
Abstract
Background
Unregulated cell proliferation or growth is a prominent characteristic of cancer. We have previously demonstrated that LMPs (cell membrane microparticles derived from apoptotic human CEM T lymphoma cells stimulated with actinomycin D) strongly suppress the proliferation of not only human endothelial cells but also mouse Lewis lung carcinoma cells.
Methods
LMPs were generated either from CEM T cells using different stimuli or from 3 different types of lymphocytes. The effects of LMPs on cancer cell proliferation were examined using cell lines from different species and tissues. The cell cycle kinetics was evaluated by FACS and the expression of cell cycle-related genes was determined using quantitative RT-PCR. The in vivo anti-tumor effect of LMPs was investigated using xenografts and allografts.
Results
LMPs at doses far above physiological levels dramatically suppressed the proliferation of cancer cells in a non species-specific manner. LMPs selectively target high proliferating cells and their anti-proliferative effect is not dependent on parental cell origin or stimuli. The anti-proliferative effect of LMPs was due to induction of cell-cycle arrest in G0/G1, with associated increases in expression of the cyclin-dependent kinase inhibitors p15INK4b, p16INK4a, and p21Cip1. In vivo, LMPs significantly suppressed tumor growth in animal tumor models.
Conclusion
These results highlight the potential role of LMPs in modulating the growth of high proliferating cells. Given that cell-based therapies are considered less toxic than pharmacologic approaches and have the potential to target multiple pathways in a synergistic manner, LMPs may serve as a veritable option for cancer treatment.
Microparticles are membrane-derived vesicles (0.1–1 µm in diameter) that are released from all eukaryotic cells during biological processes of considerable diversity. Microparticles represent more than just a miniature version of the specific cell of origin, as certain components of microparticles are selectively enriched compared to their parent cell. The composition and function of microparticles depends not only on the cellular origin but also on the agonist responsible for their formation (Citation1). Mounting evidence has recently revealed that microparticles constitute protagonists of a communication network for the intercellular exchange of biological signals and information. Most of these processes are associated with vascular dysfunction and alterations in homeostatic parameters (Citation1).
Lymphocyte-derived microparticles (LMPs), generated by actinomycin D treatment of human CEM T cells or circulating LMPs isolated from either diabetic or HIV-positive patients, impair endothelial function and vascular contraction (Citation2, Citation3). The proteomic analysis revealed that LMPs display a broad spectrum of bioactive substances and receptors on their surface (Citation4). Regarding the effects of LMPs on angiogenesis, controversial results are reported (Citation5–Citation7), which is possibly caused by the different stimulation at their origin (Citation7).
Proliferation and angiogenesis are crucial steps in tumour growth. In addition to the anti-angiogenic effect of LMPs, we have observed that LMPs strongly suppress tumour growth, cell proliferation in the animal models of Lewis lung carcinoma (LLC) (Citation6). It is well known that proliferation of mammalian cells is regulated by complexes of cyclins and cyclin-dependent kinases (CDKs), and the activity of CDKs can be limited by specific cyclin-dependent kinase inhibitors (CDKIs) (Citation8, Citation9). The deregulation of CDKs is widespread in cancer cells (Citation10, Citation11). Nonetheless, the role of LMPs in cancer is poorly understood. This study was designed to characterize the anti-proliferation effect of LMPs on various cancer cell types using LMPs generated from different stimuli and from lymphocytes of different origin. We found that LMPs exert anti-proliferative and anti-tumour effects in vitro and in vivo. These inhibitory effects operated across species lines and were not specific to tumour type or stimuli. On the molecular level, LMPs specifically induce cancer cells arrest in G1 phase associated with the upregulation of CDKIs expression.
Materials and methods
Cell lines and cell proliferation assay
Immortalized rat retinal ganglion cells (RGC-5) were kindly provided by Neeraj Agarwal (University of North Texas Health Science Center). RGC-5 cells were induced to differentiate in serum-free medium with 1.0 µM staurosporine (Citation12, Citation13). Cell lines were purchased from Applied Cell Biology Research Institute – human retinal endothelial cells (HRECs), and from American Type culture collection (ATCC, Manassas, VA) – mouse LLC, mouse neuroblastoma (N2a), human neuroblastoma (SH-SY5Y), HeLa, human breast cancer (M4A4, MCF-7, MDA-MB-231), lung carcinoma (H460), CEM T cell and Jurkat T lymphoma. Cells were maintained according to their recommendations.
Cell proliferation was evaluated by [3H]-thymidine incorporation assay as previously described (Citation6). Assays were performed 24 hours after LMPs treatment, unless otherwise indicated.
