Abstract
The discovery that stress and depression, as well as other psychiatric illnesses, are characterized by structural alterations, and that these changes result from atrophy and loss of neurons and glia in specific limbic regions and circuits, has contributed to a fundamental change in our understanding of these illnesses. These structural changes are accompanied by dysregulation of neuroprotective and neurotrophic signaling mechanisms that are required for the maturation, growth, and survival of neurons and glia. Conversely, behavioral and therapeutic interventions can reverse these structural alterations by stimulating neuroprotective and neurotrophic pathways and by blocking the damaging, excitotoxic, and inflammatory effects of stress. Lifetime exposure to cellular and environmental stressors and interactions with genetic factors contribute to individual susceptibility or resilience. This exciting area of research holds promise and potential for further elucidating the pathophysiology of psychiatric illness and for development of novel therapeutic interventions.
El haber descubierto que el estrés y la depresión, al igual que otras enfermedades psiquiáiricas, se caracterizan por alteraciones estructurales, y que estos cambios se deben a atrofía y pérdida de neuronas y glía en regiones y circuitos límbicos específicos ha contribuido a un cambío fundamental en la comprensión de estas enfermedades, Estos cambios estructurales se acompañan de una falta de regulación de los mecanismos de señales neuroprotectoras y neurotróficas, los cuales son requeridos para la maduración, crecimiento y supervivencia de las neuronas y la glía. A la inversa, las intervenciones conductualesy terapéuticas pueden revertir estas alteraciones estructurales mediante la estimulación de las vías neuroproiectoras y neurotróficas y a través del bloqueo de los efecios dañinos, excitotóxicos e inflamatorios del estrés. La exposición a lo largo de la vida a estresores celula res y ambientales, y las interacciones con factores genéticos contribuyen a la susceptibilidad o a la resiliencia. Esta inieresanie área de investigación abríga potenciales esperanzas para favorecer la dilucidación de la fisiopatología de las enfermedades psiquiátricas y el desarrollo de nuevas intervenciones terapéuticas.
Notre compréhension du stress et de la dépression, comme d'autres maladies psychiatriques, a été profondément transformée en découvrant que ces maladies sont caractérisées par des modifications structurales résultant de l'atrophie et de la perte des neurones et de la qlie dans des régions et des circuits limbiques spécifiques. Ces modifications structurales s'accompagnent d'une dysrégulaiion des mécanismes de signalisation neurotrophiques et neuroprotecteurs nécessaires à la maturation, la croissance et la survie des neurones et de la glie. À l'opposé, des interventions comportementales et thérapeutiques peuvent inverser ces modifications structurales en stimulant les voies ne uro protectrices et neurotrophiques et en bloquant les effets lésionnels, excitotoxiques et inflammatoires du stress. L'exposition durant la vie aux agents stressants cellulaires et environnementaux et les interactions avec des facteurs génétiques participent à la susceptibilité individuelle ou à la resilience. Ce domaine de recherche passionnant est prometteur et devrait permettre d'expliquer plus précisément la physiopathologie des maladies psychiatriques et de participer au développement de nouveaux traitements.
A role for damage and protection of neurons in the pathophysiology and treatment of psychiatric illness, including major depressive disorder (MDD) is based on molecular, cellular, and morphological studies in experimental animals and in human patients. Preclinical studies demonstrate that chronic stress causes alterations to the number and shape of neurons and glia in brain regions implicated in mood disorders.Citation1-Citation4 Advances in human brain imaging have also reported decreased volumes of limbic brain regions implicated in depression.Citation5,Citation6 Preclinical and postmortem studies of signal transduction pathways and target genes have extended this work at the molecular level, demonstrating dysregulation of neurotrophic factors and neuroprotective mechanisms in response to stress and in depressed patients.Citation1,Citation2,Citation7 Conversely, chronic administration of therapeutic agents blocks the effects of stress or leads to induction of neurotrophic and neuroprotective pathways.Citation2,Citation8 Together, these findings have contributed to a fundamental shift in our understanding of the cause and treatment of psychiatric illnesses and the role of neurotrophic and neuroprotective mechanisms.
This review will present evidence demonstrating neuronal damage, atrophy, and cell loss in response to stress and depression, and the mechanisms underlying these effects. Studies demonstrating the neuroprotective actions of therapeutic agents that counteract the effects of stress and depression will also be discussed. Related aspects of this work are the effects of environment, cellular stressors, insults, and interactions with genetic factors that increase susceptibility and thereby cause damage and illness . Conversely, life history of behavior or therapies that reduce stress and enhance neuronal survival, such as exercise, diet, medications, and interactions with genetic factors that increase resilience are neuroprotective, and reverse or block the damaging effects of stress.
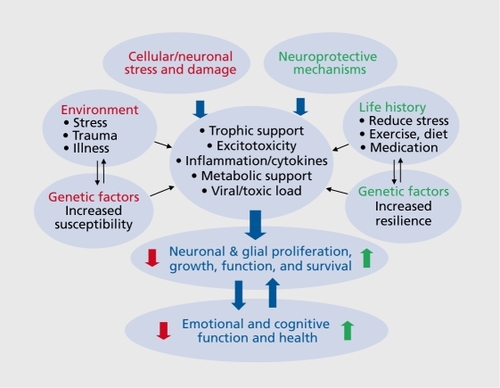
In addition, cellular growth and survival are intimately controlled by neuronal activity (Figure 1). This is due to the activity-dependent requirement for expression of neurotrophic factors and other survival pathways and mechanisms that control neurotransmission and neuroplasticity, as well as proliferation, growth, and survival.
Structural/cellular alterations in mood disorders
Depression, like most other major psychiatric illnesses, is widely accepted to be caused by neurochemical imbalances in regions of the brain that are known to control mood, anxiety, cognition, and fear. These regions include the hippocampus, prefrontal cortex (PFC), cingulate cortex, nucleus accumbens, and amygdala. In addition, brain imaging and postmortem studies have identified structural alterations in MDD patients that indicate reductions in dendrite arborization and complexity, and decreased numbers of neurons and glia in these brain regions, all of which could contribute to depressive symptoms . Together, these findings provide compelling evidence for disruption of neurotrophic factors and neuroprotective mechanisms in the pathophysiology of depression.
Structural alterations
One of the regions of interest in depression, as well as other disorders, is the hippocampus, a structure that contains high levels of receptors for glucocorticoids. Imaging studies have consistently reported that the volume of the hippocampus is decreased 10% to 20% in MDD patients.Citation9-Citation12,Citation13,Citation14 There is also evidence of a negative correlation with the length of illness and reversal with antidepressant treatment (ADT),Citation15 but additional studies are needed to further examine these relationships and to determine whether the reduction is a result or a cause of depressive illness. It is also notable that hippocampal volume reductions have been reported in other stressrelated illnesses, including post-traumatic stress disorder (PTSD)Citation16,Citation17 and schizophrenic patients.Citation18
The PFC is another ”stress-responsive“ brain region implicated in depression. The primary function of the PFC is cognition, working memory, and inhibitory control of brain regions that underlie fear and emotion. Brain imaging studies have reported a significant reduction in the volume of the PFC in MDD patients, which could underlie the reported hypofunction of this structure, most notably decreased cognition.Citation9,Citation15,Citation19,Citation20
Cellular alterations
Different types of cellular alterations could account for the volume reductions observed in the hippocampus, including reductions in the number, size, and proliferation of neurons and glia. There is one report that the size of neurons in the major subfields of the hippocampus is reduced,Citation21 suggesting a reduction in neuropil that could contribute to decreased hippocampal volume in MDD patients. There were no changes in the numbers of neurons or glia reported in this study or in other qualitative studies, although more subtle synaptic changes have been reported.Citation22
Studies of the PFC and cingulate cortex have been more extensive, and have shown a reduction in the size of neuronal cell bodies, suggestive of reduced dendritic arborization and complexity.Citation23,Citation24 In addition, the most consistent finding in studies of PFC is a decrease in the number of glia in MDD patients.Citation23-Citation25 Reductions of both astrocytesCitation26 and oligodendrocytesCitation27,Citation28 have been reported. Given the significant role of glia in providing metabolic support for neurons as well as control of neurotransmitter activity (eg, synthesis and reuptake), it is reasonable to speculate that neuronal atrophy, damage, and hypof unction of PFC could be related to the loss of glia.
Cellular alterations in animal models of depression
Animal models of depression have been used to further elucidate the ultastructural and molecular alterations that underlie the morphological changes observed in MDD patients. Most of these models are based on acute or chronic-stress paradigms, as stress is a critical factor in the etiology of depression. In addition, these models have been used to demonstrate that antidepressants can reverse or block the effects of stress on cellular morphology, which might contribute to the therapeutic actions of these agents.
