
Abstract
Paroxysmal nocturnal haemoglobinuria (PNH) is an acquired disorder of the haematopoietic stem cell that makes blood cells more sensitive to the action of complement. PNH patients experience an increased risk of arterial and venous thrombosis – major causes of death due to this disease. Though many potential interlaced mechanisms are suspected, extracellular vesicles (EVs) of various origins may play a central role. The processes possibly involved are haemolysis, platelet activation, injured endothelial cells and monocyte activation. The impact of transfusion should be evaluated. A better understanding of the mechanisms involved may help to propose guidelines for the prophylaxis and treatment of thrombosis in PNH. In this paper, we propose an updated review of the pathophysiology of the underlying mechanisms of thrombosis associated with PNH, with specific focus on the prominent role of EVs.
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare, acquired disorder of the pluripotent haematopoietic stem cell and therefore can affect erythrocytes, leukocytes, thrombocytes (Citation1) and probably some endothelial cells too (Citation2). These haematopoietic stem cells have acquired a somatic mutation in an X-linked gene: the phosphatidylinositol glycan class A (PIG-A). This gene is required for the synthesis of the glycosylphosphatidylinositol (GPI) anchor, which is necessary to attach some proteins to the cell membrane.
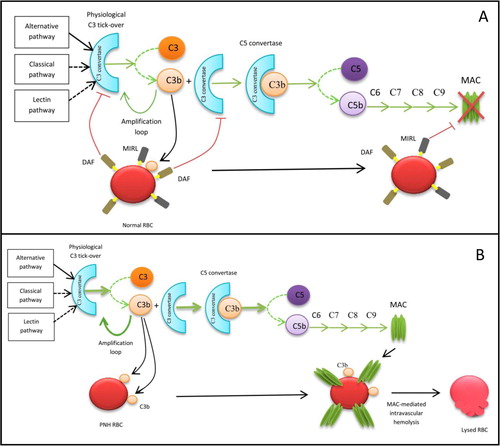
The lack of synthesis of the GPI anchor leads to the under-expression of a variety of proteins on the haematopoietic stem cell surface and on all cell lines that are generated by it. By this mechanism, lack of 2 important complement regulators is observed on the cell surface: “decay accelerating factor” (DAF), recognized by antibody CD55, and “membrane inhibitor of reactive lysis” (MIRL), recognized by antibody CD59. Consequently, red blood cells are more vulnerable to the action of complement, leading to complement-mediated intravascular haemolysis (see ) (Citation3). As a result, a high concentration of free haemoglobin is found in the plasma, responsible for nitric oxide (NO) scavenging. NO depletion causes smooth muscle dystonia, responsible for dysphagia and abdominal pain. NO depletion may also contribute to the development of arterial constriction, leading to reduced blood flow to the kidneys (with renal failure), arterial hypertension and pulmonary hypertension (associated with frequent but under-diagnosed pulmonary embolism) (Citation4).
Fig. 1 Action of complement in healthy subjects (A) and PNH patients (B). (A) Due to the presence of membrane proteins, DAF and MIRL, a normal RBC is protected from complement activation. (B) DAF and MIRL deficiency makes the RBC sensitive to complement attack, resulting in haemolysis. RBC=red blood cells, MAC=membrane attack complex.
Management and treatment of classic PNH has been dramatically revolutionized by the development of eculizumab (Soliris® by Alexion Pharmaceuticals) and its approval in 2007 by the US FDA and the European Union Commission. This humanized monoclonal antibody blocks the activation of terminal complement C5 and prevents the formation of C5a and C5b-9, preventing haemolysis. Several beneficial effects of eculizumab have been demonstrated, in terms of haemolysis, quality of life (Citation5, Citation6), renal function (Citation7) and life expectancy (Citation8). A reduction in the incidence rate of thromboembolic events (Citation9) and a decrease in markers of thrombin generation (Citation10, Citation11) were also observed during eculizumab therapy. The mechanisms involved, however, remain misunderstood.
Thrombosis in PNH
Clinical characteristics
Thrombotic events occur in 40% of PNH patients and represent the major cause of death due to this disease. Indeed, 40–67% of PNH patients will die of thrombotic complications. An initial thrombotic event increases the relative risk of death by 5- to 10-fold (Citation12). Understanding, anticipating and treating thrombosis represent a major challenge in PNH.
