
Abstract
Context: Ibuprofen is an important NSAID, however, it can cause GI disturbances when given orally, and employment of transdermal route will require permeation enhancer causing skin injury. Objective: Drug-loaded nanovesicles of ceramide-2, cholesterol, palmitic acid, and cholesteryl sulfate (ICVG) were formulated and analyzed for physicochemical and permeation properties. Materials and method: Vesicles were formulated using film hydration method and physicochemical parameters, in vitro drug release, and stability were assessed. Further, nanovesicle gels were evaluated against plain gel containing drug (CG) for ex vivo/in vivo drug permeation and anti-inflammatory activity. Results: The developed formulations showed optimal physicochemical profile and ICV-1 gave 97.24% drug release. Drug permeation was between 17.32 and 33.12 μg/cm2 for ICVG formulations and 0.27 μg/cm2 for CG. ICVG-1 and CG showed Cmax of 9.6 and 0.7 μg/ml at 8 and 4 h. ICVG-1 showed 19.9 times higher AUC than CG. Edema inhibition was 57.98% during initial hours by ICVG-1. Discussion: Ratio of ceramide 2 and palmitic acid plays a critical role in drug permeation through stratum corneum. The stability and protective effect of the formulations were due to ceramide content. Conclusion: The composition has an important role in physicochemical properties and drug permeation thereby generating an optimum formulation.
Introduction
The main goal of a controlled drug delivery system is to deliver a drug at a predefined rate to a specific site for a desired duration, for maintaining the drug level within the therapeutic window (Gaur et al. Citation2014c, Citation2013a). Transdermal drug delivery can provide for controlled input of drug and bypassing the first-pass metabolism and also protect against any gastrointestinal disturbance. But, skin, being a barrier, can pose different challenges for the drugs to be absorbed. The top layer of skin (SC) is the critical feature in permeation due to different nature of lipids present (Gaur et al. Citation2014c).
SC lipids are ceramide, free fatty acids, cholesterol, and cholesteryl sulfate in ratio of 40%, 25%, 25%, and 10% (w/w), respectively (Gaur et al. Citation2014c, Yardley and Summerly Citation1981). These are significant in protecting the lipid barrier against water loss and any other injury (Walters and Roberts Citation2002). Ceramides are the most important due to its concentration and hydrophobicity. These lipids are reported to form bilayer, and there is a detailed review on membrane properties of ceramides (Goñi and Alonso Citation2006). Ceramide 2 (Cer 2) comprises of a phytosphingosine skeleton acylated with a saturated fatty acid (stearic acid) (Wartewig and Neubert Citation2007).
The mechanisms for transdermal permeation enhancement are disruption of the skin barrier by means of delipidization, hydration and disruption of lipid packing, and changes in thermodynamic status of drug/skin. Further changes in the physiological pH of the skin, immunological reactions and chemical or pharmacological features of the drug or vehicle, excipients, duration of application, and the type of delivery device used trigger the development of irritation (Gaur et al. Citation2014c, Paudel et al. Citation2010). Presently, vast majority of materials either chemical or natural origin have been used to enhance the rate of permeation through the physiological barrier of skin.
Ibuprofen (2-(4-(2-methylpropyl) phenyl) propanoic acid) is an acid drug and has a molecular weight of 206.28 g/mol, with a log P around 4. Further, Ibuprofen is a chiral molecule and has two enantiomers; (R)-ibuprofen and (S)-ibuprofen, and among them only (S)-ibuprofen is anti-inflammatory (Lee et al. Citation2004, Evans et al. Citation1989). Ibuprofen is rapidly absorbed after oral administration and excreted in hydroxylated and carboxylated metabolites in the urine. Peak serum concentrations are reached in 1–2 h after oral administration. Its serum half-life is 2 h and it is extensively bound to plasma proteins. It accumulates slowly into the synovial spaces (Antal et al. Citation1986, Gallo et al. Citation1986).
This work was undertaken to formulate nanovesicle formulations of Ibuprofen using ceramide 2 for effective transdermal administration. The formulations were evaluated for physicochemical parameters and permeation using artificial membrane and human skin along with in vivo drug permeation, anti-inflammatory activity, and stability. These formulations were compared with a gel formulation containing free Ibuprofen. This study will be useful for development of an effective transdermal formulation of Ibuprofen.
