
Abstract
Background
Protein secretion is a fundamental process in all living cells. Gluconeogenic enzymes are secreted when Saccharomyces cerevisiae are grown in media containing low glucose. However, when cells are transferred to media containing high glucose, they are internalized. We investigated whether or not gluconeogenic enzymes were associated with extracellular vesicles in glucose-starved cells. We also examined the role that the endocytosis gene END3 plays in the internalization of extracellular proteins/vesicles in response to glucose addition.
Methods
Transmission electron microscopy was performed to determine the presence of extracellular vesicles in glucose-starved wild-type cells and the dynamics of vesicle transport in cells lacking the END3 gene. Proteomics was used to identify extracellular proteins that associated with these vesicles.
Results
Total extracts prepared from glucose-starved cells consisted of about 95% small vesicles (30–50 nm) and 5% large structures (100–300 nm). The addition of glucose caused a rapid decline in small extracellular vesicles in wild-type cells. However, most of the extracellular vesicles were still observed in cells lacking the END3 gene following glucose replenishment. Proteomics was used to identify 72 extracellular proteins that may be associated with these vesicles. Gluconeogenic enzymes fructose-1,6-bisphosphatase, malate dehydrogenase, isocitrate lyase, and phosphoenolpyruvate carboxykinase, as well as non-gluconeogenic enzymes glyceraldehyde-3-phosphate dehydrogenase and cyclophilin A, were distributed in the vesicle-enriched fraction in total extracts prepared from cells grown in low glucose. Distribution of these proteins in the vesicle-enriched fraction required the integrity of the membranes. When glucose was added to glucose-starved wild-type cells, levels of extracellular fructose-1,6-bisphosphatase, malate dehydrogenase, isocitrate lyase, phosphoenolpyruvate carboxykinase, glyceraldehyde-3-phosphate dehydrogenase, and cyclophilin A were reduced. In contrast, in cells lacking the END3 gene, levels of these proteins in the extracellular fraction remained high.
Conclusion
The END3 gene is required for the rapid decline of extracellular proteins and vesicles in response to glucose addition.
To access the supplementary material to this article, please see Supplementary files under Article Tools online.
Protein secretion is a fundamental process in both eukaryotic and prokaryotic cells. Recent large-scale proteomic/secretomic studies from bacteria, fungi, parasites, plants, murine, and human cells indicate that a large number of proteins including transcriptional regulators, mitochondrial proteins, metabolic enzymes, and heat shock proteins are secreted via the non-classical pathway (Citation1–Citation15). The secretion of glycolytic and gluconeogenic enzymes has also been described in several large-scale proteomic/secretomic studies (Citation5, Citation6) (Citation7, Citation8) (Citation10, Citation11) (Citation12, Citation13) (Citation16) . For instance, the key gluconeogenic enzyme phosphoenolpyruvate carboxykinase (Pck1p) is detected in secretomes from Clonorchis sinensis, Echinostoma caproni, and Schistosoma mansoni (Citation5, Citation6) (Citation8). Another key gluconeogenic enzyme fructose-1,6-bisphosphatase (FBPase) is also identified in secretomes from Bacillus anthracis (Citation16) and Clonorchis sinensis (Citation8), whereas malate dehydrogenase (MDH2) is in secretomes from Bacillus anthracis (Citation16), Clonorchis sinensis (Citation8), and Schistosoma mansoni (Citation5). Interestingly, Pck1p, FBPase, MDH2, and isocitrate lyase (Icl1p) are all detected in extracellular vesicles isolated from the dimorphic fungus Histoplasm capsulatum (Citation9). Using immuno-TEM, we have shown that FBPase is secreted from the yeast Saccharomyces cerevisiae (Citation17). Therefore, the secretion of gluconeogenic enzymes is widely observed from bacteria, fungi, and parasites.
Saccharomyces cerevisiae is a model system used to study global changes in transcription, translation, and protein levels in response to changes in the environment, such as the availability of glucose in the media. Glucose affects transcription, translation, and mRNA turnovers (Citation18–Citation25). Moreover, glucose also causes inactivation of gluconeogenic enzymes, mitochondria proteins, and enzymes involved in the utilization of other carbon sources (Citation20–Citation25). Gluconeogenic enzymes such as Pck1p, FBPase, MDH2, and Icl1p are inactivated when glucose is added to glucose-starved cells (Citation20, Citation21) (Citation22, Citation23) (Citation26, Citation27) (Citation28, Citation29) . This process is called catabolite inactivation (Citation20, Citation21) (Citation22, Citation23) (Citation26, Citation27) (Citation28, Citation29) . The site of degradation of gluconeogenic enzymes is influenced by the duration of starvation. These enzymes are degraded in the proteasome when glucose is added to cells that are starved of glucose for 1 day (Citation30). In contrast, when glucose is added to cells starved of glucose for 3 days, they are degraded in the vacuole (Citation30). The vacuole pathway utilizes small vesicles (Vid vesicles) that are 30–50 nm in diameter (Citation31). Vid vesicles are also known to cluster together to form large aggregates in the cytoplasm (Citation32).
We have recently shown that the key gluconeogenic enzyme FBPase is secreted into the periplasm during glucose starvation and is internalized following glucose re-feeding (Citation17, Citation33). Gluconeogenic enzymes Pck1p, MDH2, and Icl1p were also secreted in glucose-starved cells. However, whether or not these secreted enzymes associated with extracellular vesicles had not been examined. In this study, we report the identification of 2 morphologically distinct structures present in the extracellular fraction that responded differently to glucose addition. When glucose was added to glucose-starved cells, 4.5% of 30–50 nm small vesicles remained, whereas 76.3% of 100–300 nm large structures were still observed. The END3 gene is involved in the internalization of extracellular molecules into cells (Citation34–Citation36). In cells lacking the END3 gene, small vesicles were still observed following glucose addition. Furthermore, levels of extracellular FBPase, MDH2, Icl1p, Pck1p, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and cyclophilin A (Cpr1p) did not decrease following glucose addition to glucose-starved lacking the END3 gene. We conclude that the END3 gene plays an important role in the glucose-induced decline of proteins and vesicles in the extracellular fraction in Saccharomyces cerevisiae.
