
Abstract
The induction of an active immune response to control or eliminate tumours is still an unfulfilled challenge. We focused on plasmid DNA vaccines using an innovative approach whereby the antigen is expressed in association with extracellular vesicles (EVs) to facilitate antigen cross-presentation and improve induced immunity. Our two groups had independently shown previously that DNA vaccines encoding EV-associated antigens are more efficient at inducing cytotoxic T-cell responses than vaccines encoding the non-EV-associated antigen. Here, we compared our two approaches to associate the ovalbumin (OVA) antigen to EVs: (a) by fusion to the lipid-binding domain C1C2 of MFGE8(=lactadherin), which is exposed on the surface of secreted membrane vesicles; and (b) by fusion to retroviral Gag capsid protein, which is incorporated inside membrane-enclosed virus-like particles. Plasmids encoding either form of modified OVA were used as DNA-based vaccines (i.e. injected into mice to allow in vivo expression of the antigen associated to EVs). We show that both DNA vaccines induced, with similar efficiency, OVA-specific CD8+ T cells and total IgG antibodies. By contrast, each vaccine preferentially stimulated different isotypes of immunoglobulins, and the OVA-C1C2-encoding vaccine favoured antigen-specific CD4+ T lymphocyte induction as compared to the Gag-OVA vaccine. Nevertheless, both OVA-C1C2 and Gag-OVA vaccines efficiently prevented in vivo outgrowth of OVA-expressing tumours and reduced tumour progression when administered to tumour-bearing mice, although with variable efficacies depending on the tumour models. DNA vaccines encoding EV-associated antigens are thus promising immunotherapy tools in cancer but also potentially other diseases.
Antitumour vaccination is a promising means to induce systemic immunity against malignant cells during both early and metastatic cancer (Citation1). Several sources of tumour-specific antigens can be used, such as tumour cell extracts, purified proteins, or synthetic peptides known to bind to MHC class I and/or MHC class II molecules and thus activate CD8+ and/or CD4+ T cells, respectively. However, to induce strong enough immune responses, notably cytotoxic T lymphocyte (CTL) responses, these vaccine antigens must be injected together with adjuvants.
Alternatively, genetic immunization using naked plasmid DNA encoding tumour antigens is attracting increasing interest in tumour immunology due to many potential advantages, especially because no adjuvants are required to induce both T-cell and humoral immune responses (Citation2). Injection of DNA in a tissue leads to local transfection of cells, which thus express the desired antigen in a sustained manner. Local dendritic cells (DCs) either can directly express the antigen after DNA transfection and present it to CD8+ T cells, or can capture the antigen from surrounding transfected muscle or skin cells, for presentation on major histocompatibility complex (MHC) class II to CD4+ T lymphocytes, and cross-presentation on MHC class I to CD8+ T lymphocytes (Citation2, Citation3). The specific mechanisms of antigen uptake and presentation by DCs have been the subject of numerous studies, which are reviewed in Refs. (Citation4, Citation5). Soluble antigens were shown more than a decade ago to be less efficiently presented in vivo to CD4+ and, more strikingly, to CD8+ T cells than their cell-associated counterparts (Citation6). Consistently, it was shown recently that a soluble antigen fed to DCs was only presented on MHC class II molecules, whereas a liposome-encapsulated form directed to early endosomes was presented on both MHC class I and class II (Citation7) and that specific signalling pathways in DCs controlled cross-presentation of particulate but not soluble antigens (Citation8). Thus, to promote both cross-presentation on MHC class I and presentation on MHC class II molecules, especially for tumour vaccination, particulate antigens might be preferentially used.
Membrane-enclosed vesicles, such as exosomes or any type of extracellular vesicles (EVs), represent an interesting source of particulate antigens. Exosomes secreted by tumours have been shown to contain endogenous tumour antigens and to transfer them to DCs for induction of antitumour immune responses (Citation9). Immunization of mice with exosomes purified from antigen-pulsed DCs induced much more efficiently antibody and CD4+ T-cell responses than immunization with the native antigen itself (Citation10). We have shown that tumour cells secreting a model antigen as an EV-associated form induced antitumour immune responses and were controlled by the adaptive immune system, as opposed to the same tumour cells secreting the antigen as a soluble form (Citation11). Thus, inducing secretion of an antigen as an EV-associated form upon DNA vaccination represents a promising strategy for immunotherapy.
We previously validated two strategies that allow antigen secretion in association with EVs. In one approach (Citation11), antigen was fused to the lipid-binding C1C2 domain of milk fat globule – EGF Factor VIII (MFGE8), also called lactadherin, a secreted protein that is highly enriched on mouse DC-derived exosomes (Citation12). This C1C2 domain is homologous to the C-terminal domain of blood coagulation factor V and factor VIII, and binds to phosphatidylserine exposed at the surface of apoptotic cells (Citation13) or DC-derived exosomes (Citation14). As a result, antigens fused to C1C2 and coupled to a signal peptide are secreted on small EVs, including exosomes (Citation11). Consequently, we showed that a DNA vaccine encoding EV-associated ovalbumin (OVA) antigen was more efficient to induce antigen-specific CD8+ T cells in vivo and to protect mice against growth of an OVA-expressing tumour than a DNA vaccine encoding the soluble secreted OVA (Citation11). The C1C2 fusion approach has also been recently used by two other groups, in the context of prostate (Citation15) or breast (Citation16) tumour antigens.
In the second approach, the antigen is carried by recombinant virus-like particles (VLPs). VLPs, composed of one or more structural viral proteins but no genome of native viruses, mimic the organization and conformation of authentic virions but have no capability to replicate in cells, potentially yielding safe vaccine candidates. VLPs have been recently used as a platform for inducing immune responses against heterologous antigens. We have developed recombinant retrovirus-derived VLPs made of Gag from the Moloney murine leukaemia virus (MLV), which induces budding of pseudo-viruses from the plasma membrane (Citation17). Antigens can be inserted onto or into the retroviral VLPs by fusion with the transmembrane domain of vesicular stomatitis virus glycoprotein or with MLV Gag, respectively (Citation18, Citation19). These recombinant VLPs can be produced either ex vivo after cell transfection with plasmid DNA encoding wild-type or chimeric Gag proteins and envelope glycoproteins, or in vivo after injection of the same plasmid DNA. We previously demonstrated that retroVLP-encoding DNA induces higher cellular and humoral immune responses against viral antigens than a control DNA vaccine encoding viral antigens but unable to form VLPs due to a mutation in Gag (Citation18–Citation20). This strategy was initially developed and validated as an antiviral vaccine (e.g. against hepatitis C), but we recently pointed out its usefulness in oncology (Citation21).