Cell morphology
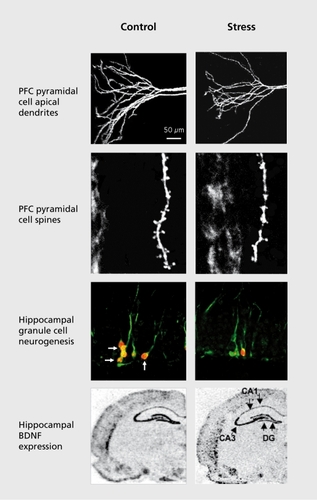
Early studies of cell morphology found that repeated stress causes atrophy of CA3 pyramidal neurons in the hippocampus, characterized by a decreased number and length of apical dendrites.Citation29,Citation30 More recent studies have shown that pyramidal neurons in the PFC undergo a similar retraction/atrophy of apical dendrites, and a reduction in spine number in response to immobilization stress (Figure 2). Citation31 Chronic exposure to high levels of exogenous corticosterone, the rodent equivalent of Cortisol, causes a similar atrophy of hippocampal and PFC neurons.Citation32,Citation33
In contrast to most neurological disorders, in which the structural alterations and loss of neurons is permanent, the stress-induced atrophy of hippocampal and PFC neurons is reversible. Most notably, removing animals from stress normalizes the dendritic arborization of pyramidal neurons over a period of several weeks.Citation3,Citation32,Citation34 Moreover, chronic administration of certain antidepressants blocks or reverses hippocampal atrophy, even with continued stress exposure.Citation29,Citation30 This reversibility supports the notion that dendritic alterations represent a type of structural plasticity that has functional consequences.
Cell proliferation
In addition to dendritic atrophy, chronic stress decreases the proliferation of new cells in the adult hippocampus and PFC. The dentate gyrus of the hippocampus is one of the few regions of the brain that continues to give rise to new neurons in adulthood, in rodents as well as nonhuman primates and humans.Citation35,Citation36 Interestingly, the rate of neurogenesis is influenced by environmental and endocrine factors, and stress is one of the most consistent and robust negative regulators (Figure 2). The proliferation of new neurons is decreased by different types of stress, including restraint, footshock, maternal separation, predator odor, psychosocial stress, and sleep deprivation, and by administration of exogenous corticosterone.Citation37 In the PFC the proliferation of glia is decreased by exposure to repeated stressCitation38 or corticosterone treatment.Citation39 Chronic stress also decreases the number of glial fibrillary acidic protein (GFAP)-positive astrocytes in the hippocampus.Citation40
In contrast, antidepressants increase the proliferation of neurons and glia in the hippocampus and/or PFC, and block or reverse the effects of stress.Citation37,Citation38,Citation41,Citation42 These effects require chronic administration (weeks), consistent with the time course for the therapeutic response to antidepressants. Different classes of antidepressant increase cell proliferation in rats, including serotonin selective transporter inhibitors, norepinephrine selective reuptake inhibitors (NSRIs), and electroconvulsive seizures (ECS),Citation41,Citation43,Citation44 indicating that this is a common target of ADT.
Behavioral consequences of altered cell morphology
A major question is whether the cellular alterations lead to changes in behavior. This has been addressed by blockade studies (ie, focused irradiation or genetic manipulation), which demonstrate that neurogenesis is required for the actions of antidepressants in certain behavioral models,Citation42,Citation45,Citation46 although there are exceptions.Citation47,Citation48 Ablation of glia in the PFC decreases sucrose consumption, a measure of anhedonia, indicating a requirement for glial function in this model.Citation49 Decreased PFC dendrite arborization in response to stress is also correlated with a reduction in attention set shifting, a PFC-dependent behavior.Citation50 These studies demonstrate a causal and/or correlative relationship between cell number and complexity with behavior.
Importance of life stress/trauma: gene-environment interactions
There is also evidence that exposure to traumatic or stressful life events can have a cumulative effect that increases susceptibility or vulnerability to mood disordersCitation51 (see Figure 1). Interactions of stress and genetic factors have also been reported, most notably for lifetime stress and the serotonin (5-HT) transporter short allele polymorphismCitation52; however, a recent meta-analysis suggests that additional studies of this polymorphism are required.Citation53 Studies of genes that increase resilience to stress and mood disorders have also been conducted.Citation54 Recent studies have also reported an interaction between early life stress or trauma and neurotrophic factors (see below).
Mechanisms underlying structural alterations and neuroprotection: gene-environment Interactions
Cellular and structural alterations in response to stress, depression, and antidepressant medications could result from a number of different mechanisms that alter the proliferation, growth, survival, and function of neurons and glia. These include altered neurotrophic/growth factor support, excitotoxicity, inflammation/cy tokines, metabolic/vascular support, viral, and toxic insults. The influence of these factors and insults on cell function and survival could occur rapidly after a single major event or could occur gradually over time with the accumulation of one or more insults, also referred to as allostatic load (Figure 1). Citation55
The effects of these cellular stressors and insults are also influenced by genetic factors that can either increase susceptibility to cellular damage, or conversely decrease susceptibility and increase resilience and neuroprotection. This complex interaction of gene -environment interactions over the lifespan is thought to contribute to the heterogeneity of depression, other psychiatric illnesses, as well as treatment of these disorders. Characterization of the molecular mechanisms and genetic factors that underlie the structural alterations and that play a key role in neuroprotection will provide important information for the diagnosis and treatment of depression.
The following sections will discuss the major molecular and cellular mechanisms underlying the actions of stress, depression, and ADT. The latter will include not only chemical antidepressants, but also other strategies that have neuroprotective actions, including exercise. As discussed above, postmortem studies demonstrate a decrease in the size, but not the number, of neurons, indicating that cell death probably does not play a major role in depression. These findings suggest that mechanisms that control maintenance of neuronal size and function, and counteract stress-induced atrophy, such as neurotrophic factors, could be critical mediators. Other important mechanisms to be discussed are glutamate excitoxicity, apoptosis, and inflammation/immune responses.
Neurotrophic/growth factors
The nerve growth factor (NGF) family has been the focus of much of the work on stress and depression, and the most widely studied member of this family is brain derived neurotrophic factor (BDNF). In addition, several other growth factors, including VEGF, IGF-1, and FGF2 have also been implicated in the effects of stress, depression, and ADT. Because these factors play a critical role in the proliferation, growth, and survival of neurons and glia in the adult brain, their altered expression or function could contribute to the cellular and morphological changes in animal models of depression and in MDD patients. This section will review key evidence demonstrating dysregulation of neurotrophic/growth factors in stress and depression.
Role of BDNF in stress, depression, and ADT
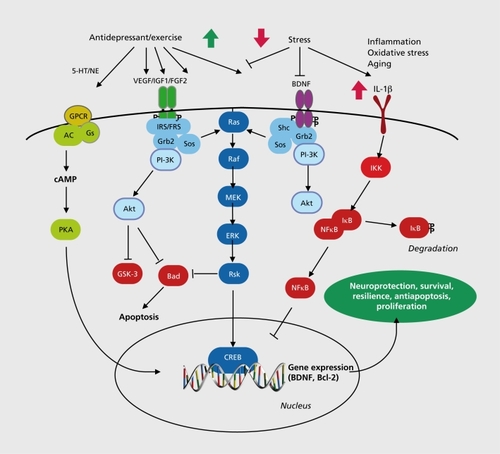
BDNF and related family members, including NGF and neurotrophin-3 (NT-3), influence the proliferation, differentiation, and growth of neurons during development, but are also expressed in the adult brain and play a critical role in the survival and function of mature neurons.Citation56 BDNF is expressed at relatively high levels in limbic brain structures implicated in mood disorders, including the hippocampus, PFC, and amygdala, and acts through a transmembrane tyrosine kinase receptor referred to as TrkB. Functional BDNF acts as a dimer to stimulate the intracellular tyrosine kinase domain of TrkB, resulting in autophosphorylation of the receptor and interactions with docking proteins that lead to activation of one of three major intracellular signaling cascades: the microtubule associated protein kinase (MAPK), the phosphatidylinositol-3 kinase (PI3K), and the phospholipase-C-γ (PLCγ) pathways .Citation8 These cascades have been linked to the neuroprotective effects of BDNF, as well as regulation of cell proliferation, differentiation, and survival.Citation56
Stress, depression, and regulation of BDNF
Smith and colleagues were the first to report that exposure to immobilization stress results in a dramatic reduction in levels of BDNF in the rodent hippocampus,Citation57 and this effect has been reported with many other types of stress.Citation1 Decreased expression of BDNF is observed in the major subfields of the hippocampus, including those layers where dendritic atrophy (CA3 pyramidal cell layer) and decreased neurogenesis (dentate gyrus granule cell layer) are observed in response to stress.Citation57 Expression of BDNF in the PFC is also decreased by chronic, but not acute stress.Citation58 Postmortem studies are consistent with the rodent work, reporting decreased levels of BDNF in the hippocampus of suicide-MDD subjects.Citation59-Citation61 These findings provide further support for the hypothesis that the morphological and behavioral abnormalities associated with MDD could result, in part, from decreased BDNF expression.
There are several possible mechanisms that could underlie the regulation of BDNF by stress. This includes a reduction of neuronal firing, as BDNF expression is dependent on activity and Ca2+-stimulated gene transcription.Citation62 BDNF expression is also decreased by adrenal-glucocorticoids, which are induced by stress and activation of the hypothalamo-pituitary-adrenal (HPA) axis.Citation57 There is also evidence that downregulation of BDNF by acute stress is mediated by interleukin-1β (IL-1β),Citation63 and epigenetic regulation of BDNF expression in response to chronic social defeat stress.Citation64
Genetic studies of BDNF and interactions with stress
A relationship between BDNF, morphology, and behavior is supported by genetic studies of BDNF. Most of this work has focused on a functional polymorphism, Val66Met, that decreases the processing and release of BDNF.Citation65 The Met allele has been associated with reduced hippocampal size and decreased memory and executive function in humans.Citation65-Citation67 The met allele has also been associated with smaller volume of cingulate cortex, and this effect is greater in patients with bipolar disorder.Citation68 There are also reports that patients carrying the Met allele, either young or aged, have an increased incidence of depression when exposed to stress or trauma.Citation69-Citation71 These latter studies highlight the importance of gene x environment interactions in complex, multifactorial illnesses such as depression.