Primary cell culture
Rat cortical neurons were isolated from adult Sprague-Dawley rats as described elsewhere (Citation14). Briefly, cortices were cleaned from their meninges and blood, and then minced and dissociated with trypsin, followed by adding DNase I and soybean trypsin inhibitor (Sigma), and triturated through a 5 mL pipet. After the tissue was settled, the supernatant was collected, and the remaining tissue pellet was retriturated. The combined supernatants were then centrifuged through a 4% BSA layer and the cell pellet was resuspended in neuronal seeding medium. Isolated cells were resuspended in neuronal seeding medium which consisted of neurobasal medium (Gibco), supplemented with 1.1% 100×antibiotic–antimycotic solution (Biofluids), 25 µM Na glutamate, 0.5 mM l-glutamine and 2% B27 Supplement (Gibco). Cells were seeded at a density of 1.5×105 cells onto each well of 24-well tissue culture plates (Corning) pre-coated with poly-d-lysine (70–150 kD, Sigma). Cells were seeded at a density of 1.5×105 cells per well in 24-well plates pre-coated with poly-d-lysine. Fully confluent cells were treated with LMPs for 24 hours.
Human retinoblastoma cells were isolated from a primary site intraocular retinoblastoma from a 2-year-old female Caucasian patient. In accordance with the CHU Sainte-Justine Ethics Committee, the specimen was obtained with the donor's informed consent and with the understanding that part of the tumour would be used for research purposes. A 50 mg retinoblastoma specimen (pathologically confirmed) was minced and subjected to collagenase enzymatic digestion. After the digestion, a low-speed centrifugation was performed to eliminate fibroblasts in the upper layer. A total of 25×104 cells were subsequently isolated and cultured in RPMI-1640 medium with 10% FBS and penicillin–streptomycin–l-glutamine. Retinoblastoma cells were characterized for syntaxin expression by fluorescence-activated cell sorting (FACS) using anti-human syntaxin-FITC (Cedarlane, Canada) (Citation15).
LMPs production
CEM T, Jurkat T lymphoma cells (ATCC, Manassas, VA) and human peripheral T cells were cultured in X-VIVO medium (Cambrex, Walkersville, MD). As described previously, LMPs were produced by stimulating T cells with actinomycin D (0.5 µg/mL) for 24 hours (Citation5). A supernatant was obtained by centrifugation at 750 g for 15 min, then 1,500 g for 5 minutes to remove cells and large debris. LMPs from the supernatant were washed after 3 centrifugation steps (50 minutes at 12,000 g) and recovered in PBS. As washing medium from the last supernatant has been widely used as control vehicle for each individual microparticle (Citation2, Citation7) (Citation16, Citation17), this particular medium was used as a control in following experiments unless otherwise noted. 1×106 T cells stimulated with actinomycin D (0.5 µg/mL) for 24 hours produce approximately 2 µg of LMPs. To investigate whether heat-denatured LMPs are good control for LMPs, we performed cell growth assay and found that heat-denatured LMPs still possess 40% effectiveness of inhibiting endothelial cell growth (unpublished data). Using the same protocol, we also generated LMPs from hyperoxia- (95% O2, 36 hours) and hypoxia- (5% O2, 36 hours) exposed T cells. Human peripheral blood lymphocytes were prepared using Ficoll-Hypaque gradient centrifugation (Citation18) and T cell populations were purified with Pan T Cell Isolation Kit II (Miltenyi Biotec). LMPs were characterized with annexin V staining by FACS analysis and gated using 1.0-µm beads in which 97% of MPs (≤1 µm) were annexin-V-Cy5 positive. The concentrations of LMPs were determined using the Bio-Rad protein assay.
Analysis of cell cycle kinetics
LLC cells in log phase of growth were exposed to basal media in the absence or presence of 10 µg/mL LMPs for 24 hours. Cells were harvested and stained with propidium iodide (Molecular Probes) and subjected to FACS analysis. The distribution of cells in G0/G1, S and G2/M phases was determined using LYSIS II software (Becton Dickinson).
RNA isolation and quantitative RT-PCR
Total RNA from monolayer LLC cells was extracted and cDNA was synthesized and amplified as previously reported (Citation19). Primers for p15INK4b, p16INK4a, p21Cip1, CDK2, CDK4 and mouse 18S rRNA were synthesized (Invitrogen) as follows (Citation20): p15INK4b: forward 5′-AAGCACCTTTGCTGCCTCC-3′, reverse 5′-GCAGCACGACAAGCCTGTCC-3′; p16INK4a: forward 5′-ACGGTGCAGATTCGA ACTG-3′, reverse 5′-TACACAAAGACCACCCAGCG-3′; p21Cip1: forward 5′-GAGCCACAGGCAC CATGTCC-3′, reverse 5′-AGACCTTGGGCAGCCCTAGC-3′; CDK2: forward 5′-TGGGCTGCAAGTACTACTCC-3′, reverse 5′-TTGTGATGCAGCCACTTCTA-3′; CDK4: forward 5′-TTGTACG GCTGATGGATG-3′, reverse 5′-CGGTCCCATTACTTGTCAC-3′; mouse housekeeping 18S rRNA was used as internal control, forward 5′-AGGGGAGAGCGGGTAAGAGA-3′, reverse 5′-GGACAGGACTAGGCGGAACA-3′. PCR amplification protocol involved 40 cycles of denaturation at 95°C for 30 seconds, primer annealing at 55°C and primer extension at 72°C for 1 minute. Each sample was analyzed in triplicate. The mRNA levels of each gene were quantified relative to 18S and the fold change in gene expression in control and LMPs-treated samples was determined according to the equation: fold change=2−ΔCt, where Δ Ct=(Ct)Experimental−(Ct)Control.