Materials and methods
Materials
Ibuprofen was a gift sample from Taj Pharmaceuticals Limited, Goregaon (West), Mumbai, (India) whereas Ceramide-2 (N-stearoyl-(2S,2R,3R)-2-amino-1,3-octadecandiol or N-stearoyl-DL-sphinganin) was gift sample from Sederma, France (Lot No.0000380664; patent No. US 2004-0120918), supplied by Croda Chemicals (India) Pvt Ltd, Thane, Mumbai, India. Cholesterol (Chol) was purchased from Himedia, Mumbai, India, Cholesteryl-3-sulphate (CS), and Hexadecanoic acid (Palmitic acid ≥ 99%, PA) were purchased from Sigma–Aldrich (New Delhi, India). All the other products were of analytical grade.
Methods
Vesicle preparation
Film hydration method was used as per the composition in for liposome preparation. The drug was dissolved in Chloroform and was mixed with lipids in the pre-decided ratio. This mixture was taken in a round-bottomed flask and evaporated to make a film (Rotavapor® R II, BUCHI India Private Ltd.). The hydration was done using acetate buffer solution (pH 5.5) by keeping the film at 200 rpm and 25°C for 1 h. The resulting dispersion was sonicated for 15 min at 100 W using a probe sonicator. The nanodispersion so obtained was extruded using a membrane (Immobilon-P Membrane, 0.22 μm pore size, Millipore Pvt. Ltd., New Delhi, India). Sephadex G-20 minicolumn was used to remove unentrapped drug from formulations (Gaur et al. Citation2014c, Citation2013b, Sinico et al. Citation2005).
Table I. Composition for ICV formulations (drug lipid ratio = 1:3).
Size and shape
Vesicle size and shape were evaluated by TEM after negative staining with phosphotungstic acid. Sample was placed over a 200-mesh copper grid and allowed to absorb. Then stain was added, allowed to soak up. The sample was dried at room temperature. The imaging was done using Philips CM-10 (Acceleration voltage: 100 kV; Magnification: upto 10,000×; Cryo-attachment).
Zeta potential and polydispersity index
Polydispersity indices and zeta potential were estimated on a Zetasizer (NanoZS, Malvern Instruments, UK), equipped with a 4 mW He–Ne laser (633 nm). The lyophilized samples were suspended in phosphate buffer, pH 7.4 and then kept in micro-centrifuge tube for determination. The number of photons (kCps) was evaluated at room temperature.
Entrapment efficiency
Entrapment efficiency was estimated using ultracentrifugation method. A known quantity of formulation was centrifuged at 12,000 rpm (Remi C-24 BL with angular R-241 rotor, Remi House, Mumbai, India). Then drug amount was determined in supernatant and the pellet independently. The entrapment efficiency was estimated as follows:
where T is the total amount of drug and C is the drug only in the supernatant (Gaur et al. Citation2014c, Citation2013b, Touitou et al. Citation2000).
Assay
HPLC assay was employed for drug analysis. The instrument possesses a Shimadzu LC-10AT VP pump, a SIL-10AF auto injector, an SPD-10A UV-VIS detector, and an SCL-10A VP system controller HPLC system (Shimadzu, Japan). The methods from Anne et al. and Gaur et al. were followed (Anne et al., Citation2000; Gaur et al., Citation2014c).
The assay was done on RP-HPLC column (Shim-pack VP-ODS) (Shimadzu, Japan) having 150 mm length and 4.6 mm i. d., packed with 5 μm particles eluted with acetonitrile/0.1 M sodium acetate/acetic acid (70:30:0.5, v/v/v). The injection volume and flow rate were 20 μl and 2.0 ml/min respectively. The detector was set at 223 nm.
Linear correlation was acquired between peak area and concentration. The calibration equation was y = 43146x − 78963 (R² = 0.999), where x is the concentration and y is the peak area.
In vitro drug release
Cellulose acetate membrane with 12 kDa cutoff was used after equilibration in saline solution in Franz diffusion cell (area = 3.14 cm2). Acceptor chamber was filled with buffer (pH 5.5) and formulation (1 g) was applied on top of the donor surface. Appropriate samples were taken at pre-decided time intervals and replaced with fresh solution. The samples were suitably diluted and analyzed using assay (Gaur et al. Citation2014c, Citation2013b, Dragicevic-Curic et al. Citation2008, An et al. Citation2011).
Stability studies
Stability determination was done as per ICH guidelines. The representative samples were kept in refrigerator for 4 ± 0.5°C and in stability chamber at 25 ± 0.5°C and 40 ± 0.5°C. The physicochemical parameters were evaluated after 30, 60, and 90 days of storage (Ishida et al. Citation2002, Gaur et al. Citation2014c, Citation2013b, Arifin and Palmer Citation2005).
