153.81. HRMS (ESI-MS, 140 eV): m/z [M + H]+ calculated for C27H28F3N3O+: 467.2106, found: 467.2116.
Molecular docking
Fentanyl and its analogues were docked to the X-ray structure of the mouse μ-opioid receptor in inactive state obtained from the protein structure repository (PDB code: 4dkl, http://www.rcsb.org). Missing residues of the receptor were restored by the MODELLER programme packageCitation19.
Docking the flexible ligands to rigid receptor model was performed by AutoDock VinaCitation20, and AutoDockToolsCitation21, using the default system setup. The size of the docking grid box was 55 Å, centered on residue Asp147. The ligand structures were drawn at pH 7.4 and energy minimized by AvogadroCitation22, using the MMFF94s molecular force field.
For visualization VMDCitation23, AutoDockTools, PMV and MSMSCitation24, program packages were used.
Data analysis was performed by RCitation25, and linux bash scripts. All calculations were performed in linux environment (Ubuntu Oneiric). To compare the binding ability of the fentanyl analogues, the “ligand efficiency” values were used by dividing the docking energy by the number of non-hydrogen atoms of the molecule.
In vitro biological assays
Chemicals
Tris-HCl, EGTA, NaCl, MgCl2 × 6H2O, GDP, the GTP analogue GTPγS, fentanyl and U-69593 was purchased from Sigma-Aldrich (Budapest, Hungary). Fatty acid free bovine serum albumin (BSA) was purchased from Serva (Heidelberg, Germany). The highly selective MOR agonist enkephalin analogue Tyr-D-Ala-Gly-(NMe)Phe-Gly-ol (DAMGO), the highly selective DOR agonist IleCitation5,Citation6-deltorphin II (IleDelt II) was synthesized in the Laboratory of Chemical Biology group of the Biological Research Centre (BRC, Szeged, Hungary). The non-selective opioid receptor antagonist naloxone was kindly provided by the company Endo Laboratories DuPont de Nemours (Wilmington, DE, USA). WIN55 212–2 was purchased from Tocris Bioscience (Bristol, UK). Ligands were dissolved in water and were stored in 1 mM stock solution at -20 °C. The radiolabelled GTP analogue, [35S]GTPγS (specific activity: 1000 Ci/mmol) was purchased from Hartmann Analytic (Braunschweig, Germany). [3H]DAMGO (specific activity: 38.8 Ci/mmol), [3H]IleDelt II (specific activity: 19,6 Ci/mmol),Citation18 and [3H]WIN55 212–2 (specific activity: 13,1 Ci/mmol) were radiolabeled by the Laboratory of Chemical Biology group in BRC (Szeged, Hungary). [3H]U-69 593 (specific activity: 43,6 Ci/mmol) was purchased from Perkin Elmer (Perkin Elmer, Waltham, MA).
Animals
For membrane homogenate preparations male and female Wistar rats (250–300 g body weight) and male guinea pigs (∼700 g body weight, LAL/HA/BR strain) were used. Rats were housed in the local animal house of BRC (Szeged, Hungary), while guinea pigs were housed in LAB-ÁLL Bt. (Budapest, Hungary). All the animals were kept in a temperature controlled room (21–24 °C) under a 12:12 light and dark cycle and were provided with water and food ad libitum. All housing and experiments were conducted according to the European Communities Council Directives (86/606/ECC) and the Hungarian Act for the Protection of Animals in Research (XXVIII.tv. 32.§). The total number of animals as well as their suffering was minimized.
Rat and guinea pig brain membrane homogenate preparation for binding assays
Rats and guinea pigs were decapitated and their brains were quickly removed. The brains were prepared for membrane preparation according to Benyhe et al.Citation26 and partly used for binding experiments and partly were further prepared for the [35S]GTPγS binding experiments according to Zádor et al.Citation27. Briefly the brains were homogenized, centrifuged in ice-cold 50 mM Tris-HCl (pH 7.4) buffer and incubated at 37 °C for 30 min in a shaking water-bathCitation28–30. After incubation the centrifugation was repeated as described before and the final pellet was suspended in 50 mM Tris-HCl pH 7.4 buffer containing 0.32 M sucrose, and stored at −80 °C. For the [35S]GTPγS binding experiments the final pellet of rat brain membrane homogenate was suspended in ice-cold TEM (Tris-HCl, EGTA, MgCl2) and stored at -80 °C for further use.