Because we had already demonstrated the superiority of these two types of EV-targeted antigens over their corresponding non-EV-targeted version in a DNA vaccination approach (Citation11, Citation18), here we compared side-by-side our two EV-targeting approaches in terms of induction of immune responses and antitumour efficacy, using OVA as a model tumour antigen. Two DNA vaccines encoding either Gag-OVA or OVA-C1C2 fusion proteins were in vivo administered, and we analysed CD4+ T, CD8+ T, and B lymphocyte–induced immune responses, and growth of OVA-expressing tumours, in prophylactic and therapeutic settings using several tumour cell lines. Our results show that the two DNA vaccines encoding an antigen secreted in association with vesicles induce efficient immune responses and antitumour activities, but with differences in the quality of these immune responses, especially in terms of resulting immunoglobulins.
Materials and methods
Plasmids and DNA vaccination
Empty pcDNA3.1/hygro (mock) and pcDNA3-OVA-C1C2 (OVA-C1C2) plasmids have been described previously (Citation11). pGag-OVA (pBL196 Gag-OVA) encodes, under cytomegalovirus promoter, a full-length OVA protein fused with MLV Gag p55 protein in the C-terminal position. The OVA-encoding cDNA (OVAL gene) was amplified by PCR from the chicken OVA cDNA (pcDNA3-OVA) with primers containing Mlu-I and Xba-I restriction sites, digested with Mlu-I and Xba-I, and then inserted by ligation into pBL36 as previously described (Citation21). All DNA plasmids were purified using the Nucleobond PC endotoxin-free kit (Macherey-Nagel) according to the manufacturer's instructions and resuspended in H2O. DNA concentration was measured using a nanodrop (LabTech France).
Cells
HEK293T (CRL-1573; ATCC), MCA-OVA [MCA101 fibrosarcoma secreting soluble OVA (Citation11)], B16F10-OVA (melanoma expressing OVA, kindly provided by K. Rock), and EL4-OVA (EG7 thymoma, ATCC, CRL-2113) cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with β-mercaptoethanol, 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin (all from LifeTechnologies, Cergy Pontoise, France), and 10% heat-inactivated foetal calf serum. Hygromycin or Geneticin® was included in the culture medium of MCA-OVA or B16F10-OVA/EL4-OVA, respectively.
OVA-containing EV purification
HEK-293T cells were transfected using a calcium phosphate transfection protocol, as previously described (Citation21) with plasmids encoding Gag-OVA or OVA-C1C2 or with pcDNA3.1, as control. Supernatants were collected, filtered through 0.45-µm pore-sized membranes, and concentrated with Centricon (Millipore, Molsheim, France). For total EV isolation, supernatants were layered on top of a sucrose step gradient (2.5 mL, 35%; 2.5 mL, 50%) and centrifuged at 100,000g for 2 hours at 4°C. Interface was collected and washed with phosphate-buffered saline (PBS) (Citation21). For separation of different types of EVs (Citation22), supernatants were centrifuged in a SW32.1 rotor (Beckman) at 300g for 5 minutes to eliminate floating cells, at 2,000g for 20 minutes to remove cell debris, and at 10,000g for 40 minutes to separate large vesicles. An ultracentrifugation step at 100,000g for 75 minutes was performed to pellet small EVs, including exosomes and VLPs, and an extra centrifugation at 200,000g for 90 minutes was performed to isolate vesicles smaller than 100 nm and protein aggregates. Each pellet was washed in PBS and centrifuged at the same speed to eliminate contaminant proteins.
Western blot
Ten percent of each pellet was diluted in LDS sample buffer containing sample-reducing agent (LifeTechnologies), boiled for 5 minutes at 95°C, and separated by SDS-PAGE (LifeTechnologies) for 75 minutes at 150 V. Proteins were transferred from gels to a nitrocellulose membrane using the iBlot® Dry Blot System (LifeTechnologies). Membranes were incubated in Western dot blocking buffer (Molecular probes) for 30 minutes. Primary anti-OVA antibody (#ABIN400491; AntibodiesOnline) was incubated in wash buffer (Molecular Probes) overnight at 4°C in a rotating shaker. Secondary anti-rabbit antibodies labelled with biotin (Molecular Probes) were incubated for 60 minutes in wash buffer. Streptavidin coupled to Qdot 655 were incubated for 60 minutes in wash buffer. Membranes were analysed under ultraviolet light camera.
Nanoparticle tracking analysis (NTA)
Suspensions containing purified Gag-OVA or OVA-C1C2 EVs were analysed using a LM10 instrument (NanoSight, UK). Each sample was diluted 1:1,000, and 5 measurements (video of 60 seconds) were done with a frame rate of 30 frames/s; the camera gain and shutter were set at 250 and 11.23 ms, respectively. Particle movement was analysed by NTA software (NanoSight) with a single detection threshold for all samples; minimal track length and minimal expected particle size were set at automatic level and 30 nm, respectively.
OVA quantification in the EVs
Anti-OVA polyclonal rabbit antibody (#ABIN400491; Antibodiesonline) was diluted in carbonate–bicarbonate buffer at 1:2,000, coated overnight at 4°C on MediSorp 96-well plates (ThermoFischer), and washed with PBS-0.05% Tween 20. Plates were incubated during 1 hour with PBS-5% bovine serum albumin, and samples were added for 2 hours at room temperature after washing. Samples were either supernatants collected from 0.5×106 plated cells 2 days after transfection with Gag-OVA, OVA-C1C2, or mock plasmids and treated with Triton-X100 detergent; or the pellets obtained after EV purification, treated or not with Triton-X100. Bound OVA was revealed by polyclonal mouse anti-OVA (#ABIN316446, at 1:2,000), followed by HRP-conjugated anti-mouse antibodies (Jackson Immunoresearch, 115-035-166) revealed by TMB-substrate solution (BD OptEIA). Reaction was stopped with 1 N HCl, and absorbance was read at 450 nm.
Mice and vaccination
Five or six-week-old female C57BL/6J mice were purchased from Charles River France, and C57BL/6 Rag2 −/− mice were bred at the Institut Curie animal facility. The care and use of animals used here strictly apply the European and National Regulation for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes in force (facility licence #C75-05-18). It complies also with internationally established principles of replacement, reduction, and refinement in accordance with Guide for the Care and Use of Laboratory Animals (NRC 2011). Mice were anesthetized with ketamine (100 mg/kg) and xylazine (7.25 mg/kg), and plasmid DNA (5 or 30 µg) was administered intramuscularly (i.m.) in one hind leg or intradermally (i.d.) at 2 sites on the lower back in 50 µl of saline buffer using a 500 µl insulin needle (Terumo). DNA injection was followed by electroporation applied with a generator (BTX, ECM30) with 20 ms/pulse, 8 pulses at 200 V/cm using specific electrodes: CUY647 or CUY650 (Sonidel Limited, Ireland) for i.m. or i.d. immunization, respectively. Conductive gel was used to ensure electrical contact between the electrodes and the skin.