Preparation of gel
The drug-loaded nanovesicles were further formulated as carbopol gels for ease in transdermal application. Carbopol 934 was dispersed in distilled water (1% solution) and shaken using mechanical stirrer. Then, this dispersion was neutralized by adding 0.5% w/w triethanolamine solution. This gel was set aside for removal of any air bubble. After 24 h, drug-loaded nanovesicles were homogeneously mixed into the carbopol gel. Plain unentrapped drug was also formulated in the form of carbopol gel (CG) (El-Leithy et al. Citation2010).
Viscosity
Viscosity was determined using Brookfield DV III ultra V6.0 RV cone and plate rheometer (Brookfield Engineering Laboratories, Inc., Middleboro, MA). Spindle # CPE40 was used at 25 ± 0.5°C for the estimation of viscosity (El-Leithy et al. Citation2010).
Ex vivo skin permeation
Human skin was obtained from plastic surgery patients for ex vivo permeation studies. Institutional ethical committee reviewed and approved the experiment protocol. Skin samples were filled in aluminum foil and kept in a polyethylene bag at − 20°C after cleaning.
The skin was thawed (37°C) and positioned onto the donor side of franz diffusion cell (area = 3.14 cm2). Acceptor chamber was filled with buffer solution (5.5) and allowed for equilibration. For permeation study, the formulation sample was applied onto the epidermal surface. There is 50 mg Ibuprofen each in 1 g sample of CG and ICVG formulations.
Suitable sample was taken at suitable interval and analyzed for drug content. After the experiment, skin was separated and stripped for 10 times with scotch crystal tape. The tapes were transferred to a glass vial for storage. Further, the epidermis was stripped from the dermis using a scalpel. Rest of the skin sample was homogenized in methanol. All the samples were analyzed for drug content (Gaur et al. Citation2014c, Dragicevic-Curic et al. Citation2008, Manconi et al. Citation2011).
In vivo skin permeation
The study was done using 18 male albino rats (6–8 weeks) in three groups. The animals were housed under standard laboratory conditions (temperature: 25 ± 2°C and relative humidity: 55 ± 5%) in polypropylene cages, with free access to diet (Lipton feed, Mumbai, India) and water ad libitum. The study was done on 7–9 weeks old animals with an average weight of 260 g. For the experiment, the animals were anesthetized by i.v. administration of ketamine hydrochloride (75 mg/kg) and xylazine (5 mg/kg) in combination. The abdominal hairs were removed and skin was cleaned with distilled water.
An area of 3.14 cm2 was selected and sample containing 50 mg drug (1 g CG formulation) and 25 mg drug (500 mg ICVG-1 formulation), respectively, were applied in Groups I and II. Group III was applied blank carbopol gel to serve as negative control. The application was protected with an open container attached to the skin with a silicon rubber. At predetermined time intervals, blood samples were removed and stored in vacutainer tubes, till plasma was separated by centrifuging at 5000 rpm for 20 min. The plasma was stored at − 21°C until drug analysis (Gaur et al. Citation2014c, Akhter et al. Citation2008). The drug content was estimated using assay.
Anti-inflammatory
Wistar albino male rats were employed for anti-inflammatory activity. Carrageenan rat paw edema model was used for anti-inflammatory activity determination. The protocol was permitted by the Institutional Animal Ethical Committee after a review. Eighteen rats (6–8 weeks) were kept under standard laboratory conditions in polypropylene cages, with free access to standard diet (Lipton feed, Mumbai, India) and water ad libitum for one week. Animals of 7–9 weeks old with an average weight of 275 g were used in study. Before the experiment, animals were fasted for 24 h with free access to water. CG and ICVG-1 were applied in Groups I and II, respectively, to the skin (3.14 cm2) as in in vivo study. Standard drug Indomethacin was administered orally at 10 mg/Kg dose in Group III. The untreated paw was considered as negative control. Initially, the treatment was applied and then after 1 h, 0.1 mL of 1% carrageenan suspension in saline was administered into the right hind paw of rat (plantar side). The edema was determined by measuring paw volume at 0, 1, 2, 3, 4, 5, and 6 h using digital plethysmometer (Water displacement method). Edema inhibition (%) was analyzed by using formula given in data analysis (Manosroi et al. Citation2008).
Skin irritation studies
The study was performed on 12 human males aged between 21 and 28 years with average weight of 73 Kg. All the participants were properly educated about the procedure and the consent forms were signed. The site of application (upper arm area) was thoroughly examined for any irregularity. 5% Sodium Lauryl Sulfate (SLS) solution was taken as positive control and untreated skin as negative control. Skin irritation was measured by visual observation and score was given as: 0, no reaction; 1, weak spotty or diffuse erythema; 2, weak but well perceptible erythema covering the total exposure area; 3, moderate erythema; 4, severe erythema with edema; and 5, very severe erythema with epidermal defects (vesicles, erosions, etc.)