Experimental procedures
Cell culture, media and antibodies
Yeast strains used in this study included wild-type strain (BY4742, MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0), wild-type strain expressing Icl1p-HA (BY4742, MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ICL1-HA-HIS3), wild-type strain expressing Pck1p-HA (BY4742, MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 PCK1-HA-HIS3), wild-type strain expressing Icl1p-HA and Pck1p-Myc (BY4742, MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ICL1-HA-HIS3, PCK1p-Myc-KanMX6), the Δend3 mutant strain (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 end3::KanMX6), the Δend3 mutant strain expressing Icl1p-HA (BY4742, MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ICL1-HA-HIS3 end3::KanMX6), and the Δend3 mutant expressing Pck1p-HA (BY4742, MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 PCK1-HA-HIS3 end3::KanMX6). Cells were grown in yeast extract-peptone-potassium acetate-glucose (YPKG) media containing 1% yeast extract, 2% peptone, 1% potassium acetate and 0.5% glucose for 3 days. Cells were harvested by centrifugation at 3,000×g for 5 min and resuspended in yeast extract-peptone-glucose (YPD) media containing 1% yeast extract, 2% peptone and 2% glucose for the indicated time points. In some experiments, wild-type cells were grown in YPKG for 1d, 2d, and 3d and harvested. Cell viability was determined using trypan blue exclusion experiments. Cells were grown in 2 ml YPKG, harvested by a low speed centrifugation at 3,000×g for 5 min, and resuspended in 100 µl phosphate buffered saline (PBS) followed by incubation with 20 µl of 0.4% trypan blue in PBS for 30 min. Cells were visualized by a Zeiss microscope. Under our experimental conditions,<0.01% of the cells were stained with trypan blue.
Extraction and western immunoblotting
Cells (OD600=10/ml) were incubated in 100 µl of extraction buffer containing 0.1M Tris pH 9.4 and 10 mM β-mercaptoethanol (β-ME) at 37°C in a shaker (215 rpm) for 15 min. After this extraction procedure, cells were pelleted. The extracellular proteins that were released into the supernatant (100 µl) were precipitated in 15% trichloroacetic acid (TCA), washed, and solubilized in 100 µl SDS-PAGE sample buffer. The remaining cell-associated (intracellular) fraction was resuspended in 100 µl PBS buffer and the same volume of glass beads. Total lysates were obtained by glass beading and proteins were solubilized in 100 µl SDS sample buffer. Intracellular and extracellular proteins (15 µl) were loaded onto SDS-PAGE gels and transferred to nitrocellulose membranes. The distribution of proteins in the intracellular and extracellular fractions was examined by immunoblotting using polyclonal antibodies directed against FBPase, MDH2, GAPDH, and Cpr1p followed by peroxidase-conjugated donkey anti-rabbit IgG diluted 1:10,000 (GE Healthcare). Icl1p-HA, Pck1p-HA, and Pck1p-Myc were detected using anti-HA (Roche) and anti-Myc (Santa Cruz Biotechnology) antibodies followed by peroxidase-conjugated donkey anti-mouse IgG (GE Healthcare). Protein bands were developed using Western Lighting Plus ECL (Perkin Elmer).
2D-LC separation and mass spectrometry
Proteomic analysis was performed using the Proteomic Core Facility at Penn State College of Medicine. Wild-type yeast cells were grown in YPKG for 3 days and subjected to the extraction procedure. Extracellular proteins (100 µg) were precipitated in 15% TCA and reduced with TCEP (tris (2-carboxyethyl) phosphine), followed by alkylation with iodoacetamide. Proteins were digested with trypsin (Promega sequencing grade) at 48°C for 16 hours. Tryptic peptides were subjected to 2D-LC separation and mass spectrometry. Peptides were dried and separated on a passivated Waters 600E HPLC system, using a 4.6×250 mm PolySULFOETHYL Aspartamide™ column (PolyLC, Columbia MD) at a flow rate of 1 ml/min. Each SCX fraction was injected onto a Chromolith CapRod column (150×0.1 mm, Merck) using a 5 µl injector loop on a Tempo LC MALDI Spotting system (ABI-MDS/Sciex). Each MALDI target plate was analyzed using ABI 5800 MALDI TOF-TOF.
Protein identification was accomplished using the Paragon Algorithm (Citation37) in ProteinPilot™ 4.0 software (ABSciex). Search parameters were set as cysteine alkylation: iodoacetamide, ID focus: biological modifications, search effort: thorough. Combined spectra were searched against the species-specific (S. cerevisiae) NCBInr database concatenated with a reversed “decoy” version of the same database plus 156 common lab contaminants. To reduce the potential number of false positive protein identifications, the Paragon Algorithm search results were further filtered through the use of a very stringent Local False Discovery Rate (FDR) estimation calculated from the Proteomics System Performance Evaluation Pipeline (PSPEP) (Citation38). We accepted 92 protein IDs with a local FDR estimate of<5%. We next removed contaminants and any protein ID based on a single peptide, leaving 72 proteins by these stringent criteria. We used the Gene Ontology Slim Mapper program in the Saccharomyces Genome Database (Stanford University) to search for the function of each protein.
Centrifugation and transmission electron microscopy
Wild-type cells and the Δend3 cells were grown in YPKG (40 ml) for 3 days and subjected to the extraction procedure. Total extracts (2 ml) were first centrifuged at 3,000×g for 5 min at 4°C and the resulting supernatants (2 ml) were centrifuged at 200,000×g for 2 hours at 4°C. Pellets from the 200,000×g centrifugation were re-suspended in 200 µl PBS buffer and aliquoted. Samples (100 µl) were incubated in the absence or presence of 2% SDS (final concentration) for 30 min followed by re-centrifugation at 200,000×g for 2 hours. The 200,000×g pellet from the second centrifugation was resuspended in 100 µl PBS and solubilized in 100 µl SDS sample buffer. The 200,000×g supernatant (100 µl) was solubilized in 100 µl SDS sample buffer. Proteins from the supernatant fraction (15 µl) and the pellet fraction (15 µl) were loaded onto SDS-PAGE gels, transferred to nitrocellulose membranes and examined by Western blotting. For transmission electron microscopy (TEM) studies, wild-type cells and the Δend3 cells (100 ml) were grown in YPKG for 3 days and transferred to 100 ml of YPD media for 30 min. Cells were subjected to the extraction procedure and pelleted. Total extracts (5 ml) were first centrifuged at 3,000×g for 5 min and subsequently at 200,000×g for 2 hours. The 200,000×g pellets were resuspended in 30 µl of PBS buffer and fixed. After washing, grids were stained with 2% uranyl acetate for 1 min and viewed on a JEOL JEM-1400 electron microscope with an Orius SC1000 CCD camera. TEM experiments were performed 3 times and 3 micrographs were used to obtain means and SD.