Receptor binding assays
Radioligand competition binding experiments
Aliquots of frozen rat brain membrane homogenates were centrifuged (18 000 rpm, 20 min, 4 °C) to remove sucrose and the pellets were suspended in 50 mM Tris-HCl buffer (pH 7.4). Membranes containing 0.3–0.5 mg/ml of protein were incubated in the presence of unlabelled fentanyl and fentanyl analogues (OMDM584–595) in crescent concentrations (10 − 10–10− 5 M) for 35 °C for 45 min with ∼1 nM [3H]DAMGO in 35 °C for 45 min. OMDM585 and 586 (10 − 10–10− 5 M) were also incubated with ∼1 nM of [3H]IleDelt II, [3H]U-69593, or [3H]WIN55 212–2 in 30 °C for 30 min, in 24 °C for 45 min and in 30 °C for 60 min, respectively in rat brain homogenates, while in case of[3H]U-69593 in guinea pig brain membrane homogenates (the guinea pig brain has significantly more kappa receptors than the rat brain). Additionally, unlabeled IleDelt II, U-69593 and WIN55 212–2 were also incubated together with their labeled counterparts in increasing concentrations (10 − 10–10 − 5 M) for control.
For experiments performed with [3H]WIN55 212–2 the incubation mixture also contained 50 mM Tris/HCl, 2.5 mM EGTA, 5 mM MgCl2 and 0.5 mg/ml fatty acid-free BSA (pH 7.4). The level of non-specific binding was determined in the presence of 10 μM unlabelled naloxone or WIN55 212–2, while total binding was determined in the absence of cold compounds. The reaction was terminated by rapid filtration under vacuum (Brandel M24R Cell Harvester; Brandel Harvesters, Gaithersburg, MD), and washed thrice with 5 ml ice-cold 50 mM Tris-HCl or 50 mM Tris/HCl, 2.5 mM EGTA, 5 mM MgCl2, 0.5% BSA (pH 7.4) in case of [3H]WIN55 212–2.
The filtration was accomplished through Whatman GF/C ([3H]DAMGO, [3H]IleDelt II or GF/B ([3H]U-69593 and [3H]WIN55 212–2) glass fibres (GE Healthcare, Little Chalfont, UK). In case of [3H]WIN55 212–2 the filter was pre-soaked in 0.1% polyethyleneimine 30 min before the filtration. The radioactivity of the filters was detected in UltimaGold™ MV aqueous scintillation cocktail (Perkin Elmer, Waltham, MA) with Packard Tricarb 2300TR liquid scintillation counter (Perkin Elmer, Waltham, MA). The competition binding assays were performed in duplicate and repeated at least three times.
Functional [35S]GTPγS binding experiments
In [35S]GTPγS binding experiments, we measure the GDP→GTP exchange of the Gαi/o protein in the presence of a given ligand to measure ligand potency and the maximal efficacy of receptors G-protein. The nucleotide exchange is monitored by a radioactive, non-hydrolysable GTP analogue, [35S]GTPγS.
The functional [35S]GTPγS binding experiments were performed as previously describedCitation31, with modifications. Briefly the rat brain membrane homogenates containing ∼10 μg/ml protein were incubated at 30 °C for 60 min in Tris-EGTA buffer (pH 7.4) composed of 50 mM Tris-HCl, 1 mM EGTA, 3 mM MgCl2, 100 mM NaCl, containing 20 MBq/0.05 cm3 [35S]GTPγS (0.05 nM) and increasing concentrations (10 − 10–10− 5 M) of fentanyl and fentanyl analogues (OMDM 584–595) in the presence or absence of 10 μM naloxone and excess GDP (30 μM) in a final volume of 1 ml. Total binding was measured in the absence of test compounds, non-specific binding was determined in the presence of 10 μM unlabelled GTPγS. The bound and unbound [35S]GTPγS was separated as described in the competition binding assays section through Whatmann GF/B glass fibres. The radioactivity of the filters was also detected as described above. [35S]GTPγS binding experiments were performed in triplicates and repeated at least three times.