The upper arm area was cleaned with swab soaked in distilled water and treatment was applied and held with a dressing. The dressing was removed after 24 h and skin was wiped with cotton before fresh application. This regimen was followed for 7 days and observations were made every day (Patel et al. Citation2009).
Group I: positive control (SLS treated)
Group II: ICVG-1
Data and statistical analysis
Ex vivo permeation. The cumulative total of drug permeated was plotted versus time and permeation profiles were generated. Steady-state drug flux (Jss) and lag time (Tlag) were calculated from slope and x-intercept of the linear portion of the profile. Further, permeability coefficient through the membrane (Kp) and diffusion parameter within the membrane (D) (Higuchi Citation1960, Aguiar and Weiner Citation1969, Dugard Citation1976, Durrheim et al. Citation1980, Haigh et al. Citation1998, Kobayashi and Saitoh Citation1999, Vasiljevic et al. Citation2006, Furuishi et al. Citation2008) and enhancement ratio (Mishra et al. Citation2007, Jukanti et al. Citation2011) were calculated using following equations.
ER = Flux of test formulation/Flux of carbopol gel containing plain drug
where D = diffusion parameter within the skin (cm2 h− 1),
h = diffusional path length, and
Cd = initial drug concentration in donor compartment.
Pharmacokinetic parameter. Plasma drug concentration (μg) was plotted against time (hrs) and plasma concentration time profile was arranged. Cmax and tmax were deduced directly from the respective profiles and AUC0→t was calculated using linear trapezoidal method in Graph pad Prism Version 4 (Akhter et al. Citation2008).
Anti-inflammatory activity. Edema inhibition (%) was estimated to establish the anti-inflammatory activity as per the following formula (Manosroi et al. Citation2008).
where, Tc is thickness of paw in control group and Tt is thickness of paw in treatment group.
Data was expressed as mean ± S.D. Six experimental values were taken for ex vivo studies and study involving live subjects. For other studies, triplicate values were used for calculation. Statistical analyses were done using the Graph pad Prism Version 4 software by using analysis of variance (ANOVA) or the paired t-test, as appropriate with statistical significance at P < 0.05.
Results
Physical characterization
Drug-entrapped vesicles demonstrated minimum vesicle size in vesicles made up with lowest palmitic acid at 119–139 nm (). Size distribution was uniform as PDI values were in between 0.144 and 0.191. The vesicle showed electrical charge on the membrane in form of zeta potential (− 33 to − 40 mV). The entrapment efficiency was good for loaded vesicles and was within 78.26–87.71%. The viscosity of the corresponding formulations was between 14743 and 17014 Cps. Maximum amount of drug was entrapped in ICV-3 which had highest palmitic acid content. All the vesicle formulation showed the presence of electric charge which was to the tune of − 33 to 40 mV. Physicochemical characterization of vesicle formulations is shown in .
Figure 1. Micrographic images of ICV-1(× 104).
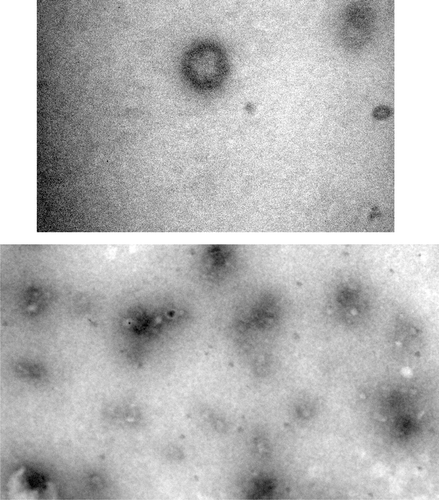
Table II. Physicochemical characterization.
In vitro drug release
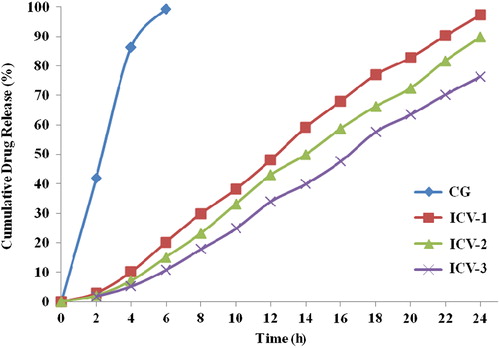
The drug amount released from vesicle formulations was estimated and plotted against time during 24-h study. The profile is shown in . Maximum drug release was in ICV-1 at 97.24%. CG showed 99.2% drug release in 6 h.