Western blot analysis
LLC cells were collected after LMPs treatment for 24 hours. Extraction of soluble proteins and fractionation by SDS-PAGE was performed as described previously (Citation5). The anti-p15, anti-p16 and anti-p21 antibodies (LifeSpan BioSciences, Inc. 1:500) were used to reveal the protein levels of p15INK4b, p16INK4a and p21 Cip1, respectively. β-Actin was used as a loading control. Proteins were visualized using the ECL western blotting detection system (PerkinElmer, Inc.). Densitometry values were measured in terms of pixel intensity by Fluorchem software.
Animal experiments
Female athymic nude mice and C57BL/6 mice (5–7 weeks old) were purchased from Charles River Laboratories International, Inc. (St-Constant, Quebec, Canada.) and used for xenografting and allografting, respectively, according to protocols approved by the CHU Sainte-Justine Animal Care Committee. The weight and general appearance of the animals were recorded every other day throughout the experiments.
Neuroblastoma xenografts and intratumoural injection of LMPs
Mice were subcutaneously injected with human neuroblastoma cells (SH-SY5Y, 20×106 cells in 0.1 mL of PBS) in the right flank. When tumours reached ~100 mm3, mice were randomized to receive 7 consecutive intratumoural injections of either 50 µL control vehicle or 2.5 mg/kg LMPs every 2 days. Tumour weights were determined at the conclusion of the treatment.
Allograft tumour growth and intravenous injection of LMPs
Allograft animal models were generated as previously described (Citation6). Briefly, 10 days after LLC inoculation, mice were randomly grouped to receive intravenous injection: control group – 100 µL control vehicle; treatment group – 50 mg/kg LMPs (in 100 µL PBS, optimal dose had been tested) every 2 days for 4 consecutive injections. Mice were weighted, and tumour size was measured and recorded every 2 days with an electronic calliper. Tumour volume was estimated based on the formula: Tumour volume=length (mm)×width2(mm2)/2. Mice were sacrificed 18 days post-LLC inoculation, and primary tumours were snap frozen.
Breast cancer xenografts and tumour explants
Human breast cancer xenografts were generated by subcutaneously inoculating mice with 2×106 M4A4 cells. Tumours were removed within 4 weeks of growth and sliced into pieces of 1–2 mm in diameter. Tumour explants were cultured on Matrigel (BD Biosciences) for 24 hours, as previously described (Citation21), and subsequently exposed to LMPs for different time periods.
Assessment of LMPs haematotoxicity
Six-week-old C57BL/6 mice received single intravenous injections of 25 or 75 mg/kg of LMPs (approximately 10 and 30 times the IC50, respectively) or control vehicles (4 mice per group). The daily assessment of body weight, activities, food consumption, skin colour, or fur was performed. Thirty days after LMPs injection, animals were euthanatized and tissues were examined for gross pathology. Peripheral blood was collected and subjected to a complete blood count.
Ki-67 immunohistochemical staining
The cell proliferation in LLC primary tumours was determined using immunohistochemical staining of Ki-67 as described (Citation22). Briefly, tumour tissues were fixed in formalin and embedded in paraffin. Sections were cut at 4 µm and baked for 4 hours at 60°C. Slides were deparaffined in xylene and brought to water through graded ethanol. An indirect immunoperoxidase staining was performed with rabbit monoclonal anti-mouse Ki-67 (clone SP6, Cell Marque, Rocklin, CA). Nuclear brown staining was considered positive. Proliferation was quantified and expressed as a proliferative index (PI) score, which was determined as the mean percentage of nuclei staining positive for Ki-67 antibody in 1,000 cells at 600×magnification. Four non-overlapping high-power fields that contained the highest average fraction of labelled cells for each tumour tissue were included in the calculation of PI score. All counts were completed by a blinded fashion.
Breast tumour explants were frozen in OCT (Sakura), sliced into 12 µm sections, fixed in 4% paraformaldehyde and permeabilized in 1.0% Triton X-100. Sections were then incubated with rabbit anti-Ki-67 antibody (1:200; ab15580, Abcam), followed by incubation with a goat anti-rabbit IgG conjugated to Alexa Fluor 488 (1:1,000; Invitrogen). Cell nuclei were stained with DAPI. Sections were observed with an epifluorescent microscope (Nikon eclipse E800) and images were taken with a Nikon DXM 1200 camera. The PI was determined as described above.