Evidence for a direct relationship between the Met allele and neuronal structure has also been reported in rodent models. In mice expressing the Met allele there is a decrease in the number and length of apical dendrites in both the hippocampusCitation72 and PFC,Citation73 similar to the actions of stress.Citation73,Citation74
Although deletion of BDNF is not sufficient to produce depressive-like behaviors, except in female mice,Citation75-Citation77 blockade or reduction of BDNF expression increases the susceptibility to the effects of stress. Exposure of BDNF heterozygous deletion mutant mice to stress or blockade of BDNF-TrkB signaling produces a depressive -like phenotype in the forced swim test.Citation78 A gene-environment interaction is also supported by the genetic association studies of the BDNF Met allele discussed above,Citation70,Citation71
A complicating factor in understanding the functions of trophic factors is the possibility that there are opposing, region-specific effects of BDNF. This is based on studies demonstrating that BDNF is increased by stress in the mesolimbic dopamine system and has a depressive effect in the social defeat model, and conversely, that ADT decreases BDNF in this reward pathway.Citation79 These findings demonstrate that the expression and function of BDNF, and possibly other trophic factors, is circuitdependent and that findings in one region cannot be extrapolated to others.
Antidepressants increase BDNF
In contrast to the effects of stress, chronic, but not acute ADT increases the expression of BDNF in the hippocampus and frontal cortex.Citation1,Citation80-Citation82 Induction of BDNF is observed with different classes of chemical antidepressants as well as electroconvulsive seizures.Citation1,Citation80,Citation82,Citation83 Other agents known to have antidepressant efficacy also increase BDNF expression in the hippocampus, including a-amino-3-hydroxyl-5-methyl-4-isoxazole -propionic acid (AMPA) receptor potentiators, NMDA receptor antagonists, transcranial magnetic stimulation, and exercise,Citation1 These findings indicate that increased expression of BDNF is a common target for different therapeutic strategies. Postmortem studies also demonstrate that BDNF levels are increased in the hippocampus of patients receiving antidepressant medication at the time of death, demonstrating the clinical relevance of ADT induction of BDNF.Citation59 These effects are thought to occur via activation of cAMP and/or Ca2+-dependent BDNF gene transcription that are activated by ADT.Citation8-Citation186
Neuroprotective, neurogenic, and behavioral actions of BDNF
The neuroprotective effects of BDNF have been well documented, primarily in cultured cell systems, but also in vivo. This includes studies demonstrating that BDNF increases survival and has neuroprotective actions in models of hypoxia, ischemia, excitotoxicity, hypoclycemia, and inflammationCitation87-Citation91; for reviews see refs 92,93. As discussed above, hippocampal pyramidal cell dendrite complexity is decreased in BDNF Met allele or heterozygous deletion mutants.Citation74 Similar effects have been observed in PFC pyramidal cells, and stress does not produce further atrophy of apical dendrites in BDNF heterozygous deletion mutants, indicating that decreased BDNF underlies the effects of stress.Citation73 These findings indicate that a full complement of functional BDNF is required for maintenance of normal dendritic arbor in both the hippocampus and PFC.
BDNF has also been shown to influence hippocampal neurogenesis. Infusions of BDNF increase hippocampal neurogenesis,Citation94,Citation95 and BDNF is necessary for the survival of new neurons in response to ADT.Citation96 The BDNF receptor, TrkB is also required for antidepressant induction of hippocampal neurogenesis, as well as the behavioral actions of antidepressants.Citation97 BDNF has also been implicated in the behavioral actions of ADT. BDNF infusions are sufficient to produce an antidepressant response in rodent behavioral models of depression,Citation98-Citation100 and mutant mouse studies demonstrate that BDNF is required for the behavioral actions of antidepressants.Citation75-Citation77 These findings are consistent with the hypothesis that induction of BDNF contributes to the neurogenic and behavioral actions of antidepressants.
Other neurotrophic/growth factors
There is now strong evidence demonstrating a role for several other growth factors in the actions of stress, depression, and ADT, including vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2), and insulin-like growth factor 1 (IGF-1).
VEGF was originally characterized as a vascular permeability factor and an endothelial cell mitogen,Citation101 but is also expressed in the brain in both neurons and glia, and has been shown to play a role in hippocampal neuroplasticity, memory, and neurogenesis.Citation102,Citation103 Chronic unpredictable stress decreases the expression of VEGF, as well as its receptor, Flk-1,Citation102 while ADT increases VEGF expression in the granule cell layer of the hippocampus.Citation104 Different classes of chemical antidepressants, including SSRI, NSRI, and ECS, increase VEGF expression in the hippocampus, indicating that VEGF is a common downstream target of these treatments.
The opposing actions of stress and ADT on VEGF suggest a possible relationship between neurogenesis and behavior. Stress has a greater effect on newborn cells associated with endothelial cells than nonvascular associated cells.Citation102 In addition, VEGF is sufficient to induce neurogenesis and produce antidepressant effects in behavioral models of depression, whereas inhibition of Flk-1 blocks the induction of adult neurogenesis and the behavioral effects of ADT.Citation104
A recent postmortem study found that the expression of FGF2 and its receptors (FGFR2 and FGFR3) are reduced in the PFC and cingulate cortex of MDD patients,Citation105 and social defeat stress decreases FGF2 in the hippocampus.Citation106 Conversely, chronic ADT increases the expression of FGF2 in cerebral cortex and hippocampus of rodentsCitation107,Citation108 and FGF2 infusions are sufficient to produce an antidepressant response in behavioral models.Citation109 The role of FGF2 in the proliferative actions of ADT, on both neurons and glia, is currently being investigated.
The expression of IGF-1 in the hippocampus is increased by chronic administration of two different monoamine oxidase inhibitor antidepessants.Citation110 In addition to expression in brain, circulating IGF-1, derived primarily from the liver, is actively transported into the brain and is required for the induction of neurogenesis in response to exercise,Citation111 Recent studies have also demonstrated that IGF-1 administration, or agents that increase IGF-1 levels, produce antidepressant-like actions in behavioral models of depression.Citation98,Citation112,Citation113 Together, these findings suggest that peripheral production and/or the central actions of IGF-1 could be novel targets for the treatment of depression.
Neuroprotective and neurotrophic effects of exercise
Exercise is reported to increase the expression of neurotrophic/growth factors, including BDNF, VEGF, FGF2, and IGF-1.Citation111,Citation114-Citation117 In addition, exercise increases neurogenesis in the adult hippocampus, an effect that is dependent on increased expression of IGF-1 and VEGF.Citation111,Citation114 IGF-1 has also been shown to underlie the neuroprotective effects of exercise against different types of brain insults.Citation118 In addition to the regulation of these growth factors, exercise has also been shown to influence other neuroprotective mechanisms.Citation119 These positive, neuroprotective actions make exercise one of the key behavioral factors for protecting, or even reversing the damage that can be caused by environmental, physical, and psychological stressors, and even the susceptibility resulting from genetic vulnerabilities (see Figure 1).
Glutamatergic excitotoxicity: stress, depression, and ADT
Excess glutamatergic excitotoxicity is one of the major mechanisms underlying neuronal damage and loss in the brain, and has been implicated in the pathophysiology of a variety of disorders, including those resulting from acute insult (eg, stroke induced ischemia or trauma) and neurodegenerative disorders (eg, amyotrophic lateral sclerosis, Huntington's chorea, epilepsy, and Alzheimer's disease.Citation120,Citation121 This section discusses evidence for excess glutamate in stress related mood disorders, the cellular mechanisms that contribute to glutamate excitotoxicity, and pharmacological strategies for intervention and treatment.