Figure 2. In vitro release profile for nanovesicles and CG formulations in 24 h.
Stability studies
ICV-1 formulation was selected for accelerated stability studies on the basis of physicochemical parameters. There are only minor alterations in physicochemical parameters and drug release of ICV-1 for the duration of study ().
Table III. Physicochemical characterization and drug release of ICV-1 after 90 days at accelerated conditions.
Ex vivo drug permeation
Human skin was employed to study the permeation profile of the vesicle formulations in ex vivo settings and obtained data was plotted against time to study the permeation profile over a period of 24 h (). For ease in application, all the vesicles were formulated in gel and compared with carbopol gel containing plain drug.
Figure 3. Permeation profile for vesicle formulations through human skin.
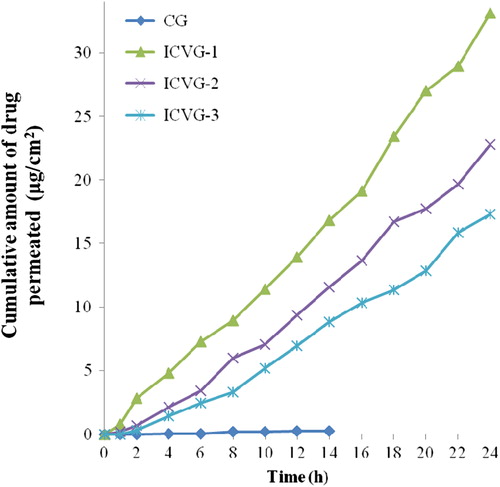
CG has shown permeation till 14 h and the maximum drug level was 0.27. Among nanovesicle gel formulation, maximum drug permeation was in ICVG-1 which was almost twice of drug amount permeated in ICVG-3. All ceramide containing formulations showed presence of drug in receptor fluid till 24 h with steady increase in drug content.
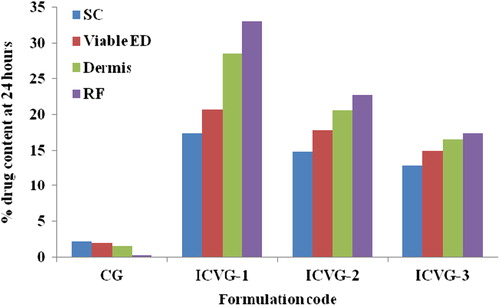
The skin sample used in ex vivo drug permeation was stratified in various skin layers using tape stripping and obtained data (in %) is presented in . CG has shown 0.27% drug content in receptor fluid which was 4.6% of the total drug content permeated to the skin by CG (5.85%). ICVG-1 has shown the maximum drug content in skin, from which close to 33% was in receptor fluid. ICVG-2 shown 30% and ICVG-3 shown 28% drug in receptor fluid from all the drug entered into the skin. CG has shown constantly declining drug content as we move to the inside skin layers whereas vesicle formulations has shown a reverse trend; however, in both the cases dermis has played the role of a drug reservoir which is in equilibrium with drug content in receptor fluid.
Figure 4. Drug content accumulated in different skin strata. SC, stratum corneum; VED, viable epidermis; D, Dermis; RF, receptor fluid.
Based on the data obtained, various permeation parameters (permeability coefficient, distribution parameter, and enhancement ration) were calculated and presented in . ICVG-1 has shown highest flux at 1.35 μg/cm2/hr and minimum lag time of 0.7 h. CG has shown flux and lag time at 0.0213 μg/cm2/hr and 2.8 h, respectively.
Table IV. Permeation parameters for CG and ICVG formulations.
In vivo peremation
ICVG-1 was selected for in vivo evaluation in rats on the basis of ex vivo drug permeation (). Cmax values for CG and ICVG-1 were 0.7 and 9.6 μg with tmax values of 4 and 8 h, respectively. The AUC value for ICVG-1 was 149.1 μg. hr/ml which was 19.9 times the AUC value of CG ().
Figure 5. Plasma concentration time profile for CG and ICVG-1.
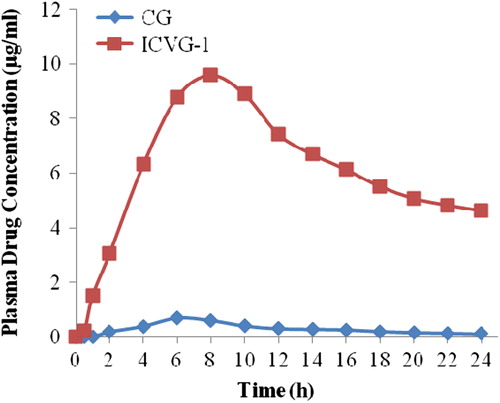
Table V. Pharmacokinetic data of the CG and ICVG-1.