Statistical analysis
All experiments were repeated at least 3 times and values are presented as means±SEM. Data were analyzed by Student's t test, one-way or two-way ANOVA followed by post-hoc Bonferroni tests for comparison among means. Statistical significance was set at P<0.05.
Results
LMPs inhibit proliferation of cancer cells originating from different tissues and species
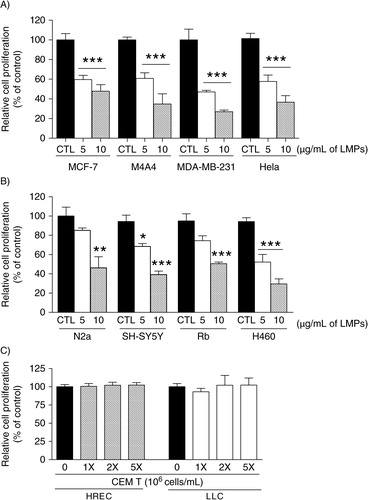
We first examined the effects of LMPs on cancer cell proliferation using cell lines from human breast cancers, cervical cancer, lung carcinoma, and human and mouse neuroblastoma. LMPs (5 and 10 µg/mL) significantly inhibited the proliferation of all tested cell lines in a concentration-dependent manner (A and B). LMPs also suppressed the proliferation of human primary retinoblastoma (Rb) cells, thus excluding the inference that LMPs only target established cancer cell lines (B). To determine whether these anti-proliferative effects were LMPs specific, we incubated HRECs and LLC cells with CEM T cells; 5 million T cells were equivalent to cell numbers producing 10 µg/mL LMPs. After 24 hours, no sustainable anti-proliferative effects were observed in cells exposed to T cells (C).
Fig. 1 LMPs inhibit cancer cell proliferation. Indicated concentrations of LMPs generated from CEM T cells via actinomycin D stimulation were added to: (A) human breast cancer cells (MCF-7, M4A4 and MDA-MB-231), HeLa cells; and (B) mouse (N2a) and human (SH-SY5Y) neuroblastoma cells, human primary retinoblastoma (Rb) cells and human non-small-cell lung cancer (H460) cells. (C) Human retinal endothelial cells (HRECs) and Lewis lung carcinoma (LLC) cells were treated with indicated concentrations of CEM T cells. Relative cell proliferation rates were determined after 24 hours of LMPs treatment and presented as percentage of control (CTL was set at 100%). *P<0.05, *P<0.01, ***P<0.001 vs. CTL.
LMPs induce cell-cycle arrest in G0/G1 phase
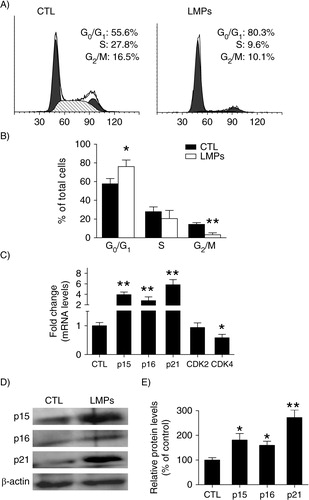
To elucidate the mechanisms underlying the anti-proliferative effect of LMPs, we evaluated the cell cycle kinetics of LLC cells by FACS. Relative to control, LMP-treated cells exhibited an increase in G0/G1-phase time; conversely, the proportion of LMPs-treated cells in G2/M markedly declined (A and B). Gene expression studies were then performed to help characterize specific mechanisms of the cell-cycle alteration. Our results showed that mRNA levels of the CDK inhibitors p15INK4b, p16INK4a and p21Cip1 were elevated by approximately 4-, 2.8- and 5.8-fold, respectively, after LMPs treatment; mRNA levels of CDK4 – a cell cycle regulator – decreased by 42% (C). Moreover, LMPs significantly augmented the protein levels of p15INK4b, p16INK4a and p21Cip1 – as indicated by a 1.8-, 1.6- and 2.7-fold induction, respectively (D and E).
Fig. 2. LMPs induce cell cycle arrest in G0/G1 phase and increase cyclin-dependent kinase (CDK) inhibitors. (A) Representative graphs depicting cell-cycle progression in LLC cells treated with or without LMPs (10 µg/mL for 24 hours). (B) The percentage of cells in each phase of the cell cycle was presented as means±SEM. The data were pooled from 4 independent experiments. (C) Gene expression in LMPs-treated Lewis lung carcinoma cells was quantified relative to the housekeeping gene and presented as fold induction compared with control (CTL). (D) LLC cells were treated with10 µg/mL LMPs for 24 hours, and CDK inhibitors p15INK4b, p16INK4a and p21Cip1 expression was detected by western blot. (E) p15INK4b, p16INK4a and p21Cip1 protein levels were normalized to β-actin, and the control condition was set to equal 100%. Values are means±SEM of 3 experiments. *P<0.05, **P<0.01 vs. CTL. P15INK4b, p16INK4a and p21Cip1 are CDK inhibitors. CDK2 and CDK4 are protein kinases of CDK family.