Data analysis
The specific binding of all radiolabeled compounds was calculated by the subtraction of non-specific binding from total binding and was given in percentage. Data were normalized to total specific binding, which was settled 100%, which in case of [35S]GTPγS also represents the basal activity of the G-protein. Experimental data were presented as means ± SEM in the function of the applied ligand concentration range in logarithm form. Points were fitted with the professional curve fitting program, GraphPad Prism 5.0 (GraphPad Prism Software Inc., San Diego, CA), using non-linear regression. In the radioligand competition binding assays the ‘One-site competition’, while in [35S]GTPγS binding assays the ‘Sigmoid dose-response’ equation was applied to determine IC50 (unlabelled ligand affinity) and the maximum G-protein efficacy (Emax) and ligand potency (EC50), respectively. For IC50 and EC50 values standard error is only given in their logarithm form by the curve fitting program due to the data representation. Statistical analyses were performed with GraphPad Prism 5.0. In case of two data sets, unpaired t-test with two-tailed p values statistical analysis was used, while for three or more data sets one-way ANOVA with Tukey’s multiple comparison tests were performed to determine the significance level. Since unlabelled ligand concentrations were presented in the logarithm form, the curve fitting programme could only calculate SEM. for the logarithm form of IC50 (logIC50) and EC50 (logEC50) values. At the same time their antilogarithm form has also been indicated. Significance was accepted at the p < 0.05 level.
Biochemical assays
Fatty acid amide hydrolase (FAAH) assays
The activity of products on the enzymatic hydrolysis of anandamide (AEA) was obtained using membranes prepared from rat brain, incubated with the test compounds and [14C] AEA (2.4 μM) in 50 mM Tris-HCl, pH 9, for 30 min at 37 °C. [14C]Ethanolamine produced from [14C]AEA hydrolysis was measured by scintillation counting of the aqueous phase after extraction of the incubation mixture with two volumes of CHCl3/CH3OH = 2/1 (by volume). Data were expressed as the concentration exerting 50% inhibition of AEA hydrolysis (IC50), calculated by GraphPad.
Assay of MAGL activity
The 10 000 g cytosolic fraction from COS-7 cells was incubated in tris HCl 50 mM, at pH 7.0 at 37 °C for 20 min, with synthetic 2-[3H] arachidonoyl-glycerol (1.0 mCi/mmol, 25 mM). After incubation, lipids were extracted with two volumes of chloroform/methanol = 2/1 (by volume), and the extracts were lyophilized under vacuum. Extracts were fractionated by TLC on silica on plastic plates using CHCl3/MeOH/NH4OH (85/15/1 by volume) as the eluting system. Bands corresponding to [3H]arachidonic acid were cut, and their radioactivity was counted with a β-counter.
In vivo nociception test
Animals
Male CD-1 mice (Harlan, Italy) weighing 25–30 g were used for all experiments. Mice were housed for at least 1 week before the experimental sessions in colony cages (7 mice in each cage) under standard light (from 7.00 a.m. to 7.00 p.m.), temperature (21 ± 1 °C), relative humidity (60 ± 10%) with food and water available ad libitum. The research protocol was approved by the Service for Biotechnology and Animal Welfare of the Istituto Superiore di Sanità and authorised by the Italian Ministry of Health, according to Legislative Decree 26/14, which implemented the European Directive 2010/63/UE on laboratory animal protection in Italy. Animal welfare was routinely checked by veterinarians from the Service for Biotechnology and Animal Welfare.
Surgery for intracerebroventricular (i.c.v.) injections
For i.c.v. injections, mice were lightly anaesthetized with isoflurane, and an incision was made in the scalp. Injections were performed using a 10 μl Hamilton microsyringe at a point 2-mm caudal and 2-mm lateral from the bregma at a depth of 3 mm in a volume of 5 μL as previously describedCitation32.
Hot-plate test
Thermal nociception (hot-plate test) was assessed with a commercially available apparatus consisting of a metal plate 25 × 25 cm (Ugo Basile, Italy) heated to a constant temperature of 55.0 ± 0.1 °C, on which a plastic cylinder (20 cm diameter, 18 cm high) was placed. The time of latency (s) was recorded from the moment the animal was placed in the cylinder on the hot plate until it licked its paws or jumped; the cut-off time was 60 s. The baseline was calculated as mean of three readings recorded before testing at intervals of 15 min, and was in the same order of magnitude in all experimental groups (mean 9.8 ± 1.2 s, N = 8–10). The time course of latency was then determined at 15, 30, 45 and 60 min after compound treatment. Experimental data were expressed as time course of the percentage of maximum effect (%MPE) = (post-drug latency – baseline latency)/(cut-off time-baseline latency) × 100. On each test day compound solutions were freshly prepared using DMSO:0.9% saline 1:5 v/v. These solutions were injected at a dose of 10 μg/mouse in a volume of 5 μL/mouse. The significance among the groups was evaluated with the analysis of variance followed by Tukey’s post-hoc comparisons using GraphPad Prism 6.03 software. Statistical significance was assumed at least for p < 0.05.
