
Abstract
Extracellular vesicles (EVs), such as exosomes and microvesicles, are released by different cell types and participate in physiological and pathophysiological processes. EVs mediate intercellular communication as cell-derived extracellular signalling organelles that transmit specific information from their cell of origin to their target cells. As a result of these properties, EVs of defined cell types may serve as novel tools for various therapeutic approaches, including (a) anti-tumour therapy, (b) pathogen vaccination, (c) immune-modulatory and regenerative therapies and (d) drug delivery. The translation of EVs into clinical therapies requires the categorization of EV-based therapeutics in compliance with existing regulatory frameworks. As the classification defines subsequent requirements for manufacturing, quality control and clinical investigation, it is of major importance to define whether EVs are considered the active drug components or primarily serve as drug delivery vehicles. For an effective and particularly safe translation of EV-based therapies into clinical practice, a high level of cooperation between researchers, clinicians and competent authorities is essential. In this position statement, basic and clinical scientists, as members of the International Society for Extracellular Vesicles (ISEV) and of the European Cooperation in Science and Technology (COST) program of the European Union, namely European Network on Microvesicles and Exosomes in Health and Disease (ME-HaD), summarize recent developments and the current knowledge of EV-based therapies. Aspects of safety and regulatory requirements that must be considered for pharmaceutical manufacturing and clinical application are highlighted. Production and quality control processes are discussed. Strategies to promote the therapeutic application of EVs in future clinical studies are addressed.
Different organisms and cell types have the capacity to release a wide variety of membrane-enclosed vesicles (e.g. exosomes, microvesicles, apoptotic bodies), ranging from approximately 40 nm to a few µm in size, into their extracellular environment. These secreted vesicles are collectively designated extracellular vesicles (EVs). EVs transmit information between cells, organs and even between organisms, and have been detected in body fluids, such as blood, urine, cerebrospinal liquid, breast milk and saliva (Citation1–Citation3). Exosomes and microvesicles comprise the most prominently described classes of EVs; they are surrounded by a phospholipid membrane and contain cell-type-specific combinations of proteins, including enzymes, growth factors, receptors and cytokines as well as lipids, coding and non-coding RNAs and metabolites (Citation1–Citation3). Exosomes are defined as 70–150 nm sized derivatives of the endosomal compartment. During endosome maturation, parts of the endosomal outer membrane, the limiting membrane, bud as intraluminal vesicles into the interior of the maturating endosomes to create multivesicular bodies (MVBs). Upon the fusion of MVBs with the plasma membrane, the intraluminal vesicles are released as exosomes into the extracellular environment (Citation4–Citation6). With average sizes of 100–1,000 nm, microvesicles represent a class of larger EVs that are formed by the outward budding of the plasma membrane (Citation7). Although the origin of exosomes and microvesicles has been defined precisely, current technology does not allow the experimental separation or even discrimination of different EV types of similar sizes (Citation8). Thus, regardless of whether cited findings claim the EVs to be exosomes, microvesicles, etc., we have elected the collective term EV in this article. It is important, however, to keep in mind that, depending on the isolation method, different EV subtypes might be enriched and, even when derived from the same cell types, may differ in their functional properties.
Already in the 1960s, the physiological functions of EVs were unveiled; for example, bone matrix vesicles play a role in bone mineralization (Citation9). The discovery that B cell–derived EVs carry functional MHC-peptide complexes on their surface and exhibit T cell stimulatory capacity led to a revival of the EV field in the mid-90s (Citation10). Furthermore, the field was massively boosted by the findings of the functional transfer of mRNA and microRNA between cells via EVs (Citation11–Citation13). Nowadays, it has become increasingly evident that EVs play a central role in many physiological and pathophysiological conditions, which have recently been comprehensively summarized (Citation2). EVs of various cell types have been shown to transfer a range of biologically active macromolecules that can effectively alter the biological properties of target cells. Due to these properties, they are considered novel agents in different therapeutic applications. The review of the main research areas addressing the therapeutic potential of EVs is followed by an overview of the current regulatory issues associated with using EVs as therapeutics. Finally, we provide a draft that should help to translate EVs into the clinic.