Quantification of CD4+ and CD8+ T-cell responses
Ten to twelve days after immunization, blood samples were collected by retro-orbital puncture. Total peripheral blood mononuclear cells (PBMC) were stained with PE-conjugated H-2Kb–SIINFEKL tetramer (Beckman Coulter), anti-CD8 and anti-T cell receptor (TCR) antibodies (BD Biosciences), followed by red blood cell lysis to quantify OVA-specific CD8+ T cells. Cells were analysed using a standard LSR-II flow cytometer (BD Biosciences), and the fluorescence-activated cell scanner (FACS) data were analysed using FlowJo software. The tetramer+ cells were gated on TCR+ CD8+ cells. Interferon (IFN)γ-producing OVA-specific CD4+ or CD8+ T cells were measured by ELISPOT on PBMC after red blood cell lysis. Briefly, microplates (Multiscreen HTS IP, Millipore) were coated with anti-murine IFNγ antibody (Diaclone). PBMC (0.2×106/well) were cultured overnight in the presence of either control medium or the 257–264 (SIINFEKL) class I–restricted OVA peptide (10 µM) or the 265–280 (TEWTSSNVMEERKIKV) class II–restricted OVA peptide (40 µM) (Polypeptide Group, Strasbourg, France) in complete medium (RPMI-Glutamax, 10% foetal calf serum, antibiotics, and β-mercaptoethanol). The detection was performed with biotinylated anti-IFNγ (matched pairs, Diaclone) followed by streptavidin–alkaline phosphatase (Mabtech) and revealed using the appropriate substrate (Biorad). Spots were counted using an ELISPOT Reader System ELR02 (AID, Germany), and results were expressed as the number of cytokine-producing cells per 1×106 PBMC.
In vivo cytotoxicity assay
CD45.1+ carboxyfluorescein succinimidyl ester (CFSE) high (CFSEhi) peptide-pulsed (5 µg/mL SIINFEKL peptide for 60 minutes at 37°C) C57BL/6 splenocytes (target cells) and CFSE low (CFSElo) unpulsed splenocytes (control cells), previously labelled with respectively 5 or 0.5 µM CFSE (LifeTechnologies) for 15 minutes at 37°C, were mixed at a 1:1 ratio, and 1×107 total cells were injected intravenously (i.v.) into CD45.2+ mice immunized 10 days before with 10 µg of DNA (i.m. or i.d.). Five hours later, splenocytes from each mouse were analysed by FACS to detect the presence of CD45.1+ cells. Antigen-specific CTL lytic activity was measured by the disappearance of peptide-pulsed targets, and expressed as clearance efficiency calculated as follows: {1−[% antigen-pulsed (CFSEhi) cells/% antigen-unpulsed (CFSElo) cells]}×100%.
Quantification of OVA-specific antibody responses
Twelve days after immunization, sera were collected by retro-orbital puncture and OVA-specific immunoglobulins were measured by standard ELISA. Briefly, Maxisorp 96-well plates were coated at 4°C with OVA (10 µg/mL) in carbonate–bicarbonate buffer. After blocking with PBS–5% milk for 2 hours, serially diluted sera were added for 2 hours at room temperature. After extensive washing, alkaline phosphatase-conjugated anti-mouse IgG, IgG1, or IgG2b (Jackson ImmunoResearch) was added to each well, and plates were incubated 1 hour at room temperature. After extensive washing, alkaline phosphatase activity was measured adding the CDP-star® Ready-to-Use substrate (Applied Biosystems). The microplates were read using a Centro LB 960 luminometer (Berthold), and sample sera were compared to a positive standard curve to express the results in arbitrary units (AU).
In vivo tumour assays
0.5×106 MCA-OVA or B16F10-OVA cells, or 1×106 EL4-OVA cells, were administered subcutaneously (s.c.) into the shaved flank of the mice. Tumour growth was measured twice a week using a caliper to determine the tumour size, calculated as length×width×[(length+width)/2]. Mice were sacrificed when the tumour reached 2 to 2.5 cm3, and the sacrifice day was recorded to plot survival of the mice. For the lung tumour model, 0.5×106 MCA-OVA or 0.2×106 B16F10-OVA cells were injected i.v. Mice were sacrificed 20 days later, and tumour burden was assessed by counting lung foci. The MCA-OVA lung nodules were counted after ink injection. For tumour prevention experiments, mice were immunized with DNA vaccines as indicated in the figures, and tumour cells were injected s.c. 10 days later. For tumour therapeutic settings, tumour cells were injected s.c. or i.v., and mice received DNA vaccines 3 or 6 days later.
Statistical analyses
For all experiments, normality of distribution of the samples and similarity of dispersions of the groups were tested. In most cases, these criteria were not reached, hence non-parametric statistical tests were used as follows: for experiments comparing a single measure of more than 2 experimental groups, Kruskal–Wallis followed by Dunn post-hoc test; for tumour growth measurement over time, one-way ANOVA with repeated measures followed by Tukey's post-hoc test; and, for survival, a log-rank test was performed.
Results
Characterization of OVA-associated EVs generated from the two DNA vaccines
We developed two different vaccine strategies whereby the antigen is expressed in association with EVs. First, OVA was fused in-frame in the C-terminal part of the MLV Gag polypeptide precursor (p55) and thus incorporated into the pseudo-particles produced after self-assembly and budding of Gag-OVA from the plasma membrane (A). Second, OVA was inserted between the mouse MFGE8-derived signal sequence and lipid-binding C1C2 domain, resulting in an antigen exposed on the surface of secreted membrane vesicles (OVA-C1C2; A). Therefore, fusion of OVA antigen to either Gag or MFGE8 domains allows the secretion of the antigen associated in a topologically different manner to EVs, predicted inside the vesicles secreted by Gag-OVA-expressing cells, but at the surface of vesicles from OVA-C1C2-expressing cells. To confirm this predicted topology, we quantified OVA by ELISA in the conditioned medium of transfected HEK293T cells, and in EVs recovered by differential ultracentrifugation from this conditioned medium, after or not, disruption of membranes by detergent. From the two plasmids, OVA was as efficiently expressed as shown by a similar overall level of OVA detected in the conditioned medium treated with detergent (B, supernatant). Similar levels of OVA were also quantified in vesicles recovered by 100,000g ultracentrifugation in the presence of Triton-X100 (B, Pellet+Det). By contrast, OVA was efficiently detected by OVA-specific antibodies in intact vesicles secreted by OVA-C1C2-transfected cells, whereas it was hardly detectable when Gag-OVA-derived vesicles were kept intact (B, Pellet), thus confirming that antigen is inside these Gag-OVA EVs, whereas antigen is outside the OVA-C1C2 EVs.