LMPs suppress tumour growth in animal models
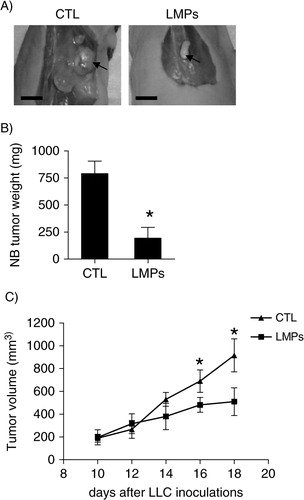
The anti-tumour effect of LMPs was investigated in vivo using human neuroblastoma xenografts and mouse LLC allografts. Compared with control, tumour growth was significantly reduced in LMPs-treated neuroblastoma xenografts (A and B). The same is true for mouse LLC allografts; the tumour volumes were significantly decreased after intravenous injection of LMPs (). No significant toxicity was observed in the mice that received systemic injection of high doses of LMPs for 30 days (data not shown).
Fig. 3. LMPs suppress tumour growth in vivo in animal models. (A) Representative images of primary neuroblastoma (NB) tumours in control and LMPs-treated xenografts at the conclusion of treatment. Bar, 5 mm. (B) NB tumour weights were measured and presented as means±SEM (n=8 mice per group). (C) Anti-tumour effect of LMPs on LLC allografts via intravenous injection. Tumour was measured every 2 days with an electronic calliper, and tumour volume was estimated based on the formula: Tumour volume=length (mm)×width2(mm2)/2. Each time point represents the mean±SEM (n=7 mice per group). *P<0.05 vs. CTL.
LMPs inhibit tumour cell proliferation in vivo and ex vivo
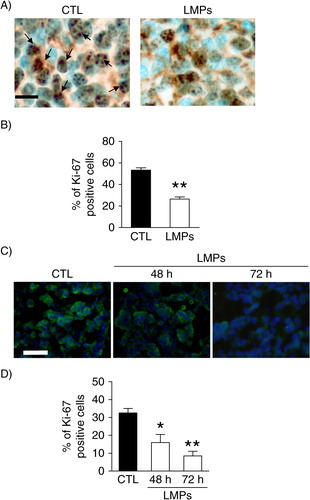
To verify whether the anti-tumour activity of LMPs was associated with the decreased tumour cell proliferation, we performed immunostaining for Ki-67, a most commonly used cell proliferation marker (Citation23–Citation25). There was a high intensity of Ki67 staining in control untreated LLC allografts, and LMPs’ treatment significantly decreased the expression of Ki-67 (A and B). In the breast tumour explants, the cell proliferation was decreased by LMPs in a time-dependent manner (C and D).
Fig. 4. LMPs inhibit tumour cell proliferation in vivo and ex vivo. (A) Representative images of Ki-67 immunostaining of paraffin-fixed LLC tumour sections. Indirect immunoperoxidase staining was performed with rabbit monoclonal anti-mouse Ki-67. Arrows indicate Ki-67-positive cells, which exhibited brown nuclei. Bar, 30 µm. (B) Cell proliferation was determined as the mean percentage of Ki-67-positive nuclei per 1,000 cells. (C) Representative pictures of Ki-67 immunostaining in human breast cancer explants treated with or without LMPs. Green florescence indicated the Ki-67-positive cells. (D) Graph illustrating the Ki-67 score for control (CTL) and LMP-treated cells. *P<0.05, **P<0.01 vs. CTL.
The anti-proliferative effect of LMPs is independent of stimuli or cell origin
Next, we questioned whether LMPs can impact the proliferation of quiescent cells and terminally differentiated cells. We therefore examined the effect of LMPs on the growth of immortalized RGC-5 cells, staurosporine-induced terminally differentiated RGC-5 cells (Citation12) and quiescent rat cortical neurons. While LMPs dose-dependently suppressed the proliferation of immortalized RGC-5 cells, they had no effect on the proliferation of differentiated RGC-5 cells or quiescent cortical neurons (A).
Fig. 5. The anti-proliferative effect of LMPs is not dependent on stimuli or cell origin. (A) Depicted are cell proliferation rates of immortalized retinal ganglion cells (RGC-5), staurosporine-induced terminally differentiated RGC-5 cells and rat cortical neurons after 24 hours of treatment with indicated concentration of LMPs. (B) Lewis lung carcinoma (LLC) cell proliferation was measured following 24 hours exposure to LMPs produced from CEM T cells using different stimuli. (C) LLC cells were treated with LMPs generated from CEM T, JurKat and human peripheral T lymphocytes using actinomycin D (ActD) stimulation. Cell proliferation is presented as a percentage of control (CTL), which was set to 100%. Values are presented as means±SEM. ***P<0.001 vs. CTL. Phytohemagglutinin (PhA).