Results
END3 is essential for the decline of small vesicles in the extracellular fraction in response to glucose addition
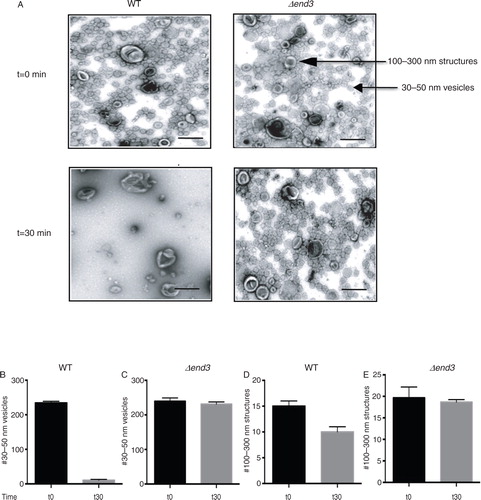
Gluconeogenic enzymes are secreted into the extracellular fraction when cells are grown in low glucose. However, whether or not these secreted enzymes associated with extracellular vesicles during glucose starvation had not been determined. We have shown previously that Vid vesicles are enriched in the 200,000×g pellet fraction (Citation31, Citation32) (Citation39). Therefore, if Vid-like vesicles are present in the extracellular fraction, they may be enriched in the 200,000×g pellet. Wild-type cells were grown in media containing low glucose for 3 days and aliquoted. Cells were harvested at t=0 min or transferred to media containing fresh glucose for 30 min. Extracellular proteins were extracted from whole cells and total extracts were centrifuged at 3,000×g for 5 min and then at 200,000×g for 2 hours. Following centrifugation, the 200,000×g pellet fraction was resuspended in PBS buffer and fixed. Samples were stained with uranyl acetate followed by observation using transmission electron microscopy (A, left panels). When total extracts were prepared from wild-type cells grown in low glucose (t=0 min), numerous small vesicles that were 30–50 nm in diameter were observed. In addition, large structures that were 100–300 nm were also detected. Quantification of these structures indicated that total extracts isolated from t=0 wild-type cells consisted of 94.6% small vesicles and 5.4% large structures. Following the addition of glucose for 30 min, very few small vesicles remained. In contrast, large structures were still observed. The number of small vesicles was 234.3±4.7 per µm2 in t=0 wild-type cells and 10.7±2.5 per µm2 in t=30 wild-type cells (B). Therefore, the addition of glucose for 30 min produced a 95.5% reduction in small vesicles. The number of 100–300 nm large structures was 13.5±1.1 per µm2 without glucose addition and 10.3±1.2 per µm2 after glucose addition (D). Hence, 76.3% of the large structures remained in total extracts following glucose replenishment for 30 min. Thus, 2 morphologically distinct structures are present in the extracellular fraction in wild-type cells and demonstrate different responses to glucose addition.
Fig. 1. The END3 gene is essential for the glucose-induced decline of small vesicles in the extracellular fraction. A, wild-type and Δend3 cells were glucose starved and re-fed with glucose for 30 min. Total extracts were obtained and centrifuged at 3,000×g for 5 min and then at 200,000×g for 2 hours. The 200,000×g pellet fraction was resuspended in PBS buffer, fixed, stained with uranyl acetate and visualized by TEM. Bars: 200 nm. B and C, quantification of the number of 30–50 nm small vesicles per µm2 before and after glucose addition in wild-type (B) and Δend3 (C) cells. D and E, quantification of the number of 100–300 nm large structures per µm2 before and after glucose addition in wild-type (D) and Δend3 (E) cells. TEM experiments were performed 3 times and images from 3 micrographs were used to obtain means and SD.
A rapid decline of small vesicles in the extracellular fraction following glucose replenishment may result from their internalization into the cytoplasm or release into the media. The later scenario is consistent with the findings that extracellular vesicles are present in culture media in Saccharomyces cerevisiae (Citation9, Citation40) (Citation41). To determine whether or not the decline of extracellular vesicles in response to glucose addition resulted from release into culture media, wild-type cells were grown in low glucose media and then transferred to high glucose media for 30 min. Culture media were collected from t=0 and t=30 min cells and centrifuged at 3,000×g for 5 min and subsequently at 200,000×g for 2 hours. Following centrifugation, the pellet fraction was resuspended in PBS buffer, fixed and stained by uranyl acetate followed by TEM. We did not observe significant amounts of extracellular vesicles in the culture media from either t=0 or t=30 wild-type cells.
We next examined the possibility that these vesicles were internalized into the cytoplasm following glucose addition. The END3 gene is involved in endocytosis in yeast (Citation34–Citation36). If the decline of vesicles in the extracellular fraction is dependent on the END3 gene, extracellular vesicles should still be observed when total extracts were prepared from Δend3 cells that were transferred from low to high glucose media for 30 min. By contrast, if this gene is not required for the decline of small vesicles from extracellular fraction in response to glucose, the number of these vesicles in the extracellular fraction should be low following glucose re-feeding of cells lacking END3. To test this, the Δend3 strain was grown in low glucose media and aliquoted. Cells were harvested at t=0 min or transferred to media containing glucose for 30 min. Total extracts were prepared and centrifuged at 3,000×g for 5 min. The resulting supernatant was further centrifuged at 200,000×g for 2 hours. The 200,000×g pellet fraction was re-suspended in buffer, fixed, stained with uranyl acetate and visualized by TEM (A, right panels). When extracts were prepared from the Δend3 cells at t=0 min, small vesicles with diameters of 30–50 nm and large structures with diameters of 100–300 nm were observed. When total extracts were prepared from Δend3 cells that were transferred to media containing high glucose for 30 min, most of the small vesicles and large structures remained. Quantification of these structures indicated that total extracts isolated from Δend3 cells contained 239.2±9.7 small vesicles per µm2 at t=0 min and 231.1±6.6 vesicles per µm2 at the t=30 min time point (C). The number of 100–300 nm large structures in total extracts isolated from Δend3 cells was 19.7±2.5 per µm2 at t=0 min and was 18.6±1.6 per µm2 at the t=30 min time point (E). Therefore, 96.6% of the small vesicles and 94.4% of the large structures were still observed when the Δend3 strain was re-fed with glucose for 30 min. Given that most of the small vesicles remained in the extracellular fraction at the t=30 min time point in the Δend3 strain, we suggest that this gene is required for the decline of small vesicles in the extracellular fraction in response to glucose replenishment.