EVs as novel therapeutics: current state of the art
EVs in anti-tumour immunotherapy
The idea to use EVs as anti-tumour vaccines arose from work published almost two decades ago. Here, EVs designated as exosomes with diameters of around 100 nm, as assessed by transmission electron microscopy, were harvested by the ultracentrifugation of the supernatant of antigen-presenting cells pulsed with antigenic peptides. These EVs contained MHC-peptide complexes capable of activating CD4 and CD8 T cells (Citation10,Citation14). EVs from dendritic cells (DCs), pulsed with tumour cell peptides, induced the rejection of a growing tumour in immune competent mice. The rejection involved the activation of tumour-specific cytotoxic T cells (Citation14). This discovery led to a phase I anti-melanoma clinical trial conducted in France and a phase I anti-non-small cell lung cancer clinical trial in the United States (Citation15,Citation16) (Table ). Both clinical trials used Good Manufacturing Practice (GMP)-compatible protocols to recover EVs from a medium conditioned by the patients’ monocyte-derived DCs (Citation17) that had been pulsed with antigenic peptides known to be expressed by the patients’ tumours. A small number of patients benefitted from the therapies of these clinical trials, mainly demonstrating the feasibility and safety of the EV administration. As a consequence, a phase II clinical trial (NCT01159288) was conducted in France, between 2012 and 2014, to treat non-small cell lung cancer patients (Citation18). EVs from mature DCs were used in this phase II clinical trial because murine models showed that the EVs of immature DCs exerted tolerogenic effects and only EVs co-injected with immune-stimulatory adjuvants or EVs from mature DCs efficiently promoted naïve T cell priming, respectively (Citation19,Citation20). In addition, patients received low-dose cyclophosphamide to inhibit regulatory immune responses and to further promote the induction of effector T cell responses (Citation21). Possibly due to their late metastatic stage, the administered EVs did not induce detectable CD4 or CD8 adaptive T cell responses in the treated patients. However, in some patients, a positive effect on natural killer (NK) cell activity was observed (Citation22). Recent results obtained in preclinical studies might help to further improve future clinical trials (Citation23). For instance, NK T cell (NKT) activating agents have improved the anti-tumour effects of DC-derived EVs (Citation24). In addition, EVs obtained from DCs pulsed with a full tumour antigen, instead of MHC class I- or class II-binding peptides, induced the activation of B cells and efficiently promoted tumour rejection in the absence of adjuvant (Citation25). Consequently, tumour-derived EVs have been considered for DC pulsing in vitro. Indeed, initial studies showed that EVs secreted by tumours constitute a source of tumour antigen that induced anti-tumour immune responses in mice (Citation26,Citation27). However, numerous subsequent studies describing the immune-suppressive effects of tumour EVs on various immune effector cells suggested caution in the use of native tumour EVs for DC pulsing (Citation28). Upon combining tumour EVs with appropriate, immune-stimulatory adjuvants the immune-inhibitory effect of tumour EVs might be successfully suppressed, thus enabling them to promote an anti-tumour response (Citation29). Taking this approach, a phase I clinical trial in China investigated tumour-derived EVs for anti-tumour immunotherapy (Citation30) (Table ). Here, EVs from the ascites fluid of colorectal cancer patients were combined with the granulocyte-macrophage colony-stimulating factor (GM-CSF) to stimulate anti-tumour DC activity. Feasibility and safety were demonstrated with a few patients benefitting from combined EVs and GM-CSF, but not from the EV alone treatment. To our knowledge, these are currently the only published studies involving EVs in immunotherapeutic anti-tumour trials.
Table I. Current and past NIH registered clinical trials investigating EV-based therapeutics
In addition to DC-EVs, human NK cell-derived EVs have been shown to exert immune stimulation. NK cell-derived EVs, purified from either cell culture supernatants or plasma of healthy volunteers, have been shown to lyse target human tumour cells in vitro (Citation31).
An alternative EV-inspired vaccination approach is based on plasmids encoding EV-associated antigens. In preclinical models, upon transfection in vivo, affected tissues were found to release EVs presenting such antigens. Plasmids encoding fusion proteins of a viral antigen with viral gag or viral envelope protein, thus leading to the secretion of the antigen in virus-like particles, were successfully used in mouse models of leukaemia virus and hepatitis C infection, as well as in human papilloma virus-induced and non-virus-induced cancer (Citation32–Citation35). In addition, plasmid DNA as well as recombinant viruses encoding antigens fused to the coding region of the phosphatidylserine-binding domain of the milk fat globule epidermal growth factor-factor VIII protein (MFGE8, also known as lactadherin), which promotes the binding of corresponding fusion proteins to EVs, have been used as anti-tumour vaccines in mouse models (Citation35–Citation38). Despite the fact that such DNA-based vaccines may represent a cost-efficient alternative to the ex vivo production of antigen-carrying EVs, testing of these approaches in clinical trials has, to our knowledge, not been described so far.
Depending on their origin and context, EVs can stimulate immune responses and promote anti-tumour responses and thus may provide important tools for novel anti-tumour therapies.
EVs as therapeutic agents against infectious diseases
Pathogens, like helminths (flat- and round-worms), fungi, bacteria as well as parasitic protozoa, including species of Plasmodium, Toxoplasma, Trypanosoma, Leishmania and Trichomonas, also secrete EVs. Both gram-positive and gram-negative bacteria can release EVs; the latter are commonly called outer membrane vesicles (OMVs) (Citation39–Citation43). Furthermore, pathogen-infected cells can release EVs containing pathogen-specific antigens. EVs carrying pathogen-specific antigens, for example, have been isolated from macrophages that have been infected with Mycobacterium tuberculosis, Mycobacterium bovis BCG, Salmonella typhimurium or Toxoplasma gondii, as well as from murine reticulocytes infected with Plasmodium yoelii (Citation39,Citation44–Citation50). Similarly, as in the anti-tumour trials, such EVs have been studied as vaccines in numerous preclinical mouse models.