Excess glutamate in depression and stress
Abnormal glutamate levels and function have been implicated in psychiatric illnesses, including schizophrenia, anxiety, and mood disorders.Citation122-Citation124 Glutamatergic abnormalities have been reported in the plasma, serum, cerebrospinal fluid (CSF), and brain tissue of individuals suffering from mood disorders.Citation123 Functional in vivo measures of glutamate content in the brain using proton magnetic resonance spectroscopy (II-MRS) show elevated glutamate levels in the occipital cortex of depressed patients, although decreases have been reported in other regions.Citation123,Citation125
Preclinical studies also demonstrate a role for glutamate in the actions of stress. Microdialysis studies have shown that stress increases extracellular levels of glutamate in the PFC and hippocampus,Citation126,Citation127 consistent with the possibility that atrophy of CA3 neurons arises in part through increased glutamate neurotransmission.Citation128,Citation129 This hypothesis is supported by studies demonstrating that N-methyl-D-aspartic acid (NMDA) receptor antagonists attenuate stress-induced atrophy of CA3-pyramidal neurons.Citation29,Citation32,Citation130 Stress or glucocorticoid treatment also increases the susceptibility to other types of neuronal insults, including excitotoxins and ischemia.Citation129,Citation131
There are several possible mechanisms that could contribute to the overactivation of glutamate in response to stress and in depression, including a decrease or loss of mechanisms for inactivation of glutamate. Glial cells are responsible for the reuptake and inactivation of glutamate from synaptic and extrasynaptic sites.Citation123 Reductions in the number or function of glia are thought to play a role in the atrophy of limbic brain regions observed in brain imaging studies, as well as decreased neuronal cell body size in postmortem brains of depressed patients.Citation123,Citation132,Citation133 Recent studies demonstrate that agents that increase glial reuptake of glutamate, such as riluzole and ceftriaxone, have antidepressant effects in rodent behavioral models and in depressed patients.Citation132-Citation134
Mechanisms of glutamate excitotoxicity
Glutamate neurotoxicity results from excessive flux of Ca2+ via ionoptopic receptors, including AMPA, kainiate, and NMDA type receptors.Citation120,Citation121,Citation123 Uncontrolled elevation of intracellular Ca2+ leads to further loss of Ca2+ buffering and homeostasis, and then to a cascade of events that contribute to cell damage and death. These include oxidative stress resulting in generation of reactive oxygen species (ROS) and nitric oxide, which results in necrotic cell death characterized by swelling, membrane damage, DNA degradation, and eventually inflammation and cell lysis.Citation120,Citation121,Citation135
There are multiple sites for controlling glutamate release and activity at pre- and postsynaptic sites, as well as for buffering intracellular Ca2+ that protects against cell damage. These mechanisms are typically overcome only by severe conditions, such as those that would occur during stroke-induced ischemia, prolonged hypoxia, uncontrolled seizures or head trauma. As discussed above, most studies do not report a loss of neurons in post-mortem tissue from depressed patients, or in animal models. However, excess glutamate is still thought to play a role in psychiatric illnesses, and this has resulted in targeting glutamatergic sites for development of therapeutic agents for mood disorders, as well as for other psychiatric, neurological, and neurodegenerative illnesses.
Glutamate and neuroprotection: therapeutic targets
Glutamate neurotransmission is controlled by a complex system of pre- and postsynaptic receptors, including ionotropic and metabotropic subtypes. In addition, regulation of tropic factor signaling cascades, including extracellular signal-related kinase (ERK), Akt, and cAMP response element binding (CREB) can serve as neuroprotective targets for excitoxicity. There is also evidence that chronic ADT regulates the phosphorylation, trafficking, and expression of glutamate receptors, providing further evidence that the actions of ADT involves this neurotransmitter system. These topics have been extensively covered by a number of recent reviews.Citation121,Citation123,Citation136,Citation137 A brief discussion of the major glutamatergic targets will be discussed here.
One of the key targets for regulation of glutamate is glial reuptake, which is the primary mechanism for inactivation of glutamate neurotransmission. Agents that increase this process, notably riluzole and ceftriaxone, are reported to have antidepressant efficacy in rodent models and in clinical trials.Citation123,Citation132,Citation138 These effects are mediated in part by increased expression of glial excitatory amino acid transporters. Riluzole also has several other interesting properties, including the ability to decrease glutamate and increase neurotrophic factor expression, making this an interesting, and potentially useful therapeutic compound. Clinical and preclinical studies are currently underway to further test the therapeutic efficacy and mechanisms underlying the actions of riluzole, Lamotragine is another compound that acts in part by decreasing glutamate release and is used for treating mood disorders, although with limited efficacy.Citation123
Blockade of the NMDA ionotropic receptor represents another primary target for neuroprotection, although this is a complex issue as glutamate is the major excitatory neurotransmitter in the brain. However, agents that block the NMDA channel, most notably memantine and ketamine, are reported to have antidepressant actions in clinical trials and rodents.Citation123-Citation137 The actions of memantine have been more modest, with greater effects when coadministered with other antidepressants. However, reports on ketamine have been extraordinary, with several studies demonstrating a rapid and sustained antidepressant response in approximately 60% of patients tested, which have all been resistant to other chemical antidepressants.Citation139,Citation140 A single intravenous dose of ketamine, which produces transient and mild psychotomimetic effects, results in an antidepressant response within 6 to 12 hours, and this effect is sustained for at least 7 days. These effects are dramatic compared with all other chemical antidepressants, which require weeks or months of treatment before a therapeutic response is observed. Further studies are needed to identify safer drugs that have rapid antidepressant effects similar to ketamine.
The most direct mechanism to explain the antidepressant action of ketamine is its direct inhibitory effect on NMDA receptors. In particular, the hypothesis that blockade of the extrasynaptic NR2B receptor subtype, which is activated by excess glutamate, underlies the therapeutic action of ketamine has received the most attention. This possibility is supported by a recent study demonstrating that a selective NR2B receptor inhibitor, CP-101,606, produces a rapid antidepressant response in treatment resistant MDD patients.Citation141 Another possible mechanism to account for the rapid actions of these agents is via blockade of NMDA receptors on GABAergic inhibitory neurons, which leads to disinhibition or activation of glutamatergic transmission. The latter possibility is supported by studies in rodents demonstrating that NMDA channel blockers increase BDNF expression in limbic structures, indicating stimulation of neuronal activity,Citation142,Citation143 and by a recent report that the behavioral actions of ketamine are blocked by inhibition of AMPA receptor activity.Citation144
The metabotropic glutamate receptors represent another interesting and diverse set of targets for drug development.Citation123,Citation136 Group I receptors, particularly mGluRl/R5 subtypes located at postsynaptic sites as well as on glia, influence both the function and release of glutamate. Drugs acting at these receptors are reported to have anxiolytic effects in rodent models. Group II receptors, mGluR2 and R3, located at presynaptic sites and on glia and regulate glutamate release, have also been targets of interest. Both agonists and antagonists of group II receptors have shown promise, with reports that mGluR2/R3 antagonists have antidepressant actions and agonists showing anxiolytic and antipsychotic effects. Most promising is a clinical report demonstrating antipsychotic efficacy of an mGluR2/3 agonist.Citation145
Allosteric AMPA receptor potentiator (ARP) agents make up another interesting group of drugs. These agents do not directly stimulate AMPA receptors, but slow the inactivation or desensitation of the receptors. The idea of using drugs that enhance AMPA receptor function would appear to be counterintuitive given the possibility of an overactive glutamate system. However, preclinical studies of these agents, which were first developed for enhancing cognition, demonstrate positive antidepressant-like effects in rodent models of depression.Citation123,Citation146
Programmed cell death (apoptosis) in stress and depression
Programmed cell death is a critical mechanism for regulation of the appropriate complement of neurons during development, but apoptotic signaling pathways are also regulated in the adult brain and influence the number and function of mature cells. Apoptosis is a highly regulated signaling process, which includes the Bcl-2 family of proteins, cytochrome C, a cytosolic adaptor protein, and caspase activation, which results in energy-dependent death.Citation135,Citation147 The Bcl-2 family includes antiapoptotic factors (ie, Bcl-2 and Bcl-xl) that antagonize proapoptotic factors (eg, Bax and Bak). Upon activation of apoptotic pathways, Bax and Bak insert into the mitochrondrial membrane and promote the release of cytochrome C, which in turn binds to the apoptotic activator factor (Apafl), leading to activation of capases 9 and 3.
Regulation of apoptosis by depression, stress, and ADT
Analsysis of postmortem tissue and rodent models has provided some evidence for apoptotic cell death and/or signaling in depression and stress.Citation135 There is a postmortem report of low levels of apoptosis in the temporal cortex and hippocampus of depressed patients.Citation148 Rodent studies demonstrate that social stress increases the number of apoptotic cells in the hippocampus and temporal cortex,Citation149 and chronic unpredictable stress increases the number of caspase 3 positive neurons in the cerebral cortex.Citation150 Maternal separation of rats is also reported to increase cell death in the dentate gyrus of hippocampus.Citation151
Genetic association studies have also provided evidence for a link between apoptosis signaling and depression. Polymorphisms of the adaptor protein Apaf1 were found to be associated with major depression.Citation152 These polymorphisms increase the activity of caspase 9 and would thereby increase the vulnerability of neurons to apoptotic cell death.
There are also several studies that have examined levels of apoptotic signaling proteins in models of stress and ADT. Chronic unpredictable stress is reported to decrease levels of the antiapoptotic factors Bcl-2 and Bcl-xl, but does not influence levels of Bax.Citation153 Administration of a high dose of adrenal-glucocorticoids reduces Bcl-2 levels, and this effect corresponds with increased sensitivity to exeitotoxic damage,Citation154 Conversely, chronic ADT increases the expression of Bcl-2 and/or Bcl-xl in limbic brain regions.Citation153,Citation155,Citation156 Chronic administration of lithium or valproate also increases Bcl-2 in the hippocampus and PFC.Citation157 The antiapoptotic actions of lithium and valproate have also been demonstrated in studies of cultured cells.Citation158-Citation160
Antidepressants also influence other signaling cascades that indirectly influence apoptotic processes. Most notable are the effects of stress and ADT on neurotrophic factors and related signaling cascades, including ERK and Akt, which increase cell survival in part via inhibition of apoptotic, and induction of antiapoptotic factors such as Bcl-2.Citation135,Citation161 Finally, it is also notable that certain members of the Bcl-2 family have also been implicated in other cellular functions, including neurotransmission, which could be involved in the actions of stress and ADT.Citation162
Mitochondria play a primary role in the storage, processing, and release of proteins involved in apoptosis, and recent studies demonstrate a role for other aspects of mitochondria function in the pathophysiology and treatment of mood disorders.Citation154,Citation163
Inflammation/immune responses
Inflammation and immune responses are major factors contributing to the etiology and pathophysiology of many medical illnesses, including depression and other psychiatric disorders. Inflammation can be caused byother medical conditions, including infection, stroke, trauma, and abnormal or autoimmune responses. However, it is also now clear that psychological stress, such as social stress, can activate the innate immune system, elevating cytokine production, and thereby stimulate inflammatory processes.Citation164,Citation167
Inflammatory and immune processes can lead to multiple actions that have acute protective actions, but that also can have damaging effects on cells and tissue. This includes many of the same actions implicated in the responses to stress and depression, including activation of the HPA axis, alterations of neurotransmitter systems, decreased neurotrophic factor expression, and increased oxidative stress.Citation167 Depending on the severity and length of the inflammatory response, these effects can result in significant actions on neuronal and glial function and cell survival or death.