Proteomic approach to identify proteins in the extracellular fraction
We next used a proteomic approach to identify proteins that were present in the extracellular fraction. This should include proteins that associate with the small vesicles as well as proteins that associate with the large structures. Furthermore, this should also contain proteins that show a rapid END3-dependent decrease in response to glucose addition. Wild-type cells were grown in media containing low glucose and extracellular proteins were extracted from these cells. Next, extracellular proteins were precipitated with TCA and digested with trypsin. The resulting tryptic peptides were then subjected to mass spectrometry for protein identification. A total of 92 extracellular proteins were identified with an estimated local FDR of<5% and 72 of these extracellular proteins were identified with more than 2 peptides with 95% confidence (Supplemental Table 1). These proteins were further classified into different functional groups as defined by the Saccharomyces genome database. About 18% of proteins were involved in carbohydrate metabolism and 14% of proteins in amino acid metabolism. We have also identified proteins involved in the metabolism of alcohol, purines, pyrimidines, fatty acids, glycerol, and acetate. Furthermore, heat shock proteins, protein disulfide isomerase, cyclophilins, anti-oxidant proteins ribosomal proteins, initiation factors, elongation factors, actin, and proteins with unknown functions were identified in the current study. Gluconeogenic enzymes MDH2, Icl1p, and Pck1p were identified. However, FBPase was not identified. GAPDH and Cpr1p are known to be secreted (Citation40, Citation42) (Citation43) and were identified in this study. Heat shock proteins Ssa1p and Ssa2p are cell-surface proteins (Citation44) that were also identified in the current study.
Distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the vesicle-enriched fraction requires membrane integrity
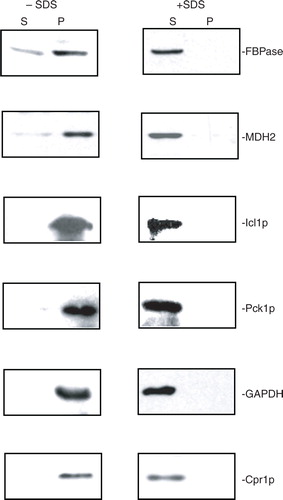
We next determined whether or not these identified proteins associated with extracellular vesicles, and whether this was dependent on membrane integrity. If these proteins are associated with extracellular vesicles, the proteins should be detectable in the 200,000×g pellet fraction in total extracts isolated from glucose-starved cells. However, if these proteins do not associate with extracellular vesicles, they should not be detected in the 200,000×g pellet fraction in total extracts. We first examined the distribution of gluconeogenic enzymes. Because antibodies against Icl1p and Pck1p were not available, we produced a wild-type strain containing HA tagged to Icl1p and Myc tagged to Pck1p. We then used HA and Myc antibodies to detect the presence of these proteins. Wild-type cells expressing Icl1p-HA and Pck1p-Myc were grown in low glucose and harvested. Total extracts were centrifuged at 3,000×g for 5 min and then 200,000×g for 2 hours. After centrifugation, the 200,000×g pellet fraction was resuspended in buffer and aliquoted. Samples were incubated in the absence and presence of detergent (2% SDS). Following incubation, samples were re-centrifuged at 200,000×g for 2 hours and the distribution of these proteins in the 200,000×g supernatant (S) and 200,000×g pellet (P) fractions was determined (). In the absence of detergent, most of the FBPase, MDH2, Icl1p, and Pck1p were in the 200,000×g pellet fraction (, left panels). When the 200,000×g pellet fraction was disrupted with 2% SDS and re-centrifuged, the majority of these proteins were in the supernatant fraction (, right panels). Therefore, the distribution of these proteins in the vesicle-enriched fraction is dependent on membrane integrity. We also determined the distribution of non-gluconeogenic enzymes such as GAPDH and Cpr1p in the vesicle-enriched fraction. In the absence of detergent, most of the GAPDH and Cpr1p were in the 200,000×g pellet fraction (, left panels). However, when membranes were disrupted by 2% SDS, these proteins were found mostly in the 200,000×g supernatant fraction (, right panels). Thus, the distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the vesicle-enriched fraction is dependent on the integrity of membranes.
Fig. 2. FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p are distributed in the vesicle-enriched fraction. Wild-type cells were starved of glucose for 3 days and harvested. Total extracts were obtained and centrifuged at 3,000×g for 5 min. The resulting supernatant was further centrifuged at 200,000×g for 2 hours. The 200,000×g pellet fraction was resuspended, aliquoted, and incubated in the absence or presence of 2% SDS for 30 min. Following incubation, samples were re-centrifuged at 200,000×g for 2 hours. The distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the 200,000×g supernatant (S) and pellet (P) fractions were examined by Western blotting. Representative data from 3 experiments are shown.