Fig. 1. Characterization of the vesicles with associated antigen generated from the two constructs. (A) Schematic representation of the expected topology of Gag-OVA– and OVA-C1C2-containing vesicles. Recombinant retroviral VLPs (top) are formed by MLV Gag capsid proteins fused with OVA antigen. Secreted vesicles (bottom) carry on their surface OVA antigens fused to the lipid-binding domains (C1C2) of MFGE8. (B) Quantification by ELISA of OVA secretion by HEK cells upon transfection with mock (non-OVA-encoding), Gag-OVA, and OVA-C1C2 plasmids. Total conditioned medium in the presence of detergent (supernatant), the pellet obtained after 100,000g ultracentrifugation, and the same pellet in the presence of detergent (Pellet+Det.) were compared. Mean±SD of 4 independent experiments are shown. (C) Western blot analysis of OVA in Gag-OVA- or OVA-C1C2-expressing cells and their supernatant after pelleting into a sucrose gradient (density, left blot) or after differential centrifugation (Diff. Ultra.: pellets obtained at 2,000g, 10,000g, 100,000g, and 200,000g). Arrows indicate the four main forms of OVA detected in the different samples. (D) Particle size distribution profiles of Gag-OVA and OVA-C1C2 vesicles measured by nanoparticle tracking analysis. Supernatant of HEK cells transfected with plasmids encoding Gag-OVA or OVA-C1C2 was submitted to ultracentrifugation at 100,000g, and pellets were analysed by nanoparticle tracking (distribution of 5 videos per sample is shown).
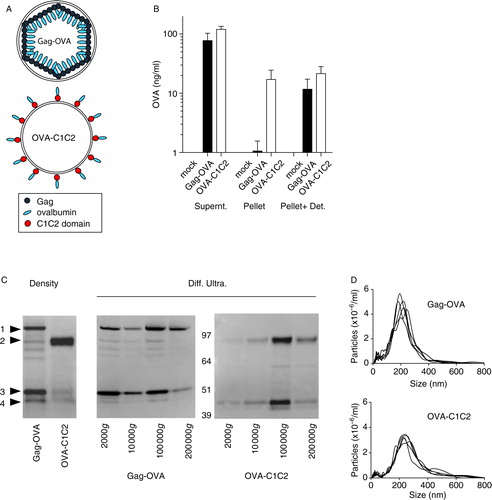
To characterize more precisely the type of EVs produced after expression of Gag-OVA or OVA-C1C2 DNA vaccines, we performed Western blot analysis of transfected cell supernatants purified by different methods. When concentrated conditioned medium was ultracentrifuged into a gradient of sucrose solutions with different concentrations, both Gag-OVA and OVA-C1C2 were detected at the interface between 35 and 50% sucrose (C, left panel), that is, in vesicles of 1.15–1.20g/mL density, classically described for EVs (Citation23). Interestingly, OVA was associated with a large range of EVs produced from Gag-OVA-expressing cells, as shown by the presence of OVA in all the pellets collected upon differential ultracentrifugation (2,000g, 10,000g, 100,000g, and 200,000g; C, right panel). Both full-length and cleaved Gag-OVA-containing fragments were observed (bands 1 and 3 in C). By contrast, in OVA-C1C2-transfected cell supernatants, OVA was recovered mainly in the 100,000g pellet (C, right panel). Again, both OVA-C1C2 and OVA-containing fragments (respectively, bands 2 and 4 in C) were detected. Interestingly, we observed that Gag-OVA and OVA-C1C2 vesicles of the 100,000g pellets were of similar and homogeneous diameters of 180–220 nm, as measured by nanoparticle tracking analysis (D), thus slightly bigger than the expected diameters of 80–100 nm and 80–120 nm, respectively. This observation is probably due to analysis of EVs and VLPs in their native conformation by NTA, rather than after fixation and dehydration as classically done by electron microscopy. Thus, both OVA fusion proteins are secreted in association with EVs, but they display different topologies, and Gag-OVA appears to be associated with more subtypes of vesicles.
CD8+ and CD4+ T-cell responses induced by the two DNA vaccines
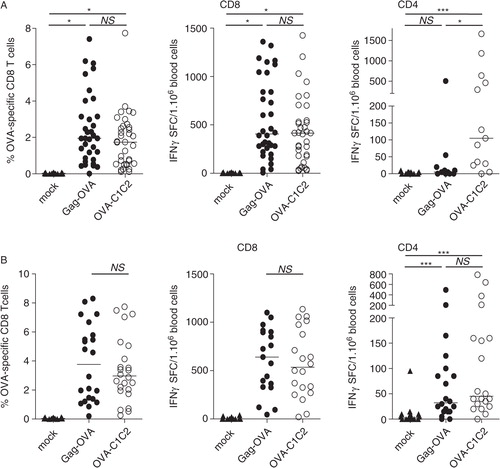
The two plasmids were used as vaccines in mice resulting in in vivo secretion of OVA in an EV-associated form. Electroporation (i.e. a mild local electric shock administered immediately after DNA injection) was used to improve efficacy of in vivo transfection (Citation24). The presence of OVA-specific T lymphocytes was quantified in the blood, 10 days after a single vaccination with 5 µg (A) or 30 µg (B) of DNA, by flow cytometry. The percentage of CD8+ T cells binding H-2Kb–SIINFEKL tetramers (, left panels) and the number of IFNγ spot-forming cells after restimulation with MHC-I or MHC-II binding peptides (, middle and right panels, respectively) were measured. We observed that Gag-OVA and OVA-C1C2 DNA vaccines induced OVA-specific CD8+ T lymphocyte expansion with identical efficacy whatever the dose used for vaccination (A and B, left panels). Interestingly, CD8+ T-cell responses were also induced at similar levels when mice were vaccinated with Gag-OVA or OVA-C1C2 by i.d. DNA injection (Supplementary Fig. 1A). These OVA-specific CD8+ T cells were similarly functional because they produced IFNγ with the same frequency in response to the specific antigenic peptide after i.m. vaccination (A–B, centre panels) or lysed in vivo with the same efficacy peptide-loaded target cells after i.d. or i.m. vaccination (Supplementary Fig. 1B). Induction of OVA-specific CD4+ T lymphocytes was also equally efficient upon vaccination with 30 µg of Gag-OVA or OVA-C1C2 (B, right panel). However, when a lower dose (5 µg) of DNA vaccine was injected, OVA-C1C2 induced significantly higher numbers of OVA-specific CD4+ T cells as compared to both mock or Gag-OVA vaccination (A, right panel).