Anti-inflammatory activity
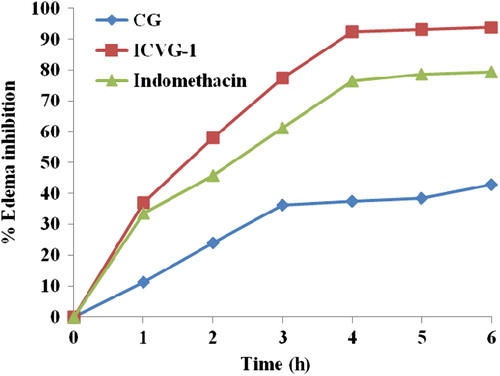
ICVG-1 was compared with CG and a standard anti- inflammatory treatment (Indomethacin, p.o.) using carrageenan rat paw edema model for determination of anti-inflammatory activity. Maximum protection against carrageenan insult was provided by ICVG-1, and it has shown 57.98% edema inhibition during initial hours of edema production (). ICVG-1 has shown 118% edema inhibition in comparison to standard drug Indomethacin albeit at a fairly low dose. The edema inhibition was 2.18 times than inhibition provided by CG highlighting the advantage of entrapment of drug inside a vesicle.
Figure 6. Anti-inflammatory activity.
Irritation potential
No irritation was observed in group treated with ceramide nanovesicle formulation whereas the group administered with SLS showed severe irritation. The application of gel containing ceramide has shown improved aesthetic appearance of skin.
Discussion
Drug-loaded vesicles devised with ceramide 2 have shown optimum physicochemical parameters. The vesicle size in nanometer range infers that the lipid composition is quite optimum for vesicle formulation. In the present study, good entrapment efficiency was obtained which could be due to the synergistic effect of the positively charged ceramide and additive effect of cholesterol. Positive charge tends to draw drug molecules which are negatively charged during the vesicle formation, whereas cholesterol increases the entrapment efficiency.
Ibuprofen has a pKa value of 4.1, similar to pKa of palmitic acid (4.8). There will be 75% ionization of drug as well as palmitic acid at skin pH. Palitic acid being a membrane component resides in membrane only whereas drug molecules tend to move inside vesicle core (hydrophilic; ionized fraction of drug) and in vesicle wall (Hydrophobic; unionized fraction of drug). Further, the ionized fraction of drug maintains a drug pool and unionized fraction residing in vesicle wall gets released. CG contained only free drug and showed burst release during drug release study.
The physical parameters show the effect of membrane components on physical properties of vesicle formulation and help in predicting the course of formulation once administered. In the present study, spherical vesicles were produced at all lipid compositions. For in vitro release, content of palmitic acid was inversely related with drug release. Palmitic acid is a saturated lipid which makes rigid vesicle layer. Further, ICV-1 has shown acceptable stability under accelerated conditions.
ICVG-1 contains almost four times the quantity of ceramide-2 with respect to palmitic acid, whereas in ICVG-3 this ratio was only 1.35. As the ratio of ceramide-2 and palmitic acid increases, the drug permeation increases. The major component of nanovesicles (Ceramide 2; 47–38% w/w) has a phase transition temperature at 61°C which can be decreased to 46°C at pH 4.0. This reduction also depends on palmitic acid content and match with the protonation of palmitic acid's anionic form (Kitagawa et al. Citation1995). The skin having a pH of 5.4 makes Tm of ceramide vesicle membrane around 51–52°C producing gel state vesicles. Further, short-chain ceramides have been reported to induce permeabilization in phosphatidylcholine membranes (Dermal cells) resulting in higher drug permeation (Touitou et al. Citation2000).
It has been reported that SC lipid liposome can transmit the encapsulated material to deeper skin layers (Fresta and Puglisi Citation1996). Zellmer et al. have shown that vesicles made up of skin lipid merged to form extended multilamellar sheets above 32.5°C and also fused with human SC liposome (Zellmer et al. Citation1998). Various workers have proposed reorganization of skin lipid vesicles inside SC and permeabilization, lateral phase separation, trans bilayer flip-flop movement in PC membrane (Goñi and Alonso Citation2006).
It has been observed that skin samples treated with ICVG formulations have shown elevated amount of drug in inner skin layers. The authors postulate that ceramide formulations fuse with corneocytes in SC to generate hybrid vesicles. Further, upon reaching dermis, these vesicles induce permeabilization and gain access into the blood circulation.