Thrombotic events are of venous origin in 85% of the cases but arterial thrombosis is not so rare (15%) (Citation4). Thrombotic events (20.5%) involve more than 1 site at the same time. Thrombosis not only affects usual sites such as deep veins of the lower limbs, pulmonary arteries, coronary arteries or central nervous system (CNS) arteries but also, more typically, unusual sites such as hepatic veins (Budd-Chiari syndrome: 40.7–44.0% of PNH patients with thrombosis (Citation12)), cavernous sinus, CNS veins, mesenteric veins or dermal veins (Citation13).
In PNH patients, the age of onset of thrombosis is lower than among the general population (46 years vs. 73 years) (Citation4). An increased risk of thrombosis was reported among African American or Latin American PNH patients compared with other PNH patients (Citation14). A lower risk was reported among Chinese and Japanese PNH patients (Citation12). The mechanism is still unknown.
Recurrences of thrombosis despite antithrombotic therapy (warfarin, unfractionated heparin, enoxaparin, aspirin, tirofiban) have been described (Citation15, Citation16).
Thrombosis during pregnancy and post-partum seems particularly frequent. Indeed, a maternal mortality of 20% and a perinatal mortality of 10% have been reported, mostly due to thrombosis. Consequently, some authors do not recommend pregnancy in patients suffering from PNH (Citation13).
The role of EVs
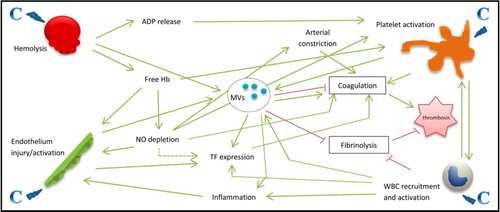
The exact pathophysiology of this hypercoagulable state in PNH is unknown but may be multifactorial, as illustrated in . Extracellular vesicles (EVs) are sub-micron-size cellular fragments released by eukaryotic cells (including platelets, leukocytes, endothelial cells and red blood cells) following activation or apoptosis. They are highly heterogeneous both in size (30–1,000 nm) and in composition. EVs are complex and ambivalent structures that not only express both activators and inhibitors of coagulation but also convey fibrinolytic properties. They are therefore considered as the main regulator of the balance between coagulation and fibrinolysis. They play a major role in cellular cross-talk, inflammation, haemostasis and thrombosis (Citation17–Citation19).
Fig. 2 Overview of the multiple mechanisms involved in the occurrence of thrombosis in PNH patients. Complement (C) activation is responsible of haemolysis, platelet activation, endothelium injury and activation, and white blood cell (WBC) recruitment and activation. From there, multiple mechanisms induce a hypercoagulable state with the occurrence of thrombosis. Note the central role of extracellular vesicles (or microvesicles: MVs). ADP=adenosine diphosphate, Hb=haemoglobin, NO=nitric oxide, TF=tissue factor.
It was demonstrated in vitro that platelet EVs have from 50 to 100-times higher pro-coagulant activity than platelets (Citation20). Indeed, an inherited defect of lipid scramblase is seen in Scott's syndrome, a rare autosomal recessive disease. This leads to reduced production of EVs, causing severe bleeding (Citation21–Citation23).
EVs are known to expose phosphatidylserine on their surface, providing a support for the assembly of the pro-coagulant enzyme complexes – prothrombinase and tenase (Citation21, Citation22, Citation24).
Tissue factor, the principal initiator of coagulation, is also exposed on the surface of some EVs. More and more evidences suggest that EVs are the main carriers of circulating tissue factor (TF) and, thereby, may contribute to normal coagulation (Citation25). However, TF activity of EVs from non-stimulated blood was not detectable in most studies (Citation26). Monocytes are thought to be the only blood cells that synthesize TF in vivo. TF is constitutively expressed in an encrypted form in monocytes but may be decrypted during cell activation. Indeed, monocyte's TF mRNA levels are increased after stimulation (Citation26). Some authors speculate that TF expressed on cholesterol-rich lipid rafts on monocyte-derived EVs may take part in the link between hypercholesterolemia and thrombosis (Citation27). The role of EV-associated TF in haemostasis is not extensively discussed here because it has been well covered in other reviews (Citation24, Citation28, Citation29) and is still under study. The synergies of phosphatidylserine and protein disulphide isomerase as well as the recycling by endothelial cells have been recently demonstrated as important in tissue factor activation (Citation30, Citation31). In addition, contrary to what was believed before, very small TF-bearing EVs (<100 nm) may have a pro-coagulant effect, especially in the presence of phospholipids (Citation32, Citation33).