END3-dependent decrease in extracellular FBPase, MDH2, Pck1p, Icl1p, GAPDH, and Cpr1p in response to glucose addition
Gluconeogenic enzymes are degraded in the vacuole when glucose is added to cells starved of glucose for 3 days (Citation32, Citation39) (Citation45, Citation46) (Citation47, Citation48) (Citation49, Citation50) . Therefore, levels of extracellular gluconeogenic enzymes should decrease following a transfer of cells from low to high glucose media. As shown in A, about 76.3% of the large structures remained in total extracts in wild-type cells at the t=30 min time point, whereas only 4.5% of the small vesicles were observed at this time point. Therefore, if these proteins associate with the 30–50 nm small vesicles, levels of these proteins in extracellular fraction should be low at the t=30 min time point following glucose addition. In contrast, if these proteins associate with the 100–300 nm large structures, substantial amounts of these proteins may still be observed in the extracellular fraction at the t=30 min time point. Wild-type cells were grown in low-glucose media for 3 days, transferred to medium containing fresh glucose for 0, 15, and 30 min. Extracellular proteins were extracted and levels of these proteins in the intracellular (I) and extracellular (E) fractions were then determined (A). In wild-type cells, FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p were in the extracellular fraction at the t=0 time point. Levels of these proteins in the extracellular fraction decreased quickly following a transfer of cells to medium containing 2% glucose for 15 and 30 min (A). A rapid decline of these proteins following glucose addition suggests that these proteins associate with small vesicles.
Fig. 3. The decline of extracellular FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in response to glucose re-feeding is dependent on END3. A, wild-type cells were starved of glucose for 3 days and transferred to media containing 2% glucose for 0, 15, and 30 min. The distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the intracellular (I) and extracellular (E) fractions was examined by Western blotting. B, The Δend3 cells were starved of glucose for 3 days and transferred to media containing glucose for 0, 15, and 30 min. The distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the intracellular (I) and extracellular (E) fractions was determined. Representative results from 3 experiments are shown.
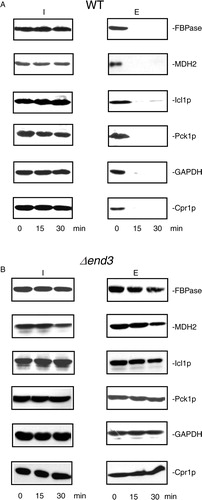
Because the vesicles were still observed in the Δend3 strain following glucose addition (see A), the decrease of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the extracellular fraction following glucose replenishment may be retarded in cells lacking the END3 gene. To test this, the Δend3 strain was starved of glucose for 3 days, transferred to medium containing high glucose for 15 and 30 min and examined for the presence of these proteins in the intracellular (I) and extracellular (E) fractions (B). At t=0 min, these proteins were in the extracellular fraction in the Δend3 strain. However, their levels in the extracellular fraction remained high following a transfer of the Δend3 strain to glucose for 30 min (B). Thus, the END3 gene is required for the glucose-induced decline of these proteins in the extracellular fraction.
Levels of extracellular FBPase, MDH2, GAPDH, and Cpr1p change depending on the duration of starvation
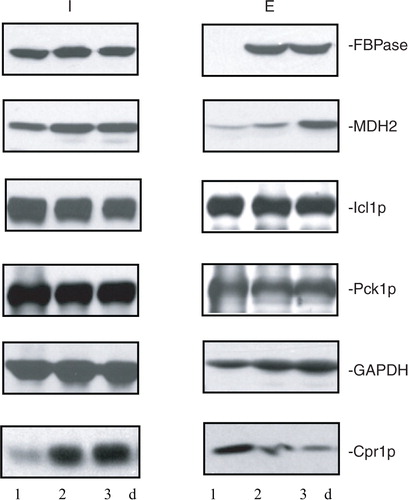
Because extracellular proteins were identified from cells that were grown in low glucose for 3 days, we next determined whether or not these proteins were present in the extracellular fraction in cells that were starved for a shorter period of time. Wild-type cells were grown in media containing low glucose for 1d, 2d, and 3d. The distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the intracellular (I) and extracellular (E) fractions was then determined (). In 1d-starved cells, FBPase was expressed, but its levels in the extracellular fraction were low. Amounts of FBPase in the extracellular fraction increased in 2d- and 3d-starved cells. In a similar manner, levels of MDH2 in the extracellular fraction were low in 1d-starved cells and increased in 2d- and 3d-starved cells (). However, levels of Icl1p and Pck1p in the extracellular fraction did not change significantly in 1d-, 2d-, and 3d-starved cells. Levels of GAPDH in the extracellular fraction were lower in 1d-starved cells and increased when cells were starved longer. In contrast, amounts of Cpr1p in the extracellular fraction were high in 1d-starved cells but decreased in 2d- and 3d-starved cells. Thus, levels of 4 out of the 6 extracellular proteins examined in this study vary depending on the duration of starvation.
Fig. 4. Levels of extracellular FBPase, MDH2, GAPDH, and Cpr1p vary depending on the duration of starvation. Wild-type cells were grown in media containing low glucose for 1d, 2d, and 3d. The distribution of FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p in the intracellular (I) and extracellular (E) fractions was determined. Representative data from 3 experiments are shown.
In summary, our results demonstrated that small vesicles were present in the extracellular fraction during glucose starvation. These small vesicles undergo a rapid decline following glucose addition in a process dependent on the END3 gene. When total extracts were prepared from cells grown in low glucose, gluconeogenic enzymes FBPase, MDH2, Icl1p, and Pck1p were present in the vesicle-enriched fraction. Non-gluconeogenic enzymes such as GAPDH and Cpr1p were also detectable in the vesicle-enriched fraction. Furthermore, their distribution in the vesicle-enriched fraction is dependent on membrane integrity. During glucose starvation, these proteins were in the extracellular fraction and their levels in the extracellular fraction decreased following glucose addition. Moreover, they all utilize an END3-dependent mechanism to reduce their levels in the extracellular fraction in response to glucose. We conclude that END3 plays an important role in the glucose-induced rapid decline of vesicles and proteins in the extracellular fraction.