Results of ex vivo studies were also replicated in in vivo permeation and anti-inflammatory activity. Edema induced by carrageenan has two phases. The first phase takes place through 1–2 h and next phase during 3–5 h. In second phase, the macrophages discharge IL-1 for inducing polymorphic nuclear cells accumulation which in turn produces the lysosomal enzymes and active oxygen. This results in damage of connective tissues and induction of paw swelling. Edema inhibition was more prominent in rat model because rat skin is thinner than human skin and time requirement is less for permeation of adequate drug amount.
Ceramide-2 is a natural lipid, commonly present in human skin. Further it is a protectant and also promotes cell regeneration giving an enhanced cosmetic appeal to the skin.
Conclusion
Present study revealed that ICVG formulations have shown the most favorable physicochemical properties along with optimal drug permeation. These formulations were stable and demonstrated better skin compatibility. The formulation ICVG-1 having composition of ceramide-2, cholesterol, cholesteryl sulfate and palmitic acid in the ratio of 4:2.5:1:1 in that order was found to be the best formulation. We recommend for the use of ceramide vesicles in transdermal delivery of Ibuprofen.
Acknowledgments
The authors want to acknowledge Croda India and Sederma, France for genuine supply of Ceramide-2 and Akums Drugs & Pharmaceuticals Ltd, Haridwar, (India) for providing Ibuprofen.
Further we acknowledge the sophisticated analytical instrumentation facility (SAIF), AIIMS, New Delhi for TEM analysis and Advanced centre for nanotechnology (ACN), National Institute of Pharmaceutical Education & Research (NIPER) for providing facility of Zetasizer nano ZS.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Aguiar AJ, Weiner MA. 1969. Percutaneous absorption studies of chloramphenicol solutions. J Pharm Sci 58:210–215.
- Akhter S, Jain GK, Ahmad FJ, Khar RK, Khan ZI, Talegaonkar S. 2008. Investigation of nanoemulsion system for transdermal delivery of domperidone: Ex-vivo and in vivo studies. Curr Nanosci 4: 381–390.
- An K, Sun Y, Xu L, Cui X. 2011. Preparation and in vitro evaluation of simvastatin ethosome. Artif Cells Blood Substit Immobil Biotechnol 39:347–350.
- Anne P, Ilkka K, Jouko Y, Per R. 2000. Controlled release injectable liposomal gel of ibuprofen for epidural analgesia. Int J Pharm 199:85–93.
- Antal E, Wright C, Brown B, Albert K, Aman L, Levin N. 1986. The influence of hemodialysis on the pharmacokinetics of ibuprofen and its major metabolites. J Clin Pharmacol 26:184.
- Arifin DR, Palmer AF. 2005. Stability of liposome encapsulated hemoglobin dispersions. Artif Cells Blood Substit Immobil Biotechnol 33:113–136.
- Dragicevic-Curic N, Scheglmann D, Albrecht V, Fahr A. 2008. Temoporfin-loaded invasomes: Development, characterization and in vitro skin permeation studies. J Control Release 127:59–69.
- Dugard PH (Ed). 1976. Advanccs in Modern Toxicology, vol. 4. New York: Wiley.
- Durrheim H, Flynn GL, Higuchi WI, Behl CR. 1980. Permeation of hairless mouse skin I: Experimental methods and comparison with human epidermal permeation by alkanols. J Pharm Sci 69:781–786.
- El-Leithy ES, Shaker DS, Ghorab MK, Abdel-Rashid RS. 2010. Evaluation of mucoadhesive hydrogels loaded with diclofenac sodium-chitosan microspheres for rectal administration. AAPS PharmSciTech 11:1695–1702.
- Evans AM, Nation RL, Sansom LN, Bochner F, Somogyi AA. 1989. Stereoselective plasma protein binding of ibuprofen enantiomers. Eur J Clin Pharmacol 36:283–290.
- Fresta M, Puglisi G. 1996. Application of liposomes as potential cutaneous drug delivery systems. In vitro and in vivo investigation with radioactively labelled vesicles. J Drug Target 4:95–101.
- Furuishi T, Io T, Fukami T, Suzuki T and Tomono K. 2008. Formulation and in vitro evaluation of pentazocine transdermal delivery system. Biol Pharm Bull 31:1439–1443.
- Gallo J, Gall E, Gillespie W, Albert K, Perrier D. 1986. Ibuprofen kinetics in plasma and synovial fluid of arthritic patients. J Clin Pharmacol 26:65.