In PNH, complement activation at the cell surface of GPI-deficient cells may stimulate the release of pro-coagulant EVs. Indeed, increased levels of circulating EVs have been measured in PNH patients, a majority of them coming from platelets. Yet, no correlation between the clone size and the number of EVs could be demonstrated (Citation34). One study (Citation35) showed increased plasmatic levels of TF antigen (measured by ELISA, after adjunction of triton) in 2 PNH patients suffering from recurrent thrombotic complications, compared to normal controls.
The role of platelet activation
Platelet membranes of PNH patients are deficient in DAF (CD55) and MIRL (CD59). It is known that complement does not directly destroy platelets but is able to induce platelet activation. Therefore, complement may induce platelet activation and vesiculation in an insufficiently regulated manner in PNH patients. Indeed, it has been found that PNH platelets, after membrane attack complex (MAC) stimulation, expose more activated factor five-binding sites and increase thrombin generation more than that of normal platelets (Citation36). One study also showed increased adherence of platelets to the abdominal vessels in PNH. This interesting feature could partly explain the particular occurrence of mesenteric vein thrombosis in these patients and suggest a peculiar mechanism involving the special distribution of a hypercoagulable state in these patients (Citation4). In contrast, other studies showed impaired function of PNH platelets as a result of a reactive down-regulation in response to their chronic hyperstimulation (Citation36).
The number of platelet extracellular vesicles (PEVs) is sometimes used as a marker of platelet activation (Citation37) and may be of particular interest in PNH. Nowadays, it is still very difficult to find a reliable plasma marker of platelet activation. P-selectin, β-thromboglobulin, platelet factor 4 (PF4) and thromboxane B2 have been studied in physiological and pathological states but all these have limitations (Citation38, Citation39). The ideal candidate should be a specific marker of platelet, resistant to the artefacts of venipuncture and collection and not influenced by antithrombotic drugs. It should be measured according to a cheap, simple and reproducible operating procedure (Citation38). Further research needs to be done in this field.
The controversial impact of haemolysis
Haemolysis has long been considered as a major contributor of thrombosis, but its role is now controversial. NO depletion can cause arterial constriction and thus play a role in arterial thrombosis, but veins do not have smooth muscle. Platelet activation may be increased by the lack of NO (Citation4) and by the release of adenosine diphosphate (ADP) (Citation40) in case of haemolysis. The impact of NO depletion in an increase of TF expression is controversial. However, no link can be made between severity of haemolysis and thrombosis or severity of anaemia and thrombosis (Citation4). One study showed that human red blood cell stroma is capable of activating the alternate complement pathway in vitro. The ensuing interaction of activated complement components with platelets may lead to activation of the coagulation system (Citation41).
Similar levels of red blood cells–derived EVs (REVs) were found in PNH patients or healthy controls, but in vitro experiments showed that PNH erythrocytes release higher amounts of pro-coagulant EVs upon complement stimulation (Citation42). This may suggest a rapid clearance of erythrocyte EVs from the circulation (Citation36). We know that vesicles are partly cleared by a scavenging receptor on the monocytes, which can induce monocyte TF expression, promoting coagulation. In turn, monocytes, after complement damage, can release EVs that contain TF. These EVs may be captured by endothelial cells, increasing their own TF expression (Citation4, Citation31).
Injured endothelial cells
Endothelial EV levels are also increased in PNH patients, showing a pro-inflammatory and pro-thrombotic phenotype (Citation43). The endothelial cells probably play an important role in the pathophysiology of thrombosis in PNH. Endothelial cells suffer from direct toxicity of free haemoglobin released (Citation44) but may also be particularly sensitive to the action of complement. It has been shown that endothelial precursor cells may be bone marrow–derived cells (Citation2). They would then be deficient in GPI-anchored proteins, making them more susceptible to complement injury. This would be of particular importance for the development of thrombosis in unusual sites (Citation4). Increased levels of endothelial cell activation markers, such as von Willebrand factor (VWF) or soluble vascular cell adhesion molecule-1 (sVCAM-1), are observed in PNH (Citation11).
The effect of complement on other cell lines
Complement injury to other cell lines can also contribute to thrombosis in PNH. Complement protein C5a binds to a receptor on granulocytes and enhances recruitment and activation of granulocytes and monocytes. This molecule is considered as a possible link between inflammation and thrombosis (Citation45). In GPI-deficient white blood cells, this mechanism may be intensified and may lead to an important release of inflammatory molecules and EVs. This inflammatory process induces damages to the endothelium and enhances TF expression, which initiates the coagulation cascade (Citation4). It seems clear that complement and coagulation systems are closely intertwined. This has been reinforced by the fact that thrombin by itself is able to initiate the alternative pathway of complement (Citation46).