Fig. 2. CD8+ and CD4+ T cell responses induced by the two DNA vaccines. Mice were immunized once with either 5 µg (A) or 30 µg (B) of DNA coding for mock, Gag-OVA, or OVA-C1C2 plasmids. Immune responses were ex vivo analysed on blood cells at day 10–12 after immunization. Frequency of OVA-specific CD8+ T cells was measured using the H-2Kb–SIINFEKL tetramer (left panels), and the number of OVA-specific IFNγ-producing CD8+ or CD4+ T cells was determined by ELISPOT after 18 hours of stimulation with the appropriate peptide (middle and right panels). Each dot represents an individual mouse, and lines indicate the median. Pooled data from 3 or 4 independent experiments are shown. *p<0.05; ***p<0.001; NS=not significantly different (Kruskal–Wallis with Dunn post-hoc test).
Antibody responses induced by the two DNA vaccines
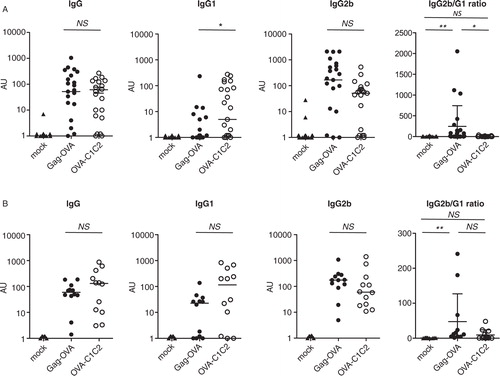
Because a difference in helper CD4+ T cell responses was observed after vaccination with the two plasmids, we then asked if antibody responses, most of which are dependent on helper T cells, were also different. When quantifying total OVA-specific IgG in blood at day 12, we observed efficient and comparable responses induced by both i.m. vaccines, whatever the amount of DNA used (A and B, left panels). However, isotypes of IgGs induced by the two vaccines were different. OVA-C1C2 induced significantly higher levels of specific IgG1 antibody as compared to Gag-OVA (at 5 µg/mouse), whereas levels of OVA-specific IgG2b antibodies were slightly higher in the Gag-OVA group at the low and high doses of vaccines, even if the difference was not statistically significant (A and B, two middle panels). The ratio of OVA-specific IgG2b to IgG1 antibodies, commonly used as an indicator of CD4+ helper T-cell bias toward either Th1 or Th2, was significantly higher after low-dose Gag-OVA than OVA-C1C2 vaccination (A, right panel), suggesting that Gag-OVA may bias more strongly immune responses toward Th1. In contrast, OVA-C1C2, which induced higher levels of IgG1 than Gag-OVA when a low dose of DNA was used (A), may induce a mixture of Th1- and Th2-type helper cells. However, these differences are not significant when high doses of DNA are used (B). Altogether, these results thus show dose-dependent specific immune properties of the two vaccines. Interestingly, the same differences in the ratio of IgG2b to IgG1 isotypes induced after Gag-OVA or OVA-C1C2 were obtained after vaccination by i.d. route (Supplementary Fig. 1C).
Fig. 3. Different types of antibody isotypes are produced in response to the two DNA vaccines. Mice were immunized once with either 5 µg (A) or 30 µg (B) of plasmids coding for mock, Gag-OVA, or OVA-C1C2. OVA-specific total IgG, IgG1, and IgG2b were measured by ELISA in sera at day 12 after immunization. Arbitrary units for individual mice and median are represented. The ratio between IG2b and IgG1 isotypes was calculated and shown on the right panels. Pooled data from 3 (A) or 2 (B) independent experiments are shown. *p<0.05; NS=non-significant (Kruskal–Wallis with Dunn post-hoc test).
Prevention of tumour outgrowth by the two DNA vaccines
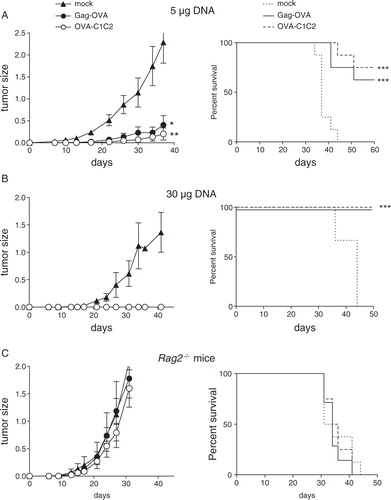
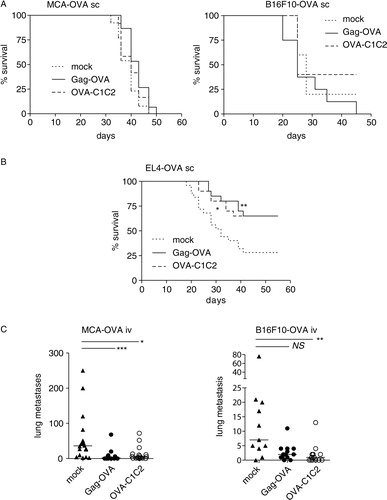
We then asked if adaptive immune responses induced by the two vaccines could affect the tumour growth. The C57BL/6-derived MCA101 fibrosarcoma secreting OVA (MCA-OVA) was used, as it is known to be poorly immunogenic (Citation11, Citation25). Mice were i.m. vaccinated with either mock or EV-associated OVA antigen encoding plasmids, 10 days before s.c. graft of the MCA-OVA tumour cells. In preventive settings, we observed that a low dose (5 µg) of either Gag-OVA or OVA-C1C2 plasmid was sufficient to strongly reduce subsequent growth of the tumour and extended the survival of tumour-bearing mice (A), whereas a high dose of DNA fully abolished tumour growth and fully protected mice (B). Importantly, this antitumour effect was not observed after vaccination in Rag2 −/− mice, which are devoid of functional T and B cells (C), demonstrating that the antitumour activity observed in A and B is due to specific adaptive immune responses generated by the vaccines.