Supplementary materials available online
IENZ_1160902_Supplementary_Material.pdf
Download PDF (431.9 KB)Acknowledgements
We are grateful to Prof. Giorgio Ortar and Prof. Enrico Morera (Department of Chemistry and Technology, “Sapienza” University of Rome) for their suggestions and mentoring activity.
Declaration of interest
This research was partly supported by an OTKA-108518 grant (S. Benyhe) provided by National Research, Development and Innovation Office (NKFIH), Budapest, Hungary.
Related Research Data
References
- Rosenblum A, Marsch LA, Joseph H, Portenoy RK. Opioids and the treatment of chronic pain: controversies, current status, and future directions. Exp Clin Psychopharmacol 2008;16:405–16
- (a) Vucković S, Prostran M, Ivanović M, et al. Pharmacological evaluation of 3-carbomethoxy fentanyl in mice. Curr Med Chem 2009;16:2468–74. (b) Casy AF, Parfitt RT. Opioid analgesics: chemistry and receptors. New York: Springer; 1986
- (a) Subramaniam A. Opioid ligands with mixed m/d opioid receptor interactions: an emerging approach to novel analgesics. AAPS J 2006;8:118–25. (b) van Rijn RM, DeFriel JN, Whistler JL. Pharmacological traits of delta opioid receptors: pitfalls or opportunities? Psychopharmacology (Berl) 2013;228:1–18. (c) Gomes I, Gupta A, Filipovska J, et al. A role for heterodimerization of mu and delta opiate receptors in enhancing morphine analgesia. Proc Natl Acad Sci USA 2004;101:5135–9
- (a) Mollica A, Pinnen F, Feliciani F, et al. New potent biphalin analogues containing p-fluoro-L-phenylalanine at the 4,4' positions and non-hydrazine linkers. Amino Acids 2010;40:1503–11. (b) Mollica A, Pinnen F, Costante R, et al. Biological active analogues of the opioid peptide biphalin: mixed α/β(3)-peptides. J Med Chem 2013;56:3419–23. (c) Mollica A, Costante R, Stefanucci A, et al. Antinociceptive profile of potent opioid peptide AM94, a fluorinated analogue of biphalin with non-hydrazine linker. J Pept Sci 2013;19:233–9. (d) Mollica A, Costante R, Novellino E, et al. Design, synthesis and biological evaluation of two opioid agonist and Cav 2.2 blocker multitarget ligands. Chem Biol Drug Des 2015;86:156–62
- (a) Petrov RR, Lee YS, Vardanyan RS, et al. Effect of anchoring 4-anilidopiperidines to opioid peptides. Bioorg Med Chem Lett 2013;23:3434–7. (b) Podolsky AT, Sandweiss A, Hu J, et al. Novel fentanyl-based dual μ/δ-opioid agonists for the treatment of acute and chronic pain. Life Sci 2013;93:1010–6. (c) Deekonda S, Wugalter L, Rankin D, et al. Design and synthesis of novel bivalent ligands (MOR and DOR) by conjugation of enkephalin analogues with 4-anilidopiperidine derivatives. Bioorg Med Chem Lett 2015;25:4683–8
- (a) Parker KE, McCall JG, McGuirk SR, et al. Effects of co-administration of 2-arachidonylglycerol (2-AG) and a selective μ-opioid receptor agonist into the nucleus accumbens on high-fat feeding behaviors in the rat. Brain Res 2015;1618:309–15. (b) Scavone JL, Sterling RC, Van Bockstaele EJ. Cannabinoid and opioid interactions: implications for opiate dependence and withdrawal. Neuroscience 2013;248:637–54
- Dvoracsko S, Stefanucci A, Novellino E, Mollica A. The design of multitarget ligands for chronic and neuropathic pain. Future Med Chem 2015;7:2469–83
- (a) Ahn K, Johnson DS, Mileni M, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol 2009;16:411–20. (b) Karlsson J, Morgillo CM, Deplano A, et al. Interaction of the N-(3-methylpyridin-2-yl)amide derivatives of flurbiprofen and ibuprofen with FAAH: enantiomeric selectivity and binding mode. PLoS One 2015;10:e0142711
- Ahn K, Johnson DS, Cravatt BF. Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders. Expert Opin Drug Discov 2009;4:763–84