Tissue factor pathway inhibitor deficiency
Tissue factor pathway inhibitor (TFPI) normally limits coagulation initiation by inhibiting TF formation. It is mainly produced by the endothelium of the microvasculature and is bound to a GPI-anchored protein. Therefore, it lacks in PNH. In previous studies, loss of TFPI expression has been associated with an increased risk of thrombosis (Citation4). In addition, a decrease of TFPI expression on circulating EVs has been observed in certain clinical situations and may induce thrombin generation due to unopposed TF activity (Citation47). This could play a role in thrombosis in PNH.
Lack of other GPI-anchored proteins
As we have already discussed, the lack of several GPI-anchored proteins at the cell surface may lead to altered coagulation. The GPI-anchors’ deficit may also interfere with coagulation in an indirect manner. For example, neutrophil proteinase-3 (PR-3) is a membrane-bound protein, which is co-localized with GPI-anchored neutrophil antigen 2a. The lack of neutrophil antigen 2a in PNH leads to loss of expression of PR-3. However, PAR-1, the predominant human platelet thrombin receptor, is a substrate for PR-3. It has been shown that impaired down-regulation of PAR-1 leads to an increased propensity for platelet activation. PR-3 also modulates coagulation in various other ways (Citation48).
Congenital and acquired thrombophilia
Frequency of congenital thrombophilia factors (Factor V Leiden, antithrombin deficiency, protein C or S deficiency, prothrombin mutations, hyperhomocysteinaemia and methylenetetrahydrofolate reductase mutations) is not increased in PNH. Testing for such factors may identify PNH patients at additional thrombotic risk, but its value for treatment decision in PNH is still unknown (Citation36). A higher rate of antiphospholipid antibodies was found in PNH patients than in healthy volunteers (Citation49). This could be a contributory factor for thrombosis in PNH.
In addition, Grünewald et al. (Citation50) have studied various markers of coagulation in PNH and noted a significant increase in pro-coagulant factor activities (factor V, fibrinogen, factor VIII, factor IX, factor X and VWF), with a positive correlation with clone size.
Impaired and/or increased fibrinolysis
About fibrinolysis in PNH, the results of various studies are conflicting. The role of urokinase-type plasminogen activator receptor (u-PAR) may however be important. U-PAR, a GPI-linked protein expressed on neutrophils, plays a central role in the regulation of haemostatic processes on cell surface. Indeed, the binding of urokinase-type plasminogen activator (u-PA) to u-PAR converts plasminogen into plasmin, mediating endogenous thrombolysis. This proteolytic activity is spatially restricted to the plasma membrane (Citation51). In PNH, studies have shown that u-PAR is under-expressed on the cellular surface of leukocytes and platelets and also that concentration of soluble u-PAR is increased in plasma of PNH patients. Furthermore, serum-soluble u-PAR concentrations correlated with PNH granulocyte clone size and were the highest in patients who later developed thrombosis (Citation52). They hypothesize that u-PAR is released from PNH haematopoietic cells to plasma (Citation53). This would be the responsibility of an impaired and displaced fibrinolytic system in PNH.
Other fibrinolytic parameters have been studied, some of them suggesting an increased activity of the fibrinolytic system in PNH. Indeed, elevated levels of d-dimers and of complexes of plasmin and plasmin inhibitors were found, as was the case for complexes of tissue-type plasminogen activator (t-PA) and its inhibitor (PAI-1). However, an inverse association with clone size was described and correlations were weak (Citation50).
In healthy subjects and in several pathological conditions, it is now suspected that EVs have a fibrinolytic activity (Citation18). The exposure of u-PA or t-PA and their receptor (u-PAR and annexin II) was demonstrated on the surface of EVs from various origins (Citation54–Citation58). Their impact on fibrinolysis in PNH patients remains to be studied.
The impact of transfusions
PNH patients who receive frequent transfusions of red blood cells may have a further increased risk of thrombosis. Indeed, the use of transfusion was isolated as an independent risk factor for thrombosis in PNH (Citation15). Unfortunately there is no prospective study that quantifies the level of this risk.
An exogenous source of PEVs or REVs in PNH could be constituted by transfusion. Indeed, patients suffering from PNH frequently need red blood cell or platelet transfusions. Anaemia induced by haemolysis is not, in this situation, a contraindication for red blood cell transfusion. On the contrary, receiving allogeneic red blood cells (in which the membrane is not GPI deficient) is good enough to limit the stimulation of erythropoiesis, decrease the production of deficient red blood cells and thus lessen haemolysis and its symptoms. Red blood cell or platelet transfusions are also required when PNH is associated with a bone marrow failure syndrome (Citation3). PNH patients, receiving frequent transfusions of red blood cells, may have a further increased risk of thrombosis. Indeed, the use of transfusion was isolated as an independent risk factor for thrombosis in PNH (Citation15). However, there is no prospective study that quantifies this risk.