- Gaur PK, Mishra S, Kumar A, Panda BP. 2014a. Development and optimization of gastroretentive mucoadhesive microspheres of gabapentin by Box-Behnken design. Artif Cells Nanomed Biotechnol 42:167–177.
- Gaur PK, Purohit S, Kumar Y, Mishra S, Bhandari A. 2014b. Development and characterization of stable nanovesicular carrier for drug delivery. Artif Cells Nanomed Biotechnol 42:296–301.
- Gaur PK, Purohit S, Kumar Y, Mishra S, Bhandari A. 2014c. Preparation, characterization and permeation studies of a nanovesicular system containing diclofenac for transdermal delivery. Pharm Dev Technol 19:48–54.
- Goñi FM, Alonso A. 2006. Biophysics of sphingolipids I. Membrane properties of sphingosine, ceramides and other simple sphingolipids. Biochimica Biophysica Acta. 1758:1902–1921.
- Haigh JM, Beyssac E, Chanet L, Aiache JM. 1998. In vitro permeation of progesterone from a gel through the shed skin of three different snake species. Int J Pharm 170:151–156.
- Higuchi T. 1960. Physical chemical analysis of percutaneous absorption process from creams and ointments. J Soc Cosmet Chem 11:85–97.
- Ishida T, Takanashi Y, Doi H, Yamamoto I, Kiwada H. 2002. Encapsulation of an antivasospastic drug, fasudil, into liposomes, and in vitro stability of the fasudil-loaded liposomes. Int J Pharm 232:59–67.
- Jukanti R, Sheela S, Bandari S, Veerareddy PR. 2011. Enhanced bioavailability of exemestane via proliposomes based transdermal delivery. J Pharm Sci 100:3208–3222.
- Kitagawa S, Yokochi N, Murooka N. 1995. pH-Dependence of phase transition of the lipid bilayer of liposomes of stratum corneum lipids. Int J Pharm 126:49–56.
- Kobayashi N, Saitoh I. 1999. Development of a test method for in vitro drug release from soluble and crystal dispersion type ointments. Chem Pharm Bull 47:199–202.
- Lee E, Williams K, Day R, Graham G, Champion D. 2004. Stereoselective disposition of ibuprofen enantiomers in man. Br J Clin Pharmacol 58:S759–S764.
- Manconi M, Caddeo C, Sinico C, Valenti D, Mostallino MC, Biggio G, Fadda AM. 2011. Ex vivo skin delivery of diclofenac by transcutol containing liposomes and suggested mechanism of vesicle-skin interaction. Eur J Pharm Biopharm 78:27–35.
- Manosroi A, Jantrawut P, Manosroi J. 2008. Anti-inflammatory activity of gel containing novel elastic niosomes entrapped with diclofenac diethylammonium. Int J Pharm 360:156–163.
- Mishra D, Garg M, Dubey V, Jain S, Jain NK. 2007. Elastic liposomes mediated transdermal delivery of an anti-hypertensive agent: propranolol hydrochloride. J Pharm Sci 96:147–155.
- Patel NA, Patel NJ, Patel RP. 2009. Formulation and evaluation of curcumin gel for topical application. Pharm Dev Technol 14:80–89.
- Paudel KS, Milewski M, Swadley CL, Brogden NK, Ghosh P, Stinchcomb AL. 2010. Challenges and opportunities in dermal/transdermal delivery. Ther Deliv 1:109–131.
- Sinico C, Manconi M, Valenti M, Fadda AM. 2005. Liposomes as carriers for dermal delivery of tretinoin: in vitro evaluation of drug permeation and vesicle-skin interaction. J Control Release 103:123–136.
- Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. 2000. Ethosomes – novel vesicular carriers for enhanced delivery: characterization and skin permeation properties. J Control Release 65:403–418.
- Vasiljevic D, Parojcic J, Primorac M, Vuleta G. 2006. An investigation into the characteristics and drug release properties ofmultiple W/O/W emulsion systems containing low concentration of lipophilic polymeric emulsifier. Int J Pharm 309:171–177.
- Walters KA, Roberts MS (Eds). 2002. The Structure and Function of Skin. New York: Marcel Dekker INC.
- Wartewig S, Neubert RH. 2007. Properties of ceramides and their impact on the stratum corneum structure: a review. Skin Pharmacol Physiol 20:220–229.
- Yardley HL, Summerly R. 1981. Lipid composition and metabolism in normal and diseased epidermis. Pharmacol Ther 13:357–383.
- Zellmer S, Zimmermann I, Selle C, Sternberg B, Pohle W, Lasch J. 1998. Physicochemical characterisation of human stratum corneum lipid liposomes. Chem Phys Lipids 94:97–108.