There are several proinflammatory cytokines of interest, including IL-1, IL-6, and tumor necrosis factor (TNF)a, that have been implicated in the pathophysiology and treatment of depression. Also of interest are studies of interferon-a, used for the treatment of hepatitis or cancer, which results in depressive-like symptoms in a large number of patients. Here we discuss a few of the most interesting targets for treatment of depression; for a more thorough review see ref 167.
TNFα and depression
One of the most consistently altered proinflammatory cytokines in depressed subjects is TNFα. An inverse correlation between levels of TNFα and treatment response has been reported.Citation168,Citation169 TNFα immunotherapy also causes depression, indicating that this cytokine may contribute to the etiology of mood disorders and is not simply a marker for depression (for reviews see refs 168,170). Moreover, a recent large clinical trial using an antibody neutralization approach demonstrated significant antidepressant effects of TNFα reduction.Citation171 This finding is supported by preclinical studies demonstrating that TNFα infusions produce a prodepressive effect,Citation172 and that TNFα receptor null mutant mice have an antidepressant phenotype in the forced swim and sucrose consumption tests.Citation173 Taken together, the preclinical and clinical studies provide strong support for TNFα receptors, particularly TNFR2, as targets for the treatment of mood disorders.
IL-1β, stress, and depression
There is also strong evidence that the proinflammatory cytokine IL-1β plays a key role in the pathophysiology of stress and depression, and that the IL-1β signaling is a relevant target for drug development.Citation174,Citation175 These findings include: i) clinical studies reporting an increase in serum levels IL-1β in MDDCitation176-Citation180;ii) reports that IL-ip produces stress like effects, including activation of the IIPA axis, regulation of monoamines, and behavioral responses in rodent modelsCitation181; iii) evidence that IL-1β contributes to conditioned fear and depressive like behavior,Citation182 and produces anhedonia and disrupts incentive motivation in rodent models183; iv) preclinical reports that IL-1β decreases hippocampal neurogenesis and underlies the decrease observed in response to stressCitation184; v) our report that CUS-induced anhedonia and decreased neurogenesis produced by is blocked by pharmacological inhibition or null mutation of IL-1β receptors.Citation184 Studies are currently underway to determine if blockade of peripheral, as well as central IL-1β signaling is sufficient to block the effects of stress and produce antidepressant actions.
Interferon and IDO
Recent studies demonstrate that one of the key factors contributing to the depressive actions of inflammation and activation of the innate immune system is the induction of a tryptophan degradative enzyme, indoleamine 2,3-dioxygenase (IDO). Chronic inflammation and infection can lead to sustained induction of interferon, which is then responsible for the increased levels of IDO. The induction of IDO then results in diversion of tryptophan from the synthesis of serotonin to kyneurenic acid, which can be further converted to toxic metabolites, most notably quinolinic acid. Evidence for IDO in depression is supported bystudies demonstrating that decreased levels of tryptophan and increased kyneurenin is associated with inflammation and depression.Citation185 Increased IDO has also been positively correlated with depression, although a direct causal relationship has not been demonstrated.
A recent study has now provided direct evidence that induction of IDO underlies the depressive behaviors caused by inflammation/activated immunologic conditions. This work was conducted using a bacterial immune activation model, Bacille Calmette-Guerin (BCM), which induces a long-lasting induction of interferon and results in depressive behaviors in animal models.Citation185,Citation186 The results demonstrate that BCM-mediated immobility in the forced swim test is reversed by an IDO inhibitor, 1-methyltryptophan, and in mice that are deficient in IDO.Citation185 In addition, BCM also increases the expression of a downstream enzyme, 3-hydroxyanthranilic acid oxygenase (3-HAO) that is involved in the synthesis of quinolinic acid. These studies indicate that an IDO inhibitor, and possibly an inhibitor of 3-HAO, could have efficacy for the treatment of depression and related mood disorders.
Summary and future directions
Significant advances have been made in characterizing the neuronal and glial damage, or structural alterations, at the cellular and anatomical levels in stress-related mood disorders and other psychiatric illnesses, and in elucidating the molecular signaling pathways and mechanisms that underlie these changes. However, this work is still at a relatively early stage, and a more complete characterization of these complex alterations and signaling mechanisms will require extensive resources and time. Moreover, identification of genetic polymorphisms that impact these pathways and systems and that influence susceptibility or resilience to illness is a major area of research that will continue to develop and unfold. When combined with studies of environmental risk factors and lifetime history of stress, this work will define and describe the mechanisms underlying individual variations of illness.
Together, the results of this work can be used to formulate a comprehensive approach for the prevention and treatment of psychiatric illnesses. Changes in lifestyle and behavior can reduce stress and exposure to environmental factors that influence cellular risk and damage and prevent illness. These approaches, as well as behavioral interventions that enhance the activity and function of specific neural circuits, and thereby provide protection, can also be used once a person has become ill. Development of therapeutic agents that target neuroprotective mechanisms, combined with genetic information will ultimately provide tailored approaches for highly specific and efficacious treatments for depression and other illnesses.
Selected abbreviations and acronyms
| ADT | = | antidepressant treatment |
| AMPA | = | α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionic acid |
| BDNF | = | brain derived neurotrophic factor |
| IDO | = | indoleamine 2,3-dioxygenase |
| MDD | = | major depressive disorder |
| NMDA | = | N-methyl-D-aspartic acid |
| PFC | = | prefrontal cortex |
| TNF | = | tumor necrosis factor |
This work is supported by USPHS grants MH45481 and 2 P01 MH25642 and by the Connecticut Mental Health Center. We would also like to thank Mr Xiaowe Su for assistance with literature research, and Drs G. Aghajanian and R-J. Li for providing images of PFC dendrites in Figure 2.
REFERENCES
- DumanR.MonteggiaLM.A neurotrophic model for stress-related mood disorders.Biol Psychiatry.2006591116112716631126
- KrishnanV.IMestlerEJ.The molecular neurobiology of depression.Nature.200845589490218923511
- McEwenB.Sex, stress and the hippocampus: allostatic load and the aging process.Neurobioi Aging.200223921939
- SahayA.HenR.Adult hippocampal neurogenesis in depression.Nat Neurosci.2007101110111517726477
- CampbellS.MacQueenG.An update on regional brain volume differences associated with mood disorders.Curr Opin Psych.2006192533
- DrevetsW.Neuroimaging studies of mood disorders.Biol Psychiatry.20004881382911063977
- ManjiH.QuirozJA.SpornJ.et al.Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for diff icult-totreat depression.Biol Psychiatry.20035370774212706957
- TanisK.NewtonSS.DumanRS.Targeting neurotrophic growth factor expression and signaling for antidepressant drug development.CNS Neurol Disorders.20076151160
- BremnerJD.VermettenE.MazureCM.Development and preliminary psychometric properties of an instrument for the measurement of childhood trauma: the Early Trauma Inventory.Depression Anxiety.200012112
- MervaalaE.FohrJ.KononenM.et al.Quantitative MRI of the hippocampus and amygdala in severe depression.Psychol Med.20003011712510722182
- ShelineY.WanyP.GadoMH.CsernanskyJG.VannierMW.Hippocampal atrophy in recurrent major depression.Proc Natl Acad Sci USA.199693390839138632988
- ShelineY.SanghaviM.MintunMA.GadoMH.Depression duration but not age predicts hippocampal volume loss in medically healthy wormen with recurrent major depression,J Neurosci.1999195034504310366636
- CampbellS.MarriottM.NahmiasC.MacQueenGM.Lower hippocampal volume in patients suffering from depression: a meta-analysis.Am J Psychiatry.200416159860715056502
- VidebechP.RavnkildeB.Hippocampal volume and depression: a meta-analysis of MRI studies.Am J Psychiatry.20041611957196615514393
- ShelineY.GadoMH.KraemerHC.Untreated depression and hippocampal volume loss.Am J Psychiatry.20031601516151812900317