Fig. 4. Prevention of tumour outgrowth by the two DNA vaccines. WT C57BL/6 (A, B) or immunodeficient Rag2 −/− C57BL/6 mice (C) were vaccinated once with Gag-OVA or OVA-C1C2 plasmid DNA at the dose of 5 µg (A) or 30 µg (B, C); and 10 days later, MCA-OVA tumour cells were grafted. Control mice were injected with the mock DNA. Results from the same experiment are shown as tumour size (left panel, mean±SD of 8 mice) and survival percentage (right panel). (A, B) are representative of 3 experiments, and (C) of 2 experiments. *p<0.05; **p<0.01 for tumour size (one-way ANOVA with repeated measures and Tukey post-hoc test); ***p=0.0005 for Gag-OVA; ***p=0.0002 for OVA-C1C2 for mice survival (log rank test).
Antitumour efficacy of the two DNA vaccines administered in therapeutic settings
Next, we sought to examine the vaccines’ potency in more relevant therapeutic models, through the treatment of mice that have already been inoculated with tumours. C57BL/6 mice grafted s.c. with OVA-expressing tumours were i.m. vaccinated with Gag-OVA, OVA-C1C2, or control plasmids. Different tumour cell lines displaying different levels of MHC class I molecules and different levels of aggressiveness were tested. Although both vaccines efficiently prevented MCA-OVA growth in a prophylactic setting (B), they did not impair progression of the already growing tumour implanted s.c. (A, left panel). Similar results were observed with B16F10-OVA tumours (A, right panel). By contrast, when a more immunogenic tumour was used (the OVA-expressing EL4 thymoma), both vaccines efficiently delayed tumour growth and even prevented the tumour growth in about 60% of surviving animals (B). For the two tumours that were not controlled by vaccination when growing s.c., we next investigated whether the EV-associated OVA-encoding vaccines affected their ability to form pseudo-metastases. Mice were injected i.v. with OVA-expressing MCA or B16F10 tumour cells, vaccinated or not 3 days later, and tumour nodules were monitored in lungs at day 20 (C and Supplementary Fig. 2). In these conditions, vaccination with the Gag-OVA plasmid efficiently reduced development of metastases in the MCA-OVA tumour model as compared to the mock vaccine, and vaccination with OVA-C1C2 significantly reduced metastases in both tumour models (C).
Fig. 5. Antitumour efficacy of the two DNA vaccines administered in therapeutic settings. Mice were subcutaneously grafted with (A) MCA-OVA (left panel) or B16F10-OVA (right panel) or (B) EL4-OVA tumour cells and immunized 3 days (MCA-OVA and EL4-OVA) or 6 days (B16F10-OVA) later with 30 µg of mock, Gag-OVA, or OVA-C1C2 plasmids. Results are shown as survival percentage. *p<0.05; **p=0.01 (log rank test). Pooled data from 2 (MCA-OVA), 1 (B16F10-OVA), or 3 (EL4-OVA) independent experiments are shown (n=20–25 per group). (C) MCA-OVA or B16F10-OVA tumour cell lines were injected intravenously, and mice were immunized 3 days later with 30 µg of mock, Gag-OVA, or OVA-C1C2 plasmids. After 20 days, tumour nodules in lungs were counted. Quantification of the lung nodules in individual mice is represented. Pooled data from 2 (B16F10-OVA) or 3 (MCA-OVA) independent experiments are shown, and horizontal bars are the medians. *p<0.05; **p<0.01; ***p<0.001; NS=not significant (Kruskal–Wallis followed by Dunn post-hoc test).
Discussion
Among the variety of vaccine formulations, immunization with plasmid DNA has several advantages compared with other approaches: DNA plasmids are easy to produce in large amounts and in clinical-grade conditions, safe, stable upon storage at various temperatures, ready to deliver, and molecularly defined. However, translation of DNA vaccines into the clinical setting has been hampered by their limited immunogenic potency in humans, especially due to the difficulty to scale up from mice to humans the injected volume for the i.m. route. In recent years, however, the development of electroporation devices designed to increase the efficacy of in vivo transfection of tissues by injected DNA (Citation26, Citation27) and authorization of these devices for clinical use in humans (Citation28, Citation29) have renewed the clinical interest in this approach, and led to several on-going clinical trials for both cancer and infectious diseases (Citation24, Citation30).
Immunogenicity of DNA vaccines can also be improved by controlling the nature of the expressed antigens. Our two groups had previously independently shown that an EV- (Citation11) or VLP-associated antigen (Citation18) induced more efficient CD8+ T-cell immune responses than the non-EV-associated counterpart, in the context of DNA vaccination. We thus compared side-by-side here these two strategies associating the same model antigen to different types of vesicles. Our results show that both induce, with similar efficacy, antigen-specific CD8+ CTLs and antibodies, as well as antitumour activity. Interestingly, however, the CD4+ helper T-cell immune responses induced by our two vaccines are different: the EV-associated antigen (OVA-C1C2) promotes efficiently CD4+ T-cell activation and induces a balanced mix of IgG1 and IgG2b, whereas the VLP-associated antigen (Gag-OVA) induces lower amounts of CD4+ T cells, and more IgG2b than IgG1 antibodies. This antibody pattern suggests that the Gag-OVA vaccine induces CD4+ Th1-polarized responses (known to promote class switching to IgG2b in B cells), whereas OVA-C1C2 induces Th1 but also Th2 immune responses (which direct the B-cell class switch to IgG1). We could not, however, directly demonstrate a specific CD4+ Th cell orientation induced by the two vaccines, because IFNγ was the only cytokine we could detect by cytokine-secretion assay (data not shown) and by ELISPOT (); all other tested cytokines (IL-4, IL-5, IL-13, IL-10, IL-2, and TNF-α) secreted by T cells were below the detection limit, as measured by cytokine secretion assay in in vitro antigen-restimulated PBMC (data not shown). Our results are consistent with previously published results of Qazi et al. (Citation10) showing induction of both IgG1 and IgG2a upon vaccination of mice with exosomes secreted by OVA-fed dendritic cells, whereas non-exosome-associated OVA induced mostly IgG1 (i.e. Th2 responses). The OVA-C1C2 plasmid used here induces similar mixed Th1 and Th2 helper responses as OVA-loaded exosomes (Citation10), whereas the Gag-OVA plasmid biases these responses more strongly toward Th1.
Our results thus highlight a difference in presentation of the OVA antigen to CD4+ T cells and B lymphocytes, but no difference in MHC class I–restricted presentation to CD8+ T cells, upon vaccination with our two different forms of OVA. The reasons for these differences are beyond the purpose of the present study. We can, however, propose some hypotheses worth exploring in future work, based on previously reported or here observed structural differences of the EVs generated by the two DNA vaccines.