Regardless of PNH, lots of observations suggest that red blood cell transfusion is associated with increased thrombotic events and mortality. Indeed, blood transfusion in the setting of acute coronary syndromes was associated with higher mortality (OR 3.94) (Citation59). Khorana et al. (Citation60) observed that red blood cell transfusion was independently associated with an increased risk of venous (OR 1.60) and arterial (OR 1.53) thrombotic events and mortality (OR 1.34) in hospitalized cancer patients. Similarly, Abu-Rustum et al. (Citation61) demonstrated an association between transfusion and increased risk of thrombotic events in women during adnexal or peritoneal cancer surgery (OR 4.80), and Nilsson et al. (Citation62) observed that perioperative blood transfusion was associated with an increased risk of venous thromboembolism in women undergoing colorectal cancer resection (OR 1.80). More recently, an increase in perioperative graft thrombosis was observed in patients transfused during a surgery of lower extremity bypass (OR 2.10 to 4.80, depending on the number of units transfused) (Citation63). Kumar et al. (Citation64) also demonstrated that red blood cell transfusion was associated with an increased risk of thrombotic events in patients with subarachnoid haemorrhage (OR 2.40).
The mechanisms that underlie these findings remain unclear. It is known that red blood cell transfusions alter viscosity by both raising haematocrit and by introducing erythrocytes with altered viscoelastic properties into the circulation (Citation62). This could increase the risk of thrombosis. Increasing the circulating red cell mass may also improve haemostasis (Citation60). The depletion of NO in stored red blood cells may promote vasoconstriction and platelet aggregation (Citation59). The delivery of redox-active iron and pro-inflammatory mediators by transfusion may also play a role (Citation60).
In addition, the delivery of pro-coagulant EVs contained in the transfused product may be able to promote haemostasis and increase the risk of thrombosis. Indeed, it has been observed that the number of EVs contained in packed red blood cells increases during storage (Citation65) and that red blood cells–derived EVs isolated from blood units are capable of initiating and propagating thrombin generation (Citation65). Moreover, duration of red blood cell storage was associated with increased incidence of deep vein thrombosis in patients with traumatic injuries in a retrospective cohort study (Citation66).
Risk factors for thrombosis
A previous history of thrombosis and a large white blood cell clone are well-known risk factors for thrombosis in PNH (Citation67). Indeed, a 10-year cumulative incidence of thrombosis of 34.5% is observed in patients having a large clone (>50%), compared with 5.3% in patients with a smaller clone (<50%) (Citation40). The risk of thromboembolic events increases by 1.64-fold for every 10% increase in the size of the white blood cell clone (Citation12). Age more than 55 years and the use of transfusion were also isolated as independent risk factors for a first thrombotic event (Citation15). Ethnicity seems to be another risk factor for thrombosis as discussed above (Citation14). The elevation of D-dimers has also been associated with an increased thrombotic risk in PNH (Citation4).
Conclusion
Even if the management of PNH has been dramatically revolutionized by the development of eculizumab, prevention and treatment of thrombotic complications remain a challenge.
A better understanding of the underlying pathophysiologic mechanisms of thrombosis in PNH should allow us to identify new potential biomarkers of thrombosis, such as EVs.
More and more studies have indicated a potential role of EVs in thrombosis associated with PNH. However, published data were produced using low-sensitive and not standardized flow cytometry. But progress has been performed in the field of flow cytometry (Citation68–Citation71). Therefore, the role of EVs should be investigated using high-sensitive and standardized techniques. Moreover, as the research focused on the pro-coagulant function of EVs, its function in fibrinolysis should also be studied in the near future.
The ultimate goal is to propose guidelines for prophylaxis and treatment of thrombosis associated with PNH. Moreover, the impact of transfusion on thrombotic risk deserves to be further studied.
Disclosure of conflict of interest
Christian Chatelain received financial support for his research work from Alexion Pharmaceuticals.
Acknowledgements
Work supported by a Grant F.R.S.-FNRS/Télévie and a Grant Fondation Mont-Godinne.
Thanks to Mr Axel Baily (Unité de Support Scientifique, CHU Dinant-Godinne UCL Namur) for his work on the figures.