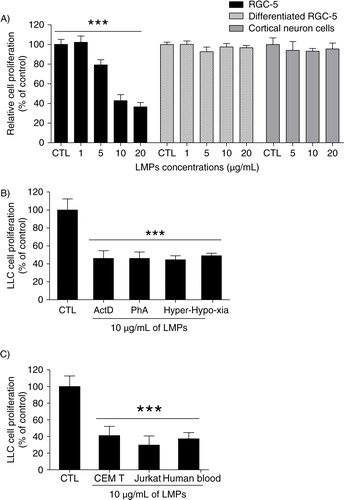
To explore whether the inhibitory effects of LMPs were stimulus dependent, we evaluated the impact of LMPs generated from CEM T cells using different stimuli: actinomycin D (induces apoptosis), phytohemagglutinin (PhA; stimulates cell division), and hyperoxia and hypoxia (trigger oxidative stress). LMPs produced under these conditions potently suppressed LLC cell proliferation by 45−49% compared with control (B).
To examine whether the anti-proliferative effect of LMPs was dependent on the parental cell origin, we evaluated LLC cell proliferation using LMPs derived from 3 types of lymphocytes: CEM T, JurKat and human peripheral T lymphocytes. Our results show that all 3 sources of LMPs (produced by actinomycin D stimulation) exerted comparable effects on LLC cell proliferation (C).
Discussion
In this report, we demonstrate for the first time that LMPs exert strong anti-proliferative effects on various cancer cells across species lines. These anti-proliferative effects are not limited to cancer cells but encompass immortalized RGCs and high-proliferating endothelial cells, as previously reported by us (Citation5). Notably, LMPs have little effect on terminally differentiated RGCs and quiescent neurons, suggesting that they selectively target high-proliferating cells, such as cancer cells and activated endothelial cells. Anti-tumour effects of LMPs in vivo were observed not only in neuroblastoma xenografts and human breast cancer explants but also in LMPs-treated LLC allografts (A). The anti-proliferative/anti-tumour properties of LMPs were further corroborated by a significantly reduced Ki-67 proliferation score in LMPs-treated tumours.
Our further studies revealed that the anti-proliferative effects of LMPs were associated with blocking of the cell cycle at G0/G1. Consistent with this observation, expression of the CDK inhibitors p15INK4b, p16INK4a and p21Cip1 were increased in LMPs-treated cells, whereas CDK4 – a pivotal cell cycle regulator of G1/S transition – was downregulated. Both p15INK4b and p16INK4a can function as tumour suppressors (Citation11, Citation26). The p16INK4a is a specific inhibitor of CDK4 and potentially blocks G1 cell-cycle progression (Citation27, Citation28). The p15INK4b-mediated pathways that control G1/S transition are frequently deregulated in tumour cells. In addition, the p21Cip1 arrests cells at the G1/S transition by reducing cyclin E/CDK2 and CDK4 activity (Citation29). Up to date, the mechanisms underlying the anti-proliferative effect of LMPs remain unclear. Although there is no evidence to support the direct inhibitory mechanism due to the fact that no cell cycle inhibitory proteins were identified from proteomic analysis of LMPs (Citation4), ours and other recent studies strongly suggested that LMPs induce cell cycle arrest through induction of oxidative stress and down-regulation of VEGF signalling (Citation5, Citation6) (Citation30, Citation31). A late experiment indicated that proteins may not be the only causative components in LMPs, lipids or other heat-stable components also contribute to the anti-cell growth effects of LMPs (unpublished data).
It has been reported that the mode of cell activation and the microenvironment where microparticles were generated relevantly determines the nature of microparticle-mediated intercellular communication (Citation1). Although previous studies investigating the role of LMPs in angiogenesis have yielded contradictory results, these conflicting angiogenic responses are probably due to different modes of stimulation of the parental lymphocytes (Citation5, Citation7) (Citation17, Citation32). Regarding the impact on cell growth, our data show that the anti-proliferative effect of LMPs is not dependent on lymphocyte origin or stimuli. This is evident from in vitro studies whereby comparable anti-proliferative effects were observed in LMPs produced from (a) lymphocytes of different origin and (b) CEM T cells exposed to 3 different stimuli. To exclude the possibility of mycoplasma contamination causing the observed effect of LMPs, a sensitive PCR-based test was performed, and it proved that LMPs and CEM T cells are mycoplasma free (data not shown).
Having previously demonstrated that low-density lipoprotein receptor (LDLR) plays a key role in mediating the anti-VEGF effect of LMPs (Citation6, Citation31), we postulate that LDLR also mediates the anti-proliferative effect of LMPs. This inference is based on observations that high LDLR activity has been linked to neoplastic cells and proliferating endothelial cells (Citation33–Citation35), and that low LDLR activity is associated with quiescent endothelial cells (Citation33). Nevertheless, we cannot exclude the involvement of other membrane receptors or proteins in this process. For example, the CD36 scavenger receptor has been shown to mediate uptake of LMPs in brain microvascular endothelial cells (unpublished data). Then again, additional studies are required to elucidate how LMPs are delivered into target cells, and to increase our understanding of the molecular biology of LMPs.