First, as shown in , OVA antigen fused with the C1C2 domain of MFGE8 is exposed onto secreted EVs, whereas when fused with the retroviral Gag capsid protein, it is incorporated inside VLPs. Therefore, secreted EVs with exposed OVA-C1C2 can bind directly to the B-cell receptor of OVA-specific B cells, thus allowing formation of MHC class II–OVA peptide complexes in the B cells and proper stimulation by helper CD4+ T cells. In contrast, the intra-VLP Gag-OVA must be released from its envelope to be recognized by B lymphocytes, and efficiently allow activation by helper CD4+ T cells and consequent IgG production. Such release could take place upon necrosis of the Gag-OVA-expressing cells in the vaccinated tissue.
Endogenous EVs and Gag-induced VLPs also differ by other characteristics. In particular, we observed (C) that, upon transient in vitro transfection, Gag-OVA is secreted in a wide range of vesicles, including large EVs pelleting at low speed, which may contain apoptotic cell-derived materials (Citation31), whereas OVA-C1C2 is not detected in this low-speed pellet. The different types of EVs and VLPs released upon Gag-OVA transfection may have opposite effects on the immune responses, and the final outcome would thus depend on the relative level of each type of EV available in vivo, which is difficult to quantify. These different EVs, however, were observed in vitro and, due to technical reasons, could not be confirmed for in vivo–produced vesicles induced by the DNA vaccines. We cannot exclude that EVs produced after in vivo Gag-OVA or OVA-C1C2 plasmid injection in muscle are not identical to those obtained in vitro, especially because the producing cells (i.e. myoblasts in vivo versus kidney-derived HEK293T cells in vitro) are different and may use different EV secretion pathways. But we can still speculate on the possible outcomes in terms of immune responses of secretion of an antigen associated with different EVs.
Presentation to CD4+ T cells of the plasmid-encoded antigen on MHC class II molecules requires capture of the antigen by antigen presenting cells (APCs), especially dendritic cells and macrophages. The way that large membrane vesicles or apoptotic cells and smaller EVs interact with these phagocytes is different, in part because of size differences: large particles are captured by phagocytosis, whereas small particles are endocytosed. Additionally, particle size determines the mechanism of trafficking to the draining lymph node, and only small particles can specifically target LN-resident cells (Citation32). In contrast, larger EVs will be more efficiently captured locally by highly phagocytic macrophages and/or neutrophils than by DCs. Thus, the large OVA-containing VLPs induced after vaccination with Gag-OVA could be captured by macrophages and/or neutrophils, in parallel with capture of small EVs and VLPs by DCs in the draining lymph node, possibly leading to different outcomes in terms of antigen presentation and induction of innate immune signalling.
In addition, Gag-OVA– and OVA-C1C2-generated EVs may display differences in surface ligands, and thus be differently targeted to DCs and macrophages. It is possible, for instance, that OVA-C1C2 competes with endogenous MFGE8 at the surface of EVs. MFGE8 is a known opsonin allowing phagocytosis of apoptotic cells by αvβ3– and αvβ5-expressing cells such as macrophages, to which it binds by its RGD-containing EGF domain (Citation13). Even though mouse DC exosomes are coated with MFGE8, we showed that it is not required for their capture by DCs in vitro (Citation14). Thus, if the EVs bearing OVA-C1C2 do not bear MFGE8, their capture by macrophages could be impaired, leaving intact their capture by DCs, whereas the Gag-OVA VLPs should be captured by both APCs. Therefore, careful examination of the kinetics and nature of cells capturing OVA-containing EVs or VLPs after vaccination will be required to fully understand the reasons for differential MHC class II–restricted presentation. Fluorescent EVs or VLPs (Citation21) have been developed and could be used to characterize the specific priming mechanisms.
Interestingly, while the types of EVs generated by the two vaccines are different in terms of structure and antigen position, both strategies induced similar levels of OVA-specific CD8+ T-cell immune responses. For presentation of an exogenous antigen on MHC class I to CD8+ T cells, and initiation of immune responses, the antigen must be internalized by DCs, the only APCs able to efficiently perform cross-presentation and cross-priming to naïve T cells (Citation5). Acquisition of antigen by DCs in vivo after DNA injection can occur via phagocytosis of dead cells or fragments of locally transfected fibroblasts or muscle cells. This pathway will also take place for DNA vaccines encoding soluble or non-secreted antigens, and will thus allow induction of the CD8+ responses observed with such vaccines (Citation11, Citation18) (Citation21). In addition, secretion of VLP- or EV-associated antigens by our modified vaccines further enhances these responses (Citation11, Citation18) (Citation21). Both VLPs and EVs have been shown to be internalized in endocytic compartments by DCs and induced efficient CD8+ T-cell responses. We previously demonstrated that tumour cells secrete exosomes carrying tumour antigens, which, after transfer to DCs, mediate CD8+ T cell–dependent antitumour effects (Citation9). Similarly, we observed in two different models that recombinant retroviral VLPs can be easily captured by DCs that cross-present the antigen fused to Gag to prime CD8+ T cells (Citation19, Citation21). Here, we observed comparable levels of CD8+ T-cell immune responses induced after Gag-OVA or OVA-C1C2 vaccination (), demonstrating that antigen localization inside or at the surface of EVs has no impact on the CTL response induction. Moreover, because the amount of antigens carried by the two types of EVs produced from transfected cells is quite similar (B), we can conclude that the two types of vesicles provide a source of antigen that can be cross-presented by DCs with the same efficacy. Our results are in line with those of others demonstrating that HIV Nef viral antigens incorporated in exosomes were cross-presented at similar levels to what was observed when the antigens were delivered by engineered lentiviral VLPs (Citation33). Simultaneously, direct presentation of OVA on their MHC class I molecules by DCs locally transfected by the plasmid in vivo could also take place. In this case, secretion of the OVA-containing EVs or VLPs would not be involved, mimicking the conditions of DNA vaccines that encode non-particulate antigens, and also accounting for the CD8+ T-cell responses observed with such vaccines (Citation11, Citation18) (Citation23).
Finally, we observed that both VLP- and secreted vesicle-bound antigen encoding DNA vaccines efficiently control in vivo outgrowth of tumours in preventive setting, and could reduce tumour progression or metastasis in therapeutic situations, with similar efficacy. Considering their specific immune properties and their equal capacity to induce antigen-specific CD8+ T-cell immune responses, our results highlight the major role of CD8+ T cells in antitumour immunity, although complementary experiments should be performed to demonstrate it formally. Antitumoral efficacy of these vaccines was evaluated here in classical models based on transplantable tumour cell lines expressing model antigen; alternative mouse models, using spontaneous or carcinogen-induced tumours, should be also developed to appreciate more precisely the efficacy of our vaccines. Nonetheless, our work highlights the interest of DNA vaccines coding for a vesicle-associated antigen in oncology and suggests that, for other applications where a specific type of CD4+ helper T-cell response must be favoured (e.g. Th2 in parasitic infection and Th1 in viral infection), one or the other of the two strategies described here should be preferentially selected.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or any for-profit organism to conduct this study.