Related Research Data
References
- Dameshek W, Fudenberg H. Paroxysmal nocturnal hemoglobinuria. AMA Arch Intern Med. 1957; 99: 202–8.
- Shi Q, Rafii S, Wu MH, Wijelath ES, Yu C, Ishida A et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998; 92: 362–7.
- Parker CJ. Management of paroxysmal nocturnal hemoglobinuria in the era of complement inhibitory therapy. Hematology Am Soc Hematol Educ Program. 2011; 2011: 21–9.
- Weitz IC. Thrombosis in patients with paroxysmal nocturnal hemoglobinuria. Semin Thromb Hemost. 2011; 37: 315–21.
- Schubert J, Hillmen P, Roth A, Young NS, Elebute MO, Szer J et al. Eculizumab, a terminal complement inhibitor, improves anaemia in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2008; 142: 263–72.
- Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood. 2008; 111: 1840–7.
- Hillmen P, Elebute M, Kelly R, Urbano-Ispizua A, Hill A, Rother RP et al. Long-term effect of the complement inhibitor eculizumab on kidney function in patients with paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2010; 85: 553–9.
- Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: sustained efficacy and improved survival. Blood. 2011; 117: 6786–92.
- Hillmen P, Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007; 110: 4123–8.
- Weitz IC, Razavi P, Rochanda L, Zwicker J, Furie B, Manly D et al. Eculizumab therapy results in rapid and sustained decreases in markers of thrombin generation and inflammation in patients with PNH independent of its effects on hemolysis and microparticle formation. Thromb Res. 2012; 130: 361–8.
- Helley D, de Latour RP, Porcher R, Rodrigues CA, Galy-Fauroux I, Matheron J et al. Evaluation of hemostasis and endothelial function in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Haematologica. 2010; 95: 574–81.
- Malato A, Saccullo G, Coco LL, Mancuso S, Santoro M, Martino S et al. Thrombotic complications in paroxysmal nocturnal haemoglobinuria: a literature review. Blood Transfus. 2012; 10: 428–35.
- Ziakas PD, Poulou LS, Rokas GI, Bartzoudis D, Voulgarelis M. Thrombosis in paroxysmal nocturnal hemoglobinuria: sites, risks, outcome. An overview. J Thromb Haemost. 2007; 5: 642–5.
- Araten DJ, Thaler HT, Luzzatto L. High incidence of thrombosis in African-American and Latin-American patients with Paroxysmal Nocturnal Haemoglobinuria. Thromb Haemost. 2005; 93: 88–91.
- de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood. 2008; 112: 3099–106.
- Audebert HJ, Planck J, Eisenburg M, Schrezenmeier H, Haberl R. Cerebral ischemic infarction in paroxysmal nocturnal hemoglobinuria report of 2 cases and updated review of 7 previously published patients. J Neurol. 2005; 252: 1379–86.
- Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011; 68: 2667–88.
- Lacroix R, Dubois C, Leroyer AS, Sabatier F, Dignat-George F. Revisited role of microparticles in arterial and venous thrombosis. J Thromb Haemost. 2013; 11: 24–35.
- Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol. 2011; 31: 15–26.
- Sinauridze EI, Kireev DA, Popenko NY, Pichugin AV, Panteleev MA, Krymskaya OV et al. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb Haemost. 2007; 97: 425–34.
- Burnier L, Fontana P, Kwak BR, Angelillo-Scherrer A. Cell-derived microparticles in haemostasis and vascular medicine. Thromb Haemost. 2009; 101: 439–51.
- Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007; 21: 157–71.
- Bevers EM, Wiedmer T, Comfurius P, Shattil SJ, Weiss HJ, Zwaal RF et al. Defective Ca(2+)-induced microvesiculation and deficient expression of procoagulant activity in erythrocytes from a patient with a bleeding disorder: a study of the red blood cells of Scott syndrome. Blood. 1992; 79: 380–8.
- Owens AP 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011; 108: 1284–97.
- Bidot L, Jy W, Bidot C Jr, Jimenez JJ, Fontana V, Horstman LL et al. Microparticle-mediated thrombin generation assay: increased activity in patients with recurrent thrombosis. J Thromb Haemost. 2008; 6: 913–9.
- Osterud B. Tissue factor expression in blood cells. Thromb Res. 2010; 125: S31–4.
- Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005; 106: 1604–11.
- Owens AP 3rd, Mackman N. Role of tissue factor in atherothrombosis. Curr Atheroscler Rep. 2012; 14: 394–401.
- Geddings JE, Mackman N. Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood. 2013; 122: 1873–80.
- Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb Haemost. 2014; 111(3): [Epub ahead of print].
- Collier ME, Mah PM, Xiao Y, Maraveyas A, Ettelaie C. Microparticle-associated tissue factor is recycled by endothelial cells resulting in enhanced surface tissue factor activity. Thromb Haemost. 2013; 110: 966–76.
- Davila M, Robles-Carrillo L, Unruh D, Huo Q, Gardiner C, Sargent IL et al. Microparticle association and heterogeneity of tumor-derived tissue factor in plasma: is it important for coagulation activation?. J Thromb Haemost. 2014; 12: 186–96.
- Gheldof D, Hardij J, Cecchet F, Chatelain B, Dogne JM, Mullier F. Thrombin generation assay and transmission electron microscopy: a useful combination to study tissue factor-bearing microvesicles. J Extracell Vesicles. 2013; 2 19728, doi: http://dx.doi.org/10.3402/jev.v2i0.19728.
- Hugel B, Socie G, Vu T, Toti F, Gluckman E, Freyssinet JM et al. Elevated levels of circulating procoagulant microparticles in patients with paroxysmal nocturnal hemoglobinuria and aplastic anemia. Blood. 1999; 93: 3451–6.
- Liebman HA, Feinstein DI. Thrombosis in patients with paroxysmal noctural hemoglobinuria is associated with markedly elevated plasma levels of leukocyte-derived tissue factor. Thromb Res. 2003; 111: 235–8.
- Van Bijnen ST, Van Heerde WL, Muus P. Mechanisms and clinical implications of thrombosis in paroxysmal nocturnal hemoglobinuria. J Thromb Haemost. 2012; 10: 1–10.
- Mullier F, Minet V, Bailly N, Devalet B, Douxfils J, Chatelain C et al. Platelet microparticle generation assay: a valuable test for immune heparin-induced thrombocytopenia diagnosis. Thromb Res. 2013. [Epub ahead of print].
- Gurney D, Lip GY, Blann AD. A reliable plasma marker of platelet activation: does it exist?. Am J Hematol. 2002; 70: 139–44.
- Yahata T, Suzuki C, Yoshioka A, Hamaoka A, Ikeda K. Platelet activation dynamics evaluated using platelet-derived microparticles in Kawasaki disease. Circ J. 2013; 78: 188–93.
- Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2003; 102: 3587–91.
- Poskitt TR, Fortwengler HP Jr, Lunskis BJ. Activation of the alternate complement pathway by autologous red cell stroma. J Exp Med. 1973; 138: 715–22.
- Kozuma Y, Sawahata Y, Takei Y, Chiba S, Ninomiya H. Procoagulant properties of microparticles released from red blood cells in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2011; 152: 631–9.
- Simak J, Holada K, Risitano AM, Zivny JH, Young NS, Vostal JG. Elevated circulating endothelial membrane microparticles in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2004; 125: 804–13.
- Schaer DJ, Buehler PW. Cell-free hemoglobin and its scavenger proteins: new disease models leading the way to targeted therapies. Cold Spring Harb Perspect Med. 2013; 3: a013433.
- Ritis K, Doumas M, Mastellos D, Micheli A, Giaglis S, Magotti P et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006; 177: 4794–802.
- Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006; 12: 682–7.
- Steppich B, Mattisek C, Sobczyk D, Kastrati A, Schomig A, Ott I. Tissue factor pathway inhibitor on circulating microparticles in acute myocardial infarction. Thromb Haemost. 2005; 93: 35–9.
- Jankowska AM, Szpurka H, Calabro M, Mohan S, Schade AE, Clemente M et al. Loss of expression of neutrophil proteinase-3: a factor contributing to thrombotic risk in paroxysmal nocturnal hemoglobinuria. Haematologica. 2011; 96: 954–62.
- Dragoni F, Iori AP, Pignoloni P, Minotti C, Chiarotti F, Mazzucconi MG et al. Thrombophilic screening in patients with paroxysmal nocturnal haemoglobinuria: a pilot study. Br J Haematol. 2010; 150: 492–4.
- Grünewald M, Siegemund A, Grünewald A, Schmid A, Koksch M, Schopflin C et al. Plasmatic coagulation and fibrinolytic system alterations in PNH: relation to clone size. Blood Coagul Fibrinolysis. 2003; 14: 685–95.
- Plesner T, Behrendt N, Ploug M. Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR. Stem Cells. 1997; 15: 398–408.