Discussion
In this study, we described the identification of 2 types of morphologically distinct structures in the extracellular fraction from Saccharomyces cerevisiae grown in low glucose. Approximately 95% of these extracellular structures were small vesicles with a 30–50 nm diameter. The presence of small vesicles in the extracellular fraction is consistent with reports showing that small vesicles are secreted from a variety of cells (Citation51–Citation53). Purified exosomes from mammalian cells are 40–100 nm in diameter and have densities of 1.1–1.2 g/ml (Citation51–Citation53). Interestingly, FBPase and MDH2 have been identified in exosomes secreted from insulinoma NIT-1 cells (Citation54). FBPase, MDH2, Icl1p, and Pck1p have also been identified in extracellular vesicles from culture media from Histoplasm capsulatum (Citation9). Small vesicles (Vid vesicles) are used to carry gluconeogenic enzymes to the vacuole for degradation when glucose is added to Saccharomyces cerevisiae grown in low glucose. Vid vesicles are 30–50 nm in diameter and have densities of 1.2 g/ml (Citation31). These vesicles exist as free and clustered forms in the cytoplasm (Citation32). In our study, about 5% of these extracellular structures were large structures that were 150–300 nm in diameter when total extracts were prepared from glucose-starved yeast cells. These large structures appeared to be heterogeneous in size and shape. Many of them contained higher electron densities in the centre that were 50–100 nm in diameter. Extracellular vesicles that are 50–250 nm have also been identified in culture media from Saccharomyces cerevisiae (Citation9, Citation40) (Citation41).
Interestingly, 30–50 nm small vesicles and 100–300 nm large structures displayed different responses to glucose addition. When glucose was added to glucose-starved cells, only 4.5% of the small vesicles remained after 30 min, whereas 76.3% of large structures remained. Rapid decline of small vesicles in the extracellular fraction in response to glucose addition is likely to result from internalization into the cytoplasm. This idea is consistent with the findings that most of these vesicles were still observed at the t=30 min in cells lacking the END3 gene that blocks endocytosis. It is also possible that END3 is involved in a process that promotes the disruption of these vesicles. However, the vacuole is the final destination for gluconeogenic enzymes following glucose addition. These enzymes that are present in the extracellular fraction prior to glucose addition should be internalized. Degradation of gluconeogenic enzymes in the vacuole during glucose re-feeding prevents energy futile cycles that may be detrimental to cells. Internalization is also consistent with previous findings that clusters of small vesicles were observed in the cytoplasm when wild-type cells were transferred to glucose for 30 min (Citation17, Citation32). Furthermore, Vid/endosomes containing aggregates of small vesicles have been purified to near homogeneity from the cytoplasm from wild-type cells that were re-fed with glucose (Citation32). Inside these Vid/endosomes, FBPase was associated with numerous small vesicles (Citation32). Therefore, these small extracellular vesicles are most likely to be internalized into the cytoplasm in response to glucose addition.
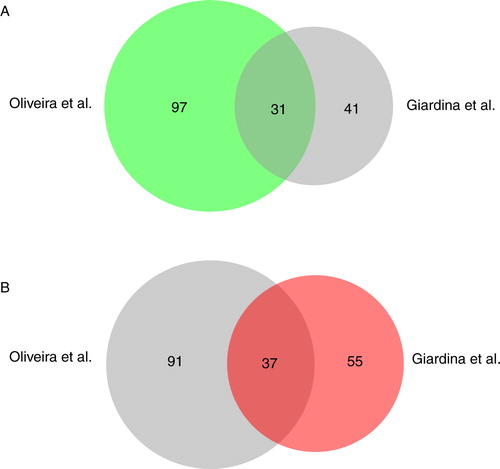
Using a proteomic approach, we have identified 72 extracellular proteins that were present in total extracts of glucose-starved cells. In a previous proteomic study by Oliveira et al., larger vesicles of 50–250 nm were isolated from culture media (Citation40). When we compared the proteins identified by Oliveira et al. with the 72 proteins identified with more than 2 peptides having 95% confidence in this study, 31 proteins overlapped (A). Common proteins included Sse1p, Cpr1p, Gpm1p, Ssb1p, Tdh1p, Adh1p, Tdh3p, Hsp104p, Eft2p, Tsa1p, Ssa1p, Hsc82p, Ssa4p, Ssb2p, Pgk1p, Rnr4p, Pgi1p, Eno2p, Sod2p, Bmh1p, Eno1, Ssa2p, Bmh2p, Tpi1p, Adh2p, Ahp1p, Pdc1p, Cdc19p, Fba1p, Aro8p, and Met6p. When we compared their proteins with the 92 proteins that were identified with one peptide or more having 95% confidence in our study, 37 proteins overlapped (B). One possible explanation for the low number of overlapping proteins is that Oliveira et al. identified more proteins associated with larger vesicles, whereas we identified more proteins associated with small vesicles.
Fig. 5. Comparison of proteins identified by Oliveira et al. and Giardina et al. The BioVenn analysis was used to compare the distribution of proteins identified by Oliveira et al. (Citation40) with the 72 proteins that were identified with more than 2 peptides having 95% confidence by Giardina et al. in this study (A) and the 92 proteins identified with more than 1 peptide having 95% confidence in this study (B).
The extracellular proteins identified in this study included proteins that associated with small vesicles and proteins that showed a rapid END3-dependent decrease in response to glucose addition. Indeed, FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p were all detectable in the vesicle-enriched fraction. Furthermore, membrane integrity is required for the distribution of these proteins in the vesicle-enriched fraction. When glucose was added to glucose-starved wild-type cells, levels of extracellular FBPase, MDH2, Icl1p, Pck1p, GAPDH, and Cpr1p decreased rapidly, while levels remained high in cells lacking the END3 gene. This suggests that these proteins utilize an END3-dependent mechanism to reduce their levels in response to glucose. Taken together, our data indicate that the END3 gene is required for the rapid decline of vesicles and proteins in the extracellular fraction in response to changes in glucose concentrations in the media.
At the present time, the molecular mechanisms for vesicle internalization are largely unknown. We propose that this is a special type of endocytosis. First, it is utilized to transport 30–50 nm vesicles across the plasma membrane. Furthermore, it is a regulated process, as it happens when cells are transferred to high glucose. Moreover, it occurs rapidly, as the decline of proteins/vesicles in the extracellular fraction is almost complete within the first 30 min of glucose addition. Rapid internalization may enable cells to adjust quickly to the changing environment. In the future, it will be desirable to purify the 100–300 nm structures for further characterization. Additionally, it will be important to identify molecules required for the biogenesis and secretion of small vesicles. It is also critical to identify additional genes involved in the internalization of small vesicles.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Supplemental Table 1
Download MS Word (26.6 KB)Acknowledgements
We thank the Mass Spectrometry and Proteomics Facility at Penn State College of Medicine for conducting 2D-LC/MALDI-MS/MS. TEM was performed at the Core Facility of the Penn State University College of Medicine. This work was supported by Penn State Bridge Fund to Hui-Ling Chiang.