- BremnerJD.RandallPScottTM.et al.MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder.Am J Psychiatry.19951529739817793467
- GurvitsT.ShentonME.HokamaH.et al.Magnetic resonoance imaging study of hippocampal volume in chronic, combat-related posttraumatic stress disorder.Biol Psychiatry.199640109110998931911
- HeckersS.Neuroimaging studies of the hippocampus in schizophrenia.Hippocampus.20011152052811732705
- BremnerJ.VythilingamM.VermettenE.et al.Reduced volume of orbitofrontal cortex in major depression.Biol Psychiatry.20025127327911958777
- DrevetsWC.PriceJLSimpsonJRet al.Subgenual prefrontal cortex abnormalities in mood disorders.Nature.19973868248279126739
- StockmeierC.MahajanGJ.KonickLC.et al.Cellular changes in the postmortem hippocampus in major depression.Biol Psychiatry.20045664065015522247
- MullerM.LucassenPJ.YassouridisA.HoogendijkJG.HolsboerF.SwabbDF.Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus.Eur J Neurosci.2001141603161211860455
- CotterD.ParianteCM.EverallIP.Glial cell abnormalities in major psychiatric disorders: the evidence and implications.Br Res Bull.200155585595
- RajkowskaG.Miguel-HidalgoJJ.WeiJ.et al.Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression.Biol Psychiatry.1999451085109810331101
- OngurD.DrevetsWC.PriceJL.Glial reduction in the subgenual prefrontal cortex in mood disorders.Proc Natl Acad Sci USA.19989513290132959789081
- SiX.Miguel-HidalgoJJ.O'DwyerG.StockmeierCA.RajkowskaG.Age-dependent reductions in the level of glial fibrillary acidic protein in the prefrontal cortex in major depression.Neuropsychopharmacoi.20042920882096
- HamidiM.DrevetsWC.PriceJL.Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes.Biol Psychiatry.20045556356915013824
- UranovaN.VostrikovVM.OrlovskayaDD.RachmanovaVI.Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study for the Stanley Neuropathology Consortium.Schizophr Res.20046726927514984887
- McEwenB.Stress and hippocampal plasticity.Curr Opin Neurobiol.199952052167620309
- WatanabeY.GouldE.DanielsDC.CameronH.McEwenBS.Tianeptine attenuates stress- induced morphological changes in the hippocampus.Eur J Pharmacol.19922221571621468492
- RadleyJ.SistiHM.HaoJ.et al.Chronic behavioral stress induces apical dendritic reorganization in pyramidal neurons of the medial prefrontal cortex.Neurosci.200412516
- MagarinosAM.McEwenBSFluggeG.FuchsEChronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews.J Neurosci.199616353435408627386
- WellmanC.Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration,J Neurobiol.20014924525311745662
- RadleyJ.RocherAB.MillerM.et al.Repeated stress induces dendrtic spine loss in the rat medial prefrontal cortex.Cereb Cortex.20061631332015901656
- GageF.Mammalian neural stem cells.Science.20002871433143810688783
- GouldE.TanapatP.Stress and hippocampal neurogenesis.Biol Psychiatry.1999461472147910599477
- Warner-SchmidtJ.DumanRS.Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment.Hippocampus.20061623924916425236
- BanasrM.ValentineGW.LiXY.GourleyS.TaylorJ.DumanRS.Chronic stress decreases cell proliferation in adult cerebral cortex of rat: reversal by antidepressant treatment.Biol Psychiatry.20076249650417585885
- AlonsoG.Prolonged corticosterone treatment of adult rats inhibits the proliferation of oligodendrocyte progenitors present throughout white and gray matter regions of the brain.Glia.20003121923110941148
- CzehB.SimonM.SchmeltingB.HiemkeC.FuchsE.Astroglial plasticity in the hippocampus is sffected by chronic psychosocial stress and concomitant fluoxetine treatment.Neuropsychopharmacoi.20063116161626
- MalbergJ.EischAJ.NestlerEJ.DumanRS.Chronic antidepressant treatment increases neurogenesis in adult hippocampus.J Neurosci.2000209104911011124987
- SantarelliL.SaxeM.GrossC.et al.Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants.Science.200330180580912907793
- KodamaM.FujiokaT.DumanRS.Chronic olanzapine or fluoxetine treatment increases cell proliferation in rat hippocampus and frontal cortex.J Biol Psych.200456570580
- MadsenT.NewtonSS.EatonME.RussellDS.DumanRS.Chronic electroconvulsive seizure up-regulates b-catenin expression in rat hippocampus: role in adult neurogenesis.Biol Psychiatry.2003541006101414625142
- AiranR.MeltzerLA.RoyM.GongY.ChenH.DeisserothK.High-speed imaging reveals neurophysiological links to behavior in an animal model of depression.Science.200731775775817690279
- JiangX.XuK.HobermanJ.TianF.et al.BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects.Neuropsychopharmacoi.20053013531361
- BessaJ.FerreiraD.MeloI.et al.Hippocampal neurogenesis induced by antidepressant drugs: an epiphenomenon in their mood-improving actions.Mol Psych.200914739
- DavidD.KlemenhagenKC.HolickKA.et al. Efficacy of the MCHR1 antagonist N-{3-(1-{[4-(3,4- difluorophenoxy) phenyl]methyl}(4-piperidyl))4methylphenyl]-2-rnethylproanamide (SNAP 94847) in mouse models of anxiety and depression following acute and chronic administration is independent of hippocampal neurogenesis.J Pharmacol Exp Ther.200732123724817237257
- BanasrM.DumanRS.Glial loss in prefrontal cortex is sufficient to induce depressive-like behaviors in rodent models.Biol Psychiatry.20086486387018639237
- ListonC.MillerMM.GoldwaterDS.et al.Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting.J Neurosci.2006267870787416870732
- LupienS.McEwenBS.GunnarMR.HeimC.Effects of stress throughout the lifespan on the brain, behaviour and cognition. WatRev Neurosci.200910434445
- CaspiA.SugdenK.MoffittTE.et al.Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene.Science.200330138638912869766
- RischN.herrellR.LehnerT.et al.Interaction between the serotonin transporter gene (5-HTTLPR), stressful like events, risk of depression: a meta-analysis.JAMA.20093012462247119531786
- FederA.NestlerEJ.CharneyDS.Psychobiology and molecular genetics of resilience. WatRev Neurosci.200910446457
- McEwenB.Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators.Eur J Pharmacol.200858317418518282566
- McAllisterA.Spatially restricted actions of BDNF.Neuron.20023654955012441043
- SmithMA.MakinoS.KvetnanskyR.PostRM.Stress alters the express of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus.J Neurosci.1995a15176817777891134
- XuH.ChenZ.HeJ.HaimanotS.LiX.DyckL.LiXM.Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stressinduced decrease in cell proliferation and BDNF expression in rat hippocampus.Hippocampus.20061655155916652337
- ChenB.DowlatshahiD.MacQueenGM.WangJ-F.YoungLT.Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication.Biol Psychiatry.20015026026511522260
- DwivediY.RizaviHS.ConleyRR.TammingaCA.PandeyGN.Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects.Arch Gen Psychiatry.2003b6080481512912764
- KaregeF.BondolfiG.GervasoniN.SchwaldM.AubryJM.BertschyG.Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity.Biol Psychiatry.2005571068107215860348
- MetsisM.TimmuskT.ArenasE.PerssonH.Differential usage of multiple BDNF promoters in the rat brain following neuronal activation.Proc Natl Acad Sci USA.19939088028415610
- BarrientosR.SprungerDB.CampeauS.et al.Brain-derived neurotrophic factor mRNA downregulation induced by social isolation is blocked by intrahippocampal interleukin-1 receptor anatagonist.Neurosci.2003121847853
- TsankovaN.BertonO.RenthalW.KumarA.NeveR.NestlerEJ.Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action.Nat Neurosci.2006946546616568101
- EganM.KojimaM.CallicottJH.et al.The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function.Cell.200311225726912553913
- FrodlT.SchuleC.SchmittG.et al.Association of the brain-derived neurotrophic factor Val66Met polymorphism with reduced hippocampal volumes in major depressio.Arch Gen Psych.200764410416
- HaririA.GoldbergTE.MattayVS.et al.Brain-derived neurotrophic factor val66 met polymorphism affects human memory-related hippocampal activity and predicts memory performance.J Neurosci.200323669094
- MatsuoK.Walss-BassC.NeryFG.et al.Neuronal correlates of brainderived neurotrophic factor Val66Met polymorphism and morphometric abnormalities in bipolar disorder.Neuropsychopharmacoi.20093419041913
- GattJ.NemeroffCB.Dobson-StoneC.et al.Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety.Mol Psych.200914681695
- KaufmanJ.YangBZ.Douglas-PalumberiH.et al.Brain-derived neurotrophic factor-5-HTTLPR gene interactions and environmental modifiers of depression in children.Biol Psychiatry.20065967368016458264
- KimJ.StewartR.KimSW.et al.Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders.Biol Psych.200762423428
- LeeY.DumanRS.MarekGJ.The mGlu2/3 receptor agonist LY354740 suppresses immobilization stress-induced increase in rat prefrontal cortical BDNF mRNA expression.Neurosci Lett.200639832833216469447