In recent years, bioactive compounds have emerged as potential anticancer therapies. Edelman and colleagues recently reported that secretions from quiescent endothelial cells reduce the proliferative and invasive phenotype of both lung and breast cancer cells (Citation36). In addition, Camussi's group have provided evidence that microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumour growth. The delivery of selected miRNAs by microvesicles and activation of negative regulators of cell cycle may explain these biological effects (Citation37, Citation38). Moreover, multiple inhibition components released from cancer cell–containing agarose beads have yielded profound effects on tumour growth in vivo. Therefore, the synergistic effect of multiple components helping to produce part of the effect is considered a unique aspect of cellular therapy (Citation39, Citation40). Along these lines, LMPs and bioactive compounds released from macrobeads have several characteristics in common, they: (a) are produced from living cells cultured under specific conditions; (b) contain multiple factors, including proteins with tumour inhibitory properties; and (c) exert effects that are neither species- nor tumour-type specific.
In summary, this study highlights the potential of LMPs to modulate growth of high-proliferating cells without targeting quiescent cells. An increased understanding of the multifaceted intrinsic regulatory mechanisms of LMPs, their cell interactions and active components may lead to the development of novel therapeutic strategies for cancer treatment.
Conflict of interest and funding
Fonds de recherche en santé du Québec – Vision Health Research Network. No potential conflicts of interest were disclosed.
Acknowledgements
The authors thank Carmen Gagnon and Bupe R. Mwaikambo for invaluable technical and editing assistance, respectively.
References
- Mause SF, Weber C. Microparticles: protagonists of a novel communication network for intercellular information exchange. Circ Res. 2010; 107: 1047–57.
- Martin S, Tesse A, Hugel B, Martinez MC, Morel O, Freyssinet JM et al. Shed membrane particles from T lymphocytes impair endothelial function and regulate endothelial protein expression. Circulation. 2004; 109: 1653–9.
- Tesse A, Martinez MC, Hugel B, Chalupsky K, Muller CD, Meziani F et al. Upregulation of proinflammatory proteins through NF-kappaB pathway by shed membrane microparticles results in vascular hyporeactivity. Arterioscler Thromb Vasc Biol. 2005; 25: 2522–7.
- Miguet L, Pacaud K, Felden C, Hugel B, Martinez MC, Freyssinet JM et al. Proteomic analysis of malignant lymphocyte membrane microparticles using double ionization coverage optimization. Proteomics. 2006; 6: 153–71.
- Yang C, Mwaikambo BR, Zhu T, Gagnon C, Lafleur J, Seshadri S et al. Lymphocytic microparticles inhibit angiogenesis by stimulating oxidative stress and negatively regulating VEGF-induced pathways. Am J Physiol Regul Integr Comp Physiol. 2008; 294: R467–76.
- Yang C, Gagnon C, Hou X, Hardy P. Low density lipoprotein receptor mediates anti-VEGF effect of lymphocyte T-derived microparticles in Lewis lung carcinoma cells. Cancer Biol Ther. 2010; 10: 448–56.
- Benameur T, Soleti R, Porro C, Andriantsitohaina R, Martinez MC. Microparticles carrying Sonic hedgehog favor neovascularization through the activation of nitric oxide pathway in mice. PLoS One. 2010; 5: e12688.
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995; 81: 323–30.
- Clurman BE, Roberts JM. Cell cycle and cancer. J Natl Cancer Inst. 1995; 87: 1499–501.
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007; 10: 13–29.
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995; 83: 993–1000.
- Yang C, Lafleur J, Mwaikambo BR, Zhu T, Gagnon C, Chemtob S et al. The role of lysophosphatidic acid receptor (LPA1) in the oxygen-induced retinal ganglion cell degeneration. Invest Ophthalmol Vis Sci. 2009; 50: 1290–8.
- Harvey R, Chintala SK. Inhibition of plasminogen activators attenuates the death of differentiated retinal ganglion cells and stabilizes their neurite network in vitro. Invest Ophthalmol Vis Sci. 2007; 48: 1884–91.
- Toman RE, Movsesyan V, Murthy SK, Milstien S, Spiegel S, Faden AI. Ceramide-induced cell death in primary neuronal cultures: upregulation of ceramide levels during neuronal apoptosis. J Neurosci Res. 2002; 68: 323–30.
- Laurie N, Mohan A, McEvoy J, Reed D, Zhang J, Schweers B et al. Changes in retinoblastoma cell adhesion associated with optic nerve invasion. Mol Cell Biol. 2009; 29: 6268–82.
- Vasina EM, Cauwenberghs S, Feijge MA, Heemskerk JW, Weber C, Koenen RR. Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death Dis. 2011; 2: e211.