Supplementary Figures 1-2
Download PDF (687.7 KB)Acknowledgements
This work was supported by Institut Curie, INSERM, CNRS, Université Pierre et Marie Curie, and a grant from Institut National du Cancer (2008-PL-BIO, DNAVaccVesic) to CT, BB, and OL. OL's team is “équipe labéllisée de la Ligue contre le Cancer”. We thank Claude Leclerc for useful scientific discussions. We acknowledge the staff of the Animal Facility from Institut Curie for animal husbandry and care.
Related Research Data
References
- Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003; 3: 630–41. [PubMed Abstract].
- Rice J, Ottensmeier CH, Stevenson FK. DNA vaccines: precision tools for activating effective immunity against cancer. Nat Rev Cancer. 2008; 8: 108–20. [PubMed Abstract].
- Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011; 239: 62–84. [PubMed Abstract].
- Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002; 20: 621–67. [PubMed Abstract].
- Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012; 12: 557–69. [PubMed Abstract].
- Li M, Davey GM, Sutherland RM, Kurts C, Lew AM, Hirst C et al. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J Immunol. 2001; 166: 6099–103. [PubMed Abstract].
- Belizaire R, Unanue ER. Targeting proteins to distinct subcellular compartments reveals unique requirements for MHC class I and II presentation. Proc Natl Acad Sci U S A. 2009; 106: 17463–8. [PubMed Abstract] [PubMed CentralFull Text].
- Graham DB, Stephenson LM, Lam SK, Brim K, Lee HM, Bautista J et al. An ITAM-signaling pathway controls cross-presentation of particulate but not soluble antigens in dendritic cells. J Exp Med. 2007; 204: 2889–97. [PubMed Abstract] [PubMed CentralFull Text].
- Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001; 7: 297–303. [PubMed Abstract].
- Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood. 2009; 113: 2673–83. [PubMed Abstract].
- Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008; 68: 1228–35. [PubMed Abstract].
- Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P et al. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999; 147: 599–610. [PubMed Abstract] [PubMed CentralFull Text].
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002; 417: 182–7. [PubMed Abstract].
- Veron P, Segura E, Sugano G, Amigorena S, Thery C. Accumulation of MFG-E8/lactadherin on exosomes from immature dendritic cells. Blood Cells Mol Dis. 2005; 35: 81–8. [PubMed Abstract].
- Rountree RB, Mandl SJ, Nachtwey JM, Dalpozzo K, Do L, Lombardo JR et al. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011; 71: 5235–44. [PubMed Abstract].
- Hartman ZC, Wei J, Glass OK, Guo H, Lei G, Yang XY et al. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011; 29: 9361–7. [PubMed Abstract] [PubMed CentralFull Text].
- Bellier B, Klatzmann D. Virus-like particle-based vaccines against hepatitis C virus infection. Expert Rev Vaccines. 2013; 12: 143–54. [PubMed Abstract].
- Bellier B, Dalba C, Clerc B, Desjardins D, Drury R, Cosset FL et al. DNA vaccines encoding retrovirus-based virus-like particles induce efficient immune responses without adjuvant. Vaccine. 2006; 24: 2643–55. [PubMed Abstract].
- Huret C, Desjardins D, Miyalou M, Levacher B, Amadoudji Zin M, Bonduelle O et al. Recombinant retrovirus-derived virus-like particle-based vaccines induce hepatitis C virus-specific cellular and neutralizing immune responses in mice. Vaccine. 2013; 31: 1540–7. [PubMed Abstract].
- Bellier B, Huret C, Miyalou M, Desjardins D, Frenkiel MP, Despres P et al. DNA vaccines expressing retrovirus-like particles are efficient immunogens to induce neutralizing antibodies. Vaccine. 2009; 27: 5772–80. [PubMed Abstract].
- Lescaille G, Pitoiset F, Macedo R, Baillou C, Huret C, Klatzmann D et al. Efficacy of DNA vaccines forming e7 recombinant retroviral virus-like particles for the treatment of human papillomavirus-induced cancers. Hum Gene Ther. 2013; 24: 533–44. [PubMed Abstract].
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006; Chapter 3: Unit 3 22. [PubMed Abstract].
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009; 9: 581–93. [PubMed Abstract].
- Chiarella P, Fazio VM, Signori E. Electroporation in DNA vaccination protocols against cancer. Curr Drug Metab. 2013; 14: 291–9. [PubMed Abstract].
- Zeelenberg IS, van Maren WW, Boissonnas A, Van Hout-Kuijer MA, Den Brok MH, Wagenaars JA et al. Antigen localization controls T cell-mediated tumor immunity. J Immunol. 2011; 187: 1281–8. [PubMed Abstract].
- Ahlén G, Söderholm J, Tjelle T, Kjeken R, Frelin L, Höglund U et al. In vivo electroporation enhances the immunogenicity of hepatitis C virus nonstructural 3/4A DNA by increased local DNA uptake, protein expression, inflammation, and infiltration of CD3+ T cells. J Immunol. 2007; 179: 4741–53.
- Widera G, Austin M, Rabussay D, Goldbeck C, Barnett SW, Chen M et al. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J Immunol. 2000; 164: 4635–40. [PubMed Abstract].
- Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK et al. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. 2008; 26: 5896–903. [PubMed Abstract] [PubMed CentralFull Text].
- Tjelle T, Rabussay D, Ottensmeier C, Mathiesen I, Kjeken R. Taking electroporation-based delivery of DNA vaccination into humans: a generic clinical protocol. Methods Mol. Biol. 2008; 423: 497–507.
- Senovilla L, Vacchelli E, Garcia P, Eggermont A, Fridman WH, Galon J et al. Trial watch: DNA vaccines for cancer therapy. Oncoimmunology. 2013; 2: e23803. [PubMed Abstract] [PubMed CentralFull Text].
- Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I. Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes. J Extracell Vesicles. 2013; 2: 20677.
- Manolova V, Flace A, Bauer M, Schwarz K, Saudan P, Bachmann MF. Nanoparticles target distinct dendritic cell populations according to their size. Eur J Immunol. 2008; 38: 1404–13. [PubMed Abstract].
- Lattanzi L, Federico M. A strategy of antigen incorporation into exosomes: comparing cross-presentation levels of antigens delivered by engineered exosomes and by lentiviral virus-like particles. Vaccine. 2012; 30: 7229–37. [PubMed Abstract].