- a) Ligresti A, Morera E, Van Der Stelt M, et al. Further evidence for the existence of a specific process for the membrane transport of anandamide. Biochem J 2004;15:265–72. (b) Maione S, Morera E, Marabese I, et al. Antinociceptive effects of tetrazole inhibitors of endocannabinoid inactivation: cannabinoid and non-cannabinoid receptor-mediated mechanisms. Br J Pharmacol 2008;155:775–82. (c) Morera E, Petrocellis LD, Morera L, et al. Synthesis and biological evaluation of piperazinyl carbamates and ureas as fatty acid amide hydrolase (FAAH) and transient receptor potential (TRP) channel dual ligands. Bioorg Med Chem Lett 2009;19:6806–9. (d) Gustin DJ, Ma Z, Min X, et al. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg Chem Chem Lett 2011;21:2492–6
- Jagerovic N, Fernández-Fernández C, Erdozain AM, et al. Combining rimonabant and fentanyl in a single entity: preparation and pharmacological results. Drug Des Devel Ther 2014;8:263–77
- Rinaldi-Carmona M, Barth F, Héaulme M, et al. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 1994;350:240–4
- (a) Cinar R, Szücs M. CB1 receptor-independent actions of SR141716 on G-protein signaling: coapplication with the mu-opioid agonist Tyr-D-Ala-Gly-(NMe)Phe-Gly-ol unmasks novel, pertussis toxin-insensitive opioid signaling in mu-opioid receptor-Chinese hamster ovary cells. J Pharmacol Exp Ther 2009;330:567–74. (b) Erdozain AM, Diez-Alarcia R, Meana JJ, Callado LF. The inverse agonist effect of rimonabant on G protein activation is not mediated by the cannabinoid CB1 receptor: evidence from postmortem human brain. Biochem Pharmacol 2012;83:260–8. (c) Zádor F, Kocsis D, Borsodi A, Benyhe S. Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem Int 2014;67:14–22
- Lange JHM, Kruse CG. Keynote review: medicinal chemistry strategies to CB1 cannabinoid receptor antagonists. Drug Discov Today 2005;10:693–702
- Dosen-Micovic L, Ivanovic M, Micovic V. Steric interactions and the activity of fentanyl analogs at the mu-opioid receptor. Bioorg Med Chem 2006;14:2887–95
- Jensen TS, Finnerup NB. Neuropathic pain treatment: a further step forward. Lancet 2009;374:1218–19
- Mollica A, Stefanucci A, Costante R, Hruby VJ. Rational approach to the design of bioactive peptidomimetics: recent developments in opioid agonist peptides. Elsevier; 2014, p. 27
- (a) Vardanyan RS, Hruby VJ. Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med Chem 2014;6:385–412. (b) Ioja E, Tourwé D, Kertész I, et al. Novel diastereomeric opioid tetrapeptides exhibit differing pharmacological activity. Brain Res Bull 2007;74:119–29
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010;31:455–61
- Sanner FM Python: a programming language for software integration and development. . J Mol Graph Model 1999;17:57–61
- Hanwell MD, Curtis DE, Lonie DC, et al. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform 2012;4:17
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996;14:33–8
- Sanner MF, Spehner J-C, Olson AJ. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 1996;38:305–20
- R Development Core Team (2008); R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0. Available from: http://www.R-project.org
- Benyhe S, Farkas J, Tóth G, Wollemann M. Met5-enkephalin-Arg6-Phe7, an endogenous neuropeptide, binds to multiple opioid and nonopioid sites in rat brain. J Neurosci Res 1997;48:249–58
- Zádor F, Kocsis D, Borsodi A, Benyhe S. Micromolar concentrations of rimonabant directly inhibits delta opioid receptor specific ligand binding and agonist-induced G-protein activity. Neurochem Int 2014;67:14–22
- Sim LJ, Selley DE, Childers SR. In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5'-[gamma-[35S]thio]-triphosphate binding. Proc Natl Acad Sci USA 1995;92:7242–6
- Traynor JR, Nahorski SR. Modulation by m-opioid agonists of guanosine-5'-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol 1995;47:848–54
- Foran SE, Carr DB, Lipkowski AW, et al. Inhibition of morphine tolerance development by a substance P-opioid peptide chimera. J Pharmacol Exp Ther 2000;295:1142–8
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. \Physiol Behav 1976;17:1031–6
- Porreca F, Mosberg HI, Hurst R, et al. Roles of mu, delta and kappa opioid receptors in spinal and supraspinal mediation of gastrointestinal transit effects and hot-plate analgesia in the mouse. J Pharmacol Exp Ther 1984;230:341–8