Notes
To access the supplementary material to this article, please see Supplementary files under Article Tools online.
References
- Bernal D, Carpena I, Espert AM, De la Rubia JE, Esteban JG, Toledo R et al. Identification of proteins in excretory/secretory extracts of Echinostoma friedi (Trematoda) from chronic and acute infections. Proteomics. 2006; 6: 2835–43.
- Hewitson JP, Harcus YM, Curwen RS, Dowle AA, Atmadja AK, Ashton PD et al. The secretome of the filarial parasite, Brugia malayi: proteomic profile of adult excretory-secretory products. Mol Biochem Parasitol. 2008; 160: 8–21.
- Dupont A, Corseaux D, Dekeyzer O, Drobecq H, Guihot AL, Susen S et al. The proteome and secretome of human arterial smooth muscle cells. Proteomics. 2005; 5: 585–96.
- Dowell JA, Johnson JA, Li L. Identification of astrocyte secreted proteins with a combination of shotgun proteomics and bioinformatics. J Proteome Res. 2009; 8: 4135–43.
- Cass CL, Johnson JR, Califf LL, Xu T, Hernandez HJ, Stadecker MJ et al. Proteomic analysis of Schistosoma mansoni egg secretions. Mol Biochem Parasitol. 2007; 155: 84–93.
- Sotillo J, Valero ML, Sanchez Del Pino MM, Fried B, Esteban JG, Marcilla A et al. Excretory/secretory proteome of the adult stage of Echinostoma caproni. Parasitol Res. 2010; 107: 691–7.
- Schaumburg J, Diekmann O, Hagendorff P, Bergmann S, Rohde M, Hammerschmidt S et al. The cell wall subproteome of Listeria monocytogenes. Proteomics. 2004; 4: 2991–3006.
- Zheng M, Hu K, Liu W, Hu X, Hu F, Huang L et al. Proteomic analysis of excretory secretory products from Clonorchis sinensis adult worms: molecular characterization and serological reactivity of a excretory–secretory antigen-fructose-1,6-bisphosphatase. Parasitol Res. 2011; 109: 737–44.
- Albuquerque PC, Nakayasu ES, Rodrigues ML, Frases S, Casadevall A, Zancope-Oliveira RM et al. Vesicular transport in Histoplasma capsulatum: an effective mechanism for trans-cell wall transfer of proteins and lipids in ascomycetes. Cell Microbiol. 2008; 10: 1695–710.
- Rodrigues ML, Franzen AJ, Nimrichter L, Miranda K. Vesicular mechanisms of traffic of fungal molecules to the extracellular space. Curr Opin Microbiol. 2013; 16: 414–20.
- Rodrigues ML, Nakayasu ES, Almeida IC, Nimrichter L. The impact of proteomics on the understanding of functions and biogenesis of fungal extracellular vesicles. J Proteomics. 2014; 97: 177–86.
- Rodrigues ML, Nakayasu ES, Oliveira DL, Nimrichter L, Nosanchuk JD, Almeida IC et al. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot Cell. 2008; 7: 58–67.
- Rodrigues ML, Nimrichter L, Oliveira DL, Frases S, Miranda K, Zaragoza O et al. Vesicular polysaccharide export in Cryptococcus neoformans is a eukaryotic solution to the problem of fungal trans-cell wall transport. Eukaryot Cell. 2007; 6: 48–59.
- Vallejo MC, Matsuo AL, Ganiko L, Medeiros LC, Miranda K, Silva LS et al. The pathogenic fungus Paracoccidioides brasiliensis exports extracellular vesicles containing highly immunogenic alpha-galactosyl epitopes. Eukaryot Cell. 2011; 10: 343–51.
- Vallejo MC, Nakayasu ES, Longo LV, Ganiko L, Lopes FG, Matsuo AL et al. Lipidomic analysis of extracellular vesicles from the pathogenic phase of Paracoccidioides brasiliensis. PLoS One. 2012; 7: e39463.
- Lamonica JM, Wagner M, Eschenbrenner M, Williams LE, Miller TL, Patra G et al. Comparative secretome analyses of three Bacillus anthracis strains with variant plasmid contents. Infect Immun. 2005; 73: 3646–58.
- Alibhoy AA, Giardina BJ, Dunton DD, Chiang HL. Vps34p is required for the decline of extracellular fructose-1,6-bisphosphatase in the vacuole import and degradation pathway. J Biol Chem. 2012; 287: 33080–93.
- Giardina BJ, Stanley BA, Chiang HL. Comparative proteomic analysis of transition of Saccharomyces cerevisiae from glucose-deficient medium to glucose-rich medium. Proteome Sci. 2012; 10: 40.
- Entian KD, Droll L, Mecke D. Studies on rapid reversible and non-reversible inactivation of fructose-1,6-bisphosphatase and malate dehydrogenase in wild-type and glycolytic block mutants of Saccharomyces cerevisiae. Arch Microbiol. 1983; 134: 187–92.
- Gancedo C. Inactivation of fructose-1,6-diphosphatase by glucose in yeast. J Bacteriol. 1971; 107: 401–5. [PubMed Abstract] [PubMed CentralFull Text].
- Gancedo JM. Yeast carbon catabolite repression. Microbiol Mol Biol Rev. 1998; 62: 334–61. [PubMed Abstract] [PubMed CentralFull Text].
- Gancedo JM. The early steps of glucose signalling in yeast. FEMS Microbiol Rev. 2008; 32: 673–704.
- Gancedo JM, Gancedo C. Inactivation of gluconeogenic enzymes in glycolytic mutants of Saccharomyces cerevisiae. Eur J Biochem. 1979; 101: 455–60.
- Carlson M. Regulation of glucose utilization in yeast. Curr Opin Genet Dev. 1998; 8: 560–4.
- Carlson M. Glucose repression in yeast. Curr Opin Microbiol. 1999; 2: 202–7.