- Agouni A, Mostefai HA, Porro C, Carusio N, Favre J, Richard V et al. Sonic hedgehog carried by microparticles corrects endothelial injury through nitric oxide release. FASEB J. 2007; 21: 2735–41.
- Niland B, Miklossy G, Banki K, Biddison WE, Casciola-Rosen L, Rosen A et al. Cleavage of transaldolase by granzyme B causes the loss of enzymatic activity with retention of antigenicity for multiple sclerosis patients. J Immunol. 2010; 184: 4025–32.
- Mwaikambo BR, Yang C, Chemtob S, Hardy P. Hypoxia up-regulates CD36 expression and function via hypoxia-inducible factor-1- and phosphatidylinositol 3-kinase-dependent mechanisms. J Biol Chem. 2009; 284: 26695–707.
- Kang Y, Ozbun LL, Angdisen J, Moody TW, Prentice M, Diwan BA et al. Altered expression of G1/S regulatory genes occurs early and frequently in lung carcinogenesis in transforming growth factor-beta1 heterozygous mice. Carcinogenesis. 2002; 23: 1217–27.
- Vescio RA, Connors KM, Youngkin T, Bordin GM, Robb JA, Umbreit JN et al. Cancer biology for individualized therapy: correlation of growth fraction index in native-state histoculture with tumor grade and stage. Proc Natl Acad Sci U S A. 1990; 87: 691–5.
- Vesselle H, Schmidt RA, Pugsley JM, Li M, Kohlmyer SG, Vallires E et al. Lung cancer proliferation correlates with [F-18]fluorodeoxyglucose uptake by positron emission tomography. Clin Cancer Res. 2000; 6: 3837–44.
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000; 182: 311–22.
- Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, A'Hern R et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst. 2007; 99: 167–70.
- Gerdes J. Ki-67 and other proliferation markers useful for immunohistological diagnostic and prognostic evaluations in human malignancies. Sem Cancer Biol. 1990; 1: 199–206.
- Cairns P, Mao L, Merlo A, Lee DJ, Schwab D, Eby Y et al. Rates of p16 (MTS1) mutations in primary tumors with 9p loss. Science. 1994; 265: 415–17.
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996; 85: 27–37.
- Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994; 264: 436–40.
- Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000; 35: 317–29.
- Burger D, Kwart DG, Montezano AC, Read NC, Kennedy CR, Thompson CS et al. Microparticles induce cell cycle arrest through redox-sensitive processes in endothelial cells: implications in vascular senescence. J Am Heart Assoc. 2012; 1: e001842.
- Yang C, Xiong W, Qiu Q, Shao Z, Hamel D, Tahiri H et al. Role of receptor-mediated endocytosis in the antiangiogenic effects of human T lymphoblastic cell-derived microparticles. Am J Physiol Regul Integr Comp Physiol. 2012; 302: R941–9.
- Mostefai HA, Agouni A, Carusio N, Mastronardi ML, Heymes C, Henrion D et al. Phosphatidylinositol 3-kinase and xanthine oxidase regulate nitric oxide and reactive oxygen species productions by apoptotic lymphocyte microparticles in endothelial cells. J Immunol. 2008; 180: 5028–35.
- Sanan DA, Strumpfer AE, van der Westhuyzen DR, Coetzee GA. Native and acetylated low density lipoprotein metabolism in proliferating and quiescent bovine endothelial cells in culture. Eur J Cell Biol. 1985; 36: 81–90.
- Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis. 2011; 28: 733–41.
- Davis W Jr.. The ATP-binding cassette transporter-2 (ABCA2) regulates cholesterol homeostasis and low-density lipoprotein receptor metabolism in N2a neuroblastoma cells. Biochim Biophys Acta. 2011; 1811: 1152–64.
- Franses JW, Baker AB, Chitalia VC, Edelman ER. Stromal endothelial cells directly influence cancer progression. Sci Transl Med. 2011; 3: 66ra5.
- Bruno S, Collino F, Deregibus MC, Grange C, Tetta C, Camussi G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013; 22: 758–71.
- Fonsato V, Collino F, Herrera MB, Cavallari C, Deregibus MC, Cisterna B et al. Human liver stem cell-derived microvesicles inhibit hepatoma growth in SCID mice by delivering antitumor microRNAs. Stem Cells. 2012; 30: 1985–98.
- Smith BH, Gazda LS, Conn BL, Jain K, Asina S, Levine DM et al. Hydrophilic agarose macrobead cultures select for outgrowth of carcinoma cell populations that can restrict tumor growth. Cancer Res. 2011; 71: 725–35.
- Smith BH, Gazda LS, Conn BL, Jain K, Asina S, Levine DM et al. Three-dimensional culture of mouse renal carcinoma cells in agarose macrobeads selects for a subpopulation of cells with cancer stem cell or cancer progenitor properties. Cancer Res. 2011; 71: 716–24.