- Sloand EM, Pfannes L, Scheinberg P, More K, Wu CO, Horne M et al. Increased soluble urokinase plasminogen activator receptor (suPAR) is associated with thrombosis and inhibition of plasmin generation in paroxysmal nocturnal hemoglobinuria (PNH) patients. Exp Hematol. 2008; 36: 1616–24.
- Ploug M, Eriksen J, Plesner T, Hansen NE, Dano K. A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria. Eur J Biochem. 1992; 208: 397–404.
- Ginestra A, Monea S, Seghezzi G, Dolo V, Nagase H, Mignatti P et al. Urokinase plasminogen activator and gelatinases are associated with membrane vesicles shed by human HT1080 fibrosarcoma cells. J Biol Chem. 1997; 272: 17216–22.
- Ginestra A, Miceli D, Dolo V, Romano FM, Vittorelli ML. Membrane vesicles in ovarian cancer fluids: a new potential marker. Anticancer Res. 1999; 19: 3439–45.
- Angelucci A, D'Ascenzo S, Festuccia C, Gravina GL, Bologna M, Dolo V et al. Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin Exp Metastasis. 2000; 18: 163–70.
- Lacroix R, Sabatier F, Mialhe A, Basire A, Pannell R, Borghi H et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: a mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood. 2007; 110: 2432–9.
- Kwaan HC, Rego EM. Role of microparticles in the hemostatic dysfunction in acute promyelocytic leukemia. Semin Thromb Hemost. 2010; 36: 917–24.
- Rao SV, Jollis JG, Harrington RA, Granger CB, Newby LK, Armstrong PW et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292: 1555–62.
- Khorana AA, Francis CW, Blumberg N, Culakova E, Refaai MA, Lyman GH. Blood transfusions, thrombosis, and mortality in hospitalized patients with cancer. Arch Intern Med. 2008; 168: 2377–81.
- Abu-Rustum NR, Richard S, Wilton A, Lev G, Sonoda Y, Hensley ML et al. Transfusion utilization during adnexal or peritoneal cancer surgery: effects on symptomatic venous thromboembolism and survival. Gynecol Oncol. 2005; 99: 320–6.
- Nilsson KR, Berenholtz SM, Garrett-Mayer E, Dorman T, Klag MJ, Pronovost PJ. Association between venous thromboembolism and perioperative allogeneic transfusion. Arch Surg. 2007; 142: 126–32. discussion 33.
- Tan TW, Farber A, Hamburg NM, Eberhardt RT, Rybin D, Doros G et al. Blood transfusion for lower extremity bypass is associated with increased wound infection and graft thrombosis. J Am Coll Surg. 2013; 216: 1005–14. e2;quiz 31–3.
- Kumar MA, Boland TA, Baiou M, Moussouttas M, Herman JH, Bell RD et al. Red blood cell transfusion increases the risk of thrombotic events in patients with subarachnoid hemorrhage. Neurocrit Care. 2014; 20: 84–90.
- Rubin O, Delobel J, Prudent M, Lion N, Kohl K, Tucker EI et al. Red blood cell-derived microparticles isolated from blood units initiate and propagate thrombin generation. Transfusion. 2013; 53: 1744–54.
- Spinella PC, Carroll CL, Staff I, Gross R, Mc Quay J, Keibel L et al. Duration of red blood cell storage is associated with increased incidence of deep vein thrombosis and in hospital mortality in patients with traumatic injuries. Crit Care. 2009; 13(R151):
- Moyo VM, Mukhina GL, Garrett ES, Brodsky RA. Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assays. Br J Haematol. 2004; 126: 133–8.
- Robert S, Lacroix R, Poncelet P, Harhouri K, Bouriche T, Judicone C et al. High-sensitivity flow cytometry provides access to standardized measurement of small-size microparticles--brief report. Arterioscler Thromb Vasc Biol. 2012; 32: 1054–8.
- Lacroix R, Judicone C, Poncelet P, Robert S, Arnaud L, Sampol J et al. Impact of pre-analytical parameters on the measurement of circulating microparticles: towards standardization of protocol. J Thromb Haemost. 2012; 10: 437–46.
- Lacroix R, Robert S, Poncelet P, Kasthuri RS, Key NS, Dignat-George F et al. Standardization of platelet-derived microparticle enumeration by flow cytometry with calibrated beads: results of the International Society on Thrombosis and Haemostasis SSC Collaborative workshop. J Thromb Haemost. 2010; 8: 2571–4.
- Mullier F, Bailly N, Chatelain C, Chatelain B, Dogne JM. Pre-analytical issues in the measurement of circulating microparticles: current recommendations and pending questions. J Thromb Haemost. 2013; 11: 693–6.