- LiuR-J.AghajanianGK.Stress blunts serotonin- and hypocretinevoked EPSCs in prefrontal cortex: role of corticosterone-mediated apical dendritic atrophy.Proc Natl Acad Sci USA.200810535936418172209
- ChenH.PandeyGN.DwivediY.Hippocampal cell proliferation regulation by repeated stress and antidepressants.Neuroreport.20061786386716738477
- MonteggiaL.BarrotM.PowellCM.et al.Essential role of brainderived neuroprophic factor in adult hippocampal function.Proc Natl Acad Sci USA.2004101108271083215249684
- MonteggiaL.LuikartB.BarrotM.et al.al.Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors.Biol Psych.200761187197
- SaarelainenT.HendolinP.LucasG.et al.Activation of the trkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects.J Neurosci.20032334935712514234
- DumanC.SchlesingerL.KodamaM.RussellDS.DumanRS.A role for MAPK signaling in behavioral models of depression and antidepressant treatment.Biol Psych.200761661670
- BertonO.McClungCA.DileoneRJ.et al.Essential role of BDNF in the misolimbic dopamine pathway in social defeat stress.Science.2006311868878
- CastrenE.VoikarV.RantamakiT.Role of neurotrophic factors in depression.Curr Opin Pharmacol.20077182117049922
- NestlerE.BarrotM.DiLeoneRJ.EischAJ.Gold SJ. Monteggia LM. Neurobiology of Depression.Neuron.200234132511931738
- NibuyaM.MorinobuS.DumanRSRegulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments.J Neurosci.199515753975477472505
- GrovesJ.Is it time to reassess the BDNF hypothesis of depression?Moi Psychiatry.2007121079108817700574
- NestlerE.TerwilligerRZDumanRS.Chronic antidepressant administration alters the subcellular distribution of cAMP-dependent protein kinase in rat frontal cortex.J Neurochem.198953164416472795022
- PerezJ.TinelliD.BrunelloN.RacagniG.cAMP-dependent phosphorylation of soluble and crude microtubule fractions of rat cerebral cortex after prolonged desmethylimipramine treatment.Eur J Pharmacol.19891723053162550266
- TiraboschiE.TarditoD.KasaharaJ.et al.Selective phosphorylation of nuclear CREB by fluoxetine is linked to activation of CaM kinase IV and MAP kinase cascades.Neuropsychopharm.20042918311840
- Destot-WongK.LiangK.GuptaSK.et al.The AMPA receptor positive allosteric modulator, S18986, is neuroprotctive against neonatal excitotoxic and inflammatory brain damage through BDNF synthesis.Neuropharmacol.200951277286
- HanB.HoltzmanDM.BDNF protects the neonatal brain from hypoxicischemic injury in vivo via the ERK pathway.J Neurosci.2000205775578110908618
- MiyataK.MiyataN.OmoriH.et al.Involvement of the brain-derived neurotrophic factor/TrkB pathway in neuroprotective effect of cyclosporing A in forebrain.Neurosci.2001105571578
- SunX.ZhouH.LuoX.LiS.YuD.HuaJ.MuD.MaoM.Neuroprotection of brain-derived neurotropic factor against hypoxic injury in vitro requires activation of wxtracellular signal-regulated kinase and phosphatidylinositol 3-kinase.int J Dev Neurosci.20082636337018243629
- WaltonM.WaltonB.ConnorP.et al.Neuronal death and survival in two models of hypoxic-ischemic brain damage.Brain Res Rev.19992913716810209230
- ArancioO.ChaoMV.Neurotrophins, synaptic plasticity and dementia.Curr Opin Neurobiol.20071732533017419049
- HenniganA.O'CallaghanRM.KellyAM.Neurotrophins and their receptors: roles in plasticity, neurodegeneratin and neuroprotection.Biochen Soc Trans.200735(Pt 2)424427
- PenceaV.BingamanKDWiegandSJ.LuskinMB.Infusion of brainderived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus.J Neurosci.2001216706671711517260
- ScharfmanH.MacLuskyNJ.Similarities between actions of estrogen and BDNF in the hippocampus: coincidence or clue?Trends Neurosci.200528798515667930
- SairanenM.LucasG.ErnforsP.CastrenM.CasrenE.Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus.J Neurosci.2005251089109415689544
- LiY.LuikartBW.BirnbaumS.et al.TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment.Neuron.20085939941218701066
- HoshawB.MalbergJE.LuckiI.Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects.Brain Res.20051037 20420815777771
- ShirayamaY.ChenA C-HNakagawaS.RussellRS.DumanRS.Brain derived neurotrophic factor produces antidepressant effects in behavioral models of depression.J Neurosci.2002223251326111943826
- SiuciakJA.LewisDR.WiegandSJ.LindsayR.Antidepressant-like effect of brain derived neurotrophic factor (BDNF).Pharmacol Biochem Beh.199756131137
- FerraraN.GerberHP.LeCouterJ.The biology of VEGF and its receptors.Nat Med.2003966967612778165
- HeineV.ZarenoJ.MaslamS.JoelsM.LucassenPJ.Chronic stress in the adult dentate gyrus reduces cell proliferation near the vasculature and VEGF and Flk-1 protein expression.Eur J Neurosci.2005211304131415813940
- PalmerT.WillhoiteAR.GageFH.Vascular niche for adult hippocampal neurogenesis.J Comp Neurol.200042547949410975875
- Warner-SchmidtJ.DumanRS.VEGF is an essential mediator of neurogenic and behavioral actions of antidepressants.Proc Natl Acad Sci USA.20071044647465217360578
- EvansS.ChoudaryPV.NealCR.et al.Dysregulation of the fibroblast growth factor system in major depression.Proc Natl Acad Sci USA.2004101155061551115483108
- TurnerC.GulaEL.TaylorLP.WatsonSJ.AkilH.Antidepressant-like effects of intracerebroventrivular FGF2 in rats.Brain Res.20081224636818586016
- MalleiA.ShiB.MocchettiI.Antidepressant treatments induce the expression of basic fibroblast growth factor in cortical and hippocampal neurons.Mol Pharmacol.2002611017102411961119
- MaragnoliM.FumagalliF.GennarelliM.RacagniG.RivaMA.Fluoxetine and olanzapine have synergistic effects in the modulation of fibroblast growth factor 2 expression within the rat brain.Biol Psych.20045510951102
- TurnerC.CalvoN.FrostDO.AkilH.WatsonSJ.The fibroblast growth factor system is downregulated following social defeat.Neurosci Lett.200843014715018061349
- KhawajaX.XuJ.LiangJ-J.BarrettJE.Proteomic analysis of protein changes developing in rat hippocampus after chronic antidepressant treatment: Impliations for depressive disorders and future therapies.J Neurosci.200475451460
- TrejoJ.CarroE.Torres-AlemanI.Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus.J Neurosci.2001211628163411222653
- DumanC.SchlesingerL.RussellDR.DumanRS.Peripheral IGF-1 produces antidepressant-like behavior and is required for the effect of exercise.Behav Br Res.2009198366371
- MalbergJ.PiattB.RizzoSJS.et al.Increasing the levels of insulin-like growth factor- 1 by an IGF binding inhibitor produces anxiolytic and antidepressant-like effects.Neuropsychopharmacoi.20073223602368
- FabelK.FabelK.TarnB.et al.VEGF is necessary for exercise-induced adult hippocampal neurogenesis.Eur J Neurosci.2003182803281214656329
- Gomez-PinillaF.DaoL.VannarithS.Physical exercise induces FGF-2 and its mRNA in the hippocampus.Brain Res.1997764189295187
- HunsbergerJ.NewtonSS.BennettAH.et al.Antidepressant actions of the exercise-regulated gene VGF.Nat Med.2007131476148218059283
- Russo-NeustadtA.BeardRC.CotmanCW.Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression.Neuropsychopharmacoi.199921679682
- CarroE.TrejoJL.BusiguinaS.Torres-AlemanI.Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy.J Neurosci.2001215678568411466439
- CotmanC.BerchtoldNC.ChristieLA.Exercise builds brain health: key roles of growth factor cascades and inflammation.Trends Neurosci.20073046447217765329
- DongX.WangY.QinZH.Molecular mechansms of excitotoxicity and their relevance to pathogenisis of neurodegenerative diseases.Acta Pharmacol Sin.20093037938719343058
- KaliaL.KaliaSK.SalterMW.NMDA receptors in clinical neurology: excitatory times ahead.Lancet Neurol.2008774275518635022
- AmielJ.MathewSJ.Glutamate and anxiety disorders.Curr Psych Rep.20079278283
- SanacoraG.ZarateCA.KrystalJH.ManjiHK.Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders.Nat Rev Drig Discov.20087426437
- SodhiM.WoodKH.Meador-WoodruffJ.Role of glutamate in schizophrenia: integrating excitatory avenues of research.Exp Rev Neurother.2008813891406
- SanacoraG.GueorguievaR.EppersonCN.et al.Subtype-specific alterations of y-aminobutyric acid and glutamate in patients with major depression.Arch Gen Psychiatry.2004b6170571315237082
- LowryM.WittenbergL.YamamotoB.Effect of acute stress on hippocampal glutamate levels and spectrin proteolysis in young and aged rats.J Neurochem.1995652682747790870
- MoghaddamB.Stress preferentially increases extraneuronal levies of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia.J Neurochem.199360165016578097232
- SapolskyR.Glucocorticoids and atrophy of the human hippocampus.Science.19962737497508701325
- SapolskyR.The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death.Biol Psychiatry.20004875576511063972
- McEwenB.Estrogens effects on the brain: multiple sites and molecular mechanisms.J Appl Physiol.2001912785280111717247
- RoyM.SapolskyRM.The exacerbation of hippocampal excitotoxicity by glucocorticoids is not mediated by apoptosis.Neuroendocrinoi.2003772431