- Gamo FJ, Navas MA, Blazquez MA, Gancedo C, Gancedo JM. Catabolite inactivation of heterologous fructose-1,6-bisphosphatases and fructose-1,6-bisphosphatase-beta-galactosidase fusion proteins in Saccharomyces cerevisiae. Eur J Biochem. 1994; 222: 879–84.
- Minard KI, McAlister-Henn L. Glucose-induced degradation of the MDH2 isozyme of malate dehydrogenase in yeast. J Biol Chem. 1992; 267: 17458–64. [PubMed Abstract].
- Minard KI, McAlister-Henn L. Glucose-induced phosphorylation of the MDH2 isozyme of malate dehydrogenase in Saccharomyces cerevisiae. Arch Biochem Biophys. 1994; 315: 302–9.
- Holzer H. Proteolytic catabolite inactivation in Saccharomyces cerevisiae. Revis Biol Celular. 1989; 21: 305–19. [PubMed Abstract].
- Hung GC, Brown CR, Wolfe AB, Liu J, Chiang HL. Degradation of the gluconeogenic enzymes fructose-1,6-bisphosphatase and malate dehydrogenase is mediated by distinct proteolytic pathways and signaling events. J Biol Chem. 2004; 279: 49138–50.
- Huang PH, Chiang HL. Identification of novel vesicles in the cytosol to vacuole protein degradation pathway. J Cell Biol. 1997; 136: 803–10.
- Brown CR, Dunton D, Chiang HL. The vacuole import and degradation pathway utilizes early steps of endocytosis and actin polymerization to deliver cargo proteins to the vacuole for degradation. J Biol Chem. 2010; 285: 1516–28.
- Giardina BJ, Chiang HL. The key gluconeogenic enzyme fructose-1,6-bisphosphatase is secreted during prolonged glucose starvation and is internalized following glucose re-feeding via the non-classical secretory and internalizing pathways in Saccharomyces cerevisiae. Plant Signal Behav. 2013; 8(8):
- Tang HY, Xu J, Cai M. Pan1p, End3p, and S1a1p, three yeast proteins required for normal cortical actin cytoskeleton organization, associate with each other and play essential roles in cell wall morphogenesis. Mol Cell Biol. 2000; 20: 12–25.
- Engqvist-Goldstein AE, Drubin DG. Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol. 2003; 19: 287–332.
- Galletta BJ, Cooper JA. Actin and endocytosis: mechanisms and phylogeny. Curr Opin Cell Biol. 2009; 21: 20–7.
- Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP et al. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol Cell Proteomics. 2007; 6: 1638–55.
- Tang WH, Shilov IV, Seymour SL. Nonlinear fitting method for determining local false discovery rates from decoy database searches. J Proteome Res. 2008; 7: 3661–7.
- Brown CR, Wolfe AB, Cui D, Chiang HL. The vacuolar import and degradation pathway merges with the endocytic pathway to deliver fructose-1,6-bisphosphatase to the vacuole for degradation. J Biol Chem. 2008; 283: 26116–27.
- Oliveira DL, Nakayasu ES, Joffe LS, Guimaraes AJ, Sobreira TJ, Nosanchuk JD et al. Characterization of yeast extracellular vesicles: evidence for the participation of different pathways of cellular traffic in vesicle biogenesis. PLoS One. 2010; 5: e11113.
- Oliveira DL, Nakayasu ES, Joffe LS, Guimaraes AJ, Sobreira TJ, Nosanchuk JD et al. Biogenesis of extracellular vesicles in yeast: many questions with few answers. Commun Integr Biol. 2010; 3: 533–5.
- Delgado ML, Gil ML, Gozalbo D. Candida albicans TDH3 gene promotes secretion of internal invertase when expressed in Saccharomyces cerevisiae as a glyceraldehyde-3-phosphate dehydrogenase-invertase fusion protein. Yeast. 2003; 20: 713–22.
- Delgado ML, O'Connor JE, Azorin I, Renau-Piqueras J, Gil ML, Gozalbo D. The glyceraldehyde-3-phosphate dehydrogenase polypeptides encoded by the Saccharomyces cerevisiae TDH1, TDH2 and TDH3 genes are also cell wall proteins. Microbiology. 2001; 147: 411–17. [PubMed Abstract].
- Lopez-Ribot JL, Chaffin WL. Members of the Hsp70 family of proteins in the cell wall of Saccharomyces cerevisiae. J Bacteriol. 1996; 178: 4724–6. [PubMed Abstract] [PubMed CentralFull Text].
- Brown CR, Chiang HL. A selective autophagy pathway that degrades gluconeogenic enzymes during catabolite inactivation. Commun Integr Biol. 2009; 2: 177–83. [PubMed Abstract] [PubMed CentralFull Text].
- Brown CR, Cui DY, Hung GG, Chiang HL. Cyclophilin A mediates Vid22p function in the import of fructose-1,6-bisphosphatase into Vid vesicles. J Biol Chem. 2001; 276: 48017–26. [PubMed Abstract].
- Brown CR, Hung GC, Dunton D, Chiang HL. The TOR complex 1 is distributed in endosomes and in retrograde vesicles that form from the vacuole membrane and plays an important role in the vacuole import and degradation pathway. J Biol Chem. 2010; 285: 23359–70.
- Brown CR, Liu J, Hung GC, Carter D, Cui D, Chiang HL. The Vid vesicle to vacuole trafficking event requires components of the SNARE membrane fusion machinery. J Biol Chem. 2003; 278: 25688–99.
- Brown CR, McCann JA, Chiang HL. The heat shock protein Ssa2p is required for import of fructose-1, 6-bisphosphatase into Vid vesicles. J Cell Biol. 2000; 150: 65–76.
- Brown CR, McCann JA, Hung GG, Elco CP, Chiang HL. Vid22p, a novel plasma membrane protein, is required for the fructose-1,6-bisphosphatase degradation pathway. J Cell Sci. 2002; 115: 655–66. [PubMed Abstract].
- Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010; 73: 1907–20.
- Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics. 2009; 9: 4997–5000.
- Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics. 2009; 6: 267–83.
- Lee HS, Jeong J, Lee KJ. Characterization of vesicles secreted from insulinoma NIT-1 cells. J Proteome Res. 2009; 8: 2851–62.