- BanasrM.ChowdhuryGM.NewtonSS.et al.Glial pathology in an animal model of depression: Reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate modulating drug riluzole.Mol Psychol.In press
- ValentineG.SanacoraG.Targeting glial physiology and glutamate cycling in the treatment of depression.Biochem Pharmacol.20097843143919376090
- PittengerC.DumanRS.Stress, depression, and neuroplasticity: A convergence of mechanisms.Neuropsychopharmacoi.20083388109
- McKernanD.DinanTG.CryanJF.“Killing the Blues:” A role for cellular suicide (apoptosis) in depression and the antidepressant response?Prog Neurobiol.20098824626319427352
- ConnP.JonesCK.Promise of mGluR2/3 activators in psychiatry.Neuropsychopharmacoi.200934248249
- LiptonS.Pathologically-activated therapeutics for neuroprotection: mechanism of NMDA receptor block by memantine and S-nitrosylation.Curr Drug Targets.2007862163217504105
- MineurY.SomenziO.PicciottoMR.Cytosine, a partial agonist of high affinity nicotinic acetylcholine receptors, has antidepressant-like properties in male C57BL/6J mice.Neuropharmacol.20075212561262
- BermanR.CappielloA.AnandA.et al.Antidepressant effects of ketamine in depressed patients.Biol Psychiatry.20004735135410686270
- ZarateC.SinghJ.ManjiHK.Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder.Biol Psych.20065910061020
- PreskomS.BakerB.OmoK.KolluriS.MennitiFS.LandenJW.A placebo-controlled trial of the NR2B subunit specific NMDA antagonist CP101,606 plus paroxetine for treatment resistant depression (TRD). Paper presented at: APA. 2007
- CastrenE.da Pennha BerzaghiM.LindholmD.ThoenenH.Differential effects of MK-801 on brain-derived neurotrophic factor mRNA levels in different regions of the rat brain.Exp Neurol.19931222442528405262
- TakahashiM.KakitaA.FutamuraT.et al.Sustained brain-derived neurotrophic factor up-regulation and sensorimotor gating abnormality induced by postnatal exposure to phencyclidine: comparison with adult treatment.J Neurochem.20069977078016903871
- MaengS.ZarateCA.DuJ.et al.Cellular mechanisms underlying the antidepressant effects of ketamine: Role of alpha-amino-3-hydroxy-5methylisoxazole-4-propionic acid receptors.Biol Psych.200763349352
- PatilS.ZhangL.MartenyiF.et al.Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial.Nat Med.2007131102110717767166
- SuX.LiX-Y.BanasrM.KooJW.ShahidM.HenryB.DumanRS.Chronic treatment with AMPA receptor potentiator Org 16576 increases neuronal cell proliferation and survival in adult rodent hippocampus.Psychopharrnacol.2009, In press
- CoryS.AdamsJM.The Bcl2 family: regulators of the cellular life-ordeath switch.Nat Rev Cancer.2002264765612209154
- LucassenP.MullerMB.HolsboerF.et al.Hippocampal apoptosis in major depression is a minor event and absent from sub-areas at risk for glucocorticoid overexposure.Am J Pathology.2001158453468
- LucassenP.Vollmann-HonsdorfGK.GleisbergM.CzéhB.De KloetER.FuchsE.Chronic psychosocial stress differentially affects apoptosis in hippocampal subregions and cortex of the adult tree shrew.Eur J Neurosci.20011416116611488960
- BachisA.MalleiA.CruzMMI.WellsteinA.MocchettiI.Chronic antidepressant treatments increase basic fibroblast growth factor and fibroblast growth factor-binding protein in neurons.Neuropharmacol.20085511141120
- LeeJ.DuanW.MattsonMP.Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice.J Neurochem.2002821367137512354284
- HarlanJ.ChenY.GubbinsE.MuellerR.et al.Variants in Apaf-1 segregating with major depression promote apoptosome function.Mol Psych.2006117685
- KostenT.GallowayMP.DumanRS.RussellDS.D'SaC.Repeated unpredictable stress and antidepressants differentially regulate expression of the Bcl-2 family of apoptotic genes in rat cortical, hippocampal, and limbic brain structures.Neuropsychopharmacoi.20083315451558
- DuJ.WangY.HunterR.WeiY.et al.Dynamic regulation of mitochondrial function by glucocorticoids.PNAS.20091063543354819202080
- MurrayF.HutsonPH.Hippocampal Bcl-2 expression is selectively increased following chronic but not acute treatment with antidepressants, 5-HT(1A0 or 5-HT(2C/2B0 receptor antagonists.Eur J Pharmacol.2007569414717582397
- XuB.GouldingEH.ZangK.et al.Brain-derived neurotropohic factor regulates energy balance downstream of melanocortin-4 receptor. WatNeurosci.20036736742
- ChenR.ChuangDM.Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression. A prominent role in neuroprotection against excitotoxicity.J Biol Chem.19992746039604210037682
- BieleckaA.ObuchowiczE.Antiapoptotic action of lithium and valproate.Pharmacol Rep.20086077178219211968
- ChuangD.The antiapoptotic actions of mood stabilizers: molecular mechanisms and therapeutic potentials.Ann N Y Acad Sci.2005105319520416179524
- ChuangD.ManjiHK.In search of the Holy Grail for the treatment of neurodegenerative disorders: has a simple cation been overlooked?Biol Psych.20076246
- CoyleJ.DumanRSFinding the intracellular signaling pathways affected by mood disorder treatments.Neuron.20033815716012718851
- JonasE.HoitD.HickmanJA.et al.Modulation of synaptic transmission by the BCL-2 family protein BCL-xL.J Neurosci.2003238423843112968005
- BachmanR.WangY.YuanP.et al.Common effects of lithium and valproate on mitochondrial functions: protection against methamphetamine-induced mitochondrial damage,int J Neuropsychopharmacoi.200912805822
- AnismanH.Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder.J Psych Neurosci.200934420
- GrippoA.JohnsonAK.Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models.Stress.20091211219116888
- KhairovaR.machado-VieiraR.DuJ.ManjiHK.A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder.Int J Neuropsychopharmacoi.200912561578
- MillerA.MaleticV.RaisonCL.Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression.Biol Psych.200965732741
- AnismanH.MeraliZ.Cytokines, stress and depressive illness.Brain Behav linmun.200216513524
- LevineJ.BarakY.ChengappaKN.RapoportA.RebeyM.BarakV.Cerebrospinal cytokine levels in patients with acute depression.Neuropsychobiology.19994017117610559698
- AnismanH.HayleyS.TurrinN.MeraliZ.Cytokines as a stressor: implications for depressive illness.Int J Neuropharmacol.20025357373
- TyringS.GottliebA.PappK.et al.Etanercept and clinical outcomes, fatique, and depression in psoriasis: double-blind placebo-controlled randomized phase III trial.Lancet.2006367293516399150
- ReynoldsJ.IgnatowskiTA.SudR.SpenglerRN.An antidepressant mechanism of desipramine is to decrease tumor necrosis factor-alpha production culminating in increases in noradrenergic neurotransmission.Neurosci.2005133519531
- SimenB.DumanCH.SimenAA.DumanRS.TNF alpha signaling in depression and anxiety: behavioral consequences of individual receptor targeting.Biol Psychiatry.20065977578516458261
- KooJ.DumanRS.IL-1B is an essential mediator of the anti-neurogenic and anhedonic effects of sress.Proc Natl Acad Sci USA.200810575175618178625
- KooJ.DumanRS.IL-1 receptor null mice show decreased anxiety and enhanced fear memory.Neurosci Lett.2009 In press
- LanquillonS.KriegJC.Bening-Abu-ShachU.VedderH.Cytokine production and treatment response in major depressive disorder.Neuropsychopharm.200022370379
- MaesM.MeltzerHY.BosmansE.BergmansR.VandoolaegheE.RanjanR.Increased plasma concentrations of interleukin-6, soluble interleukin-6, soluble interleukin-2 and transferrin receptor in major depression.J Affect Dis.1995343013098550956
- MikovaO.YakimovaR.BosmansE.KenisG.MaesM.Increased serum tumor necrosis factor alpha concentrations in major depression and multiple sclerosis.Eur Neuropsychopharm.200111203208
- SluzewskaA.RybakowskiJK.LaciakM.MackiewiczA.SobieskaM.WiktorowiczK.Interleukin-6 serum levels in depressed patients before and after treatment with fluoxetine.Annals NY Acad Sci.1995762474476
- TugluC.KaraSH.CaliyurtO.VardarE.AbayE.Increased serum tumor necrosis factor-alpha levels and treatment response in major depressive disorder.Psychopharmacology (Berl).200317042943312955291
- ConnorT.LeonardBE.Depression, stress and immunological activation: the role of cytokines in depressive disorders.Life Sci.1998625836069472719
- MaierS.WatkinsLR.Intracerebroventrivular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock.Brain Res.19956952792828556346
- MeraliZ.BrennanK.BrauP.AnismanH.Dissociating anorexia and anhedonia elicited by interleukin-1beta: antidepressant and gender effects on responding for “free chow” and “earned” sucrose intake.Psychopharm.2003165413418
- KooJ.DumanRS.IL-1B is an essential mediator of the anti-neurogenic and anhedonic effects of sress.Soc Neurosci Abst.2008105751756
- O'ConnorJ.LawsonMA.AndreC.et al.Induction of IDO by bacille Calmette-Guerin is responsible for development of murine depressive-like behavior.J Immunol.20091823202321219234218
- O'ConnorJ.LawsonMA.AndreC.et al.Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2, 3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin.J Neurosci.2009294200420919339614