Mainly, two different strategies are under investigation that will just briefly be mentioned in this paragraph: (a) EVs from in vitro pulsed DCs and (b) EVs released by the pathogen or infected cells. The regulatory concerns for such vaccination studies differ from those for the other therapeutic EV applications. Consequently, an independent ISEV position paper will be prepared that will give a more comprehensive overview of EVs in infectious diseases, and a discussion of the underlying regulatory issues.
Proof-of-principle studies to pulse DCs in vitro with antigens of the obligate intracellular parasite Toxoplasma gondii showed that – similar to tumour biology – EVs released by these DCs could induce an immune response conferring protection against subsequent infections (Citation51–Citation53). Alternatively, EVs released from pathogens or infected cells, respectively, have been directly used as vaccines in numerous preclinical mouse models (Citation44,Citation47–Citation50,Citation54–Citation62). Notably, Novartis generated a vaccine named Bexsero that consists of OMVs derived from Neisseria meningitidis. This is used as a vaccine against serogroup B meningococcal diseases in children (Citation63,Citation64). Nanovesicles derived from bacterial protoplasts devoid of bacterial outer membrane components have also been tested as a vaccine in preclinical models. These were found to induce protection against bacterial sepsis in mice (Citation65).
This plethora of studies highlights the potential of EVs as vaccines against infectious diseases. Apart from qualifying EVs as vaccines for humans, efforts to use EV-based vaccines in animal health are exploited. For animal farming, new vaccination strategies are highly desired and are of great economic interest. So far, most of the vaccine approaches currently used in animal health rely on modifications of the original pathogen, either by attenuation, inactivation or as subunit vaccines (Citation66). The potential use of EVs as novel therapeutic agents in animal health was recently shown for the PRRSV virus, as in vitro infections were partially inhibited by EV-delivered artificial microRNAs (Citation67).
The pros of using EVs rather than whole cells as carriers of MHC-peptides complexes for vaccination (both for immunotherapy and infectious diseases), is that EVs are more stable upon freezing and thawing than cells, which always undergo a degree of mortality. Furthermore, EVs bear a defined repertoire of preformed MHC-peptide complexes, not prone to the alteration that has been observed in living cells that, after thawing, can generate new MHC-peptide complexes in the absence of relevant antigenic peptides. The cons of the approach using peptide-pulsed secreting cells or EVs is that the repertoire of MHC-peptide complexes presented is limited, and thus may not be enough to generate a neutralizing immune response against a complex tumour or pathogen. This caveat may be overcome by using EVs produced by cells pulsed with full-length antigens or extracts of tumour or infected cells (Citation25). Since EVs cannot multiply, they should provide a safer source of tumour- or infected cell-derived antigens than whole cells. However, tumour-derived EV fractions have been shown to contain and transfer oncogenic molecules to non-tumoural cells (Citation68), and it is often difficult to separate EVs from certain pathogens, for example, retroviruses which display similar biophysical properties (Citation69). Consequently, EVs may not be as safe as more inert antigen-sources, such as cell lysates. The balance between advantages (the more efficient capture of EVs rather than the soluble molecules of antigen-presenting cells (Citation36)) and inconveniences (the potential transfer of oncogenic or viral activity by EVs but not by the soluble or extracted cell-derived molecules) needs to be carefully evaluated, for each vaccination approach.
EVs carrying pathogen-specific antigens may provide useful vehicles for the development of new vaccination strategies against infectious diseases in human and animals.
Unmodified EVs in immune-modulatory and regenerative therapies
Increasing evidence suggests that EVs are important players in mediating the therapeutic effects of cells being used as therapeutics, such as mesenchymal stem/stromal cells (MSCs) or endothelial cells. Before highlighting the therapeutic potential of such EVs, some background information about such EV-releasing cells and their therapeutic impact will be provided.
Originally, MSCs were described as a subpopulation of stromal bone marrow cells with osteogenic potential (Citation70,Citation71). Following the description of such cells as holding multi-lineage potential (Citation72), MSCs emerged as one of the most intensively studied non-haematopoietic adult stem cell entities (Citation73,Citation74). MSCs can be isolated from different tissues, including bone marrow, adipose tissue and umbilical cord blood (Citation75–Citation78), and some MSC-subtypes were originally considered to contain pluripotent developmental capabilities (Citation79,Citation80). In addition, MSCs exert strong immune-modulating activities. In 2002, it was initially reported that they are able to suppress the proliferation of mitogen-stimulated T cells (Citation81). Meanwhile, MSCs have been found to inhibit DC maturation and activation, modulate B cell and NK cell functions, promote regulatory T cell formation and regulate the polarization of M1-like (classically activated) pro-inflammatory to M2-like (alternatively activated) anti-inflammatory macrophages (Citation82–Citation88).
A number of clinical trials have been initiated to assess the therapeutic value of MSCs in various diseases (Citation89). Up to now, more than 500 such studies have been registered in the www.ClinicalTrials.gov database. Many of these studies were designed as cell replacement strategies, based on the hypothesis that MSCs home to and become integrated into affected tissues to replace lost cell types and thus restore tissue and organ functions (Citation90). Other studies focused on MSCs as immune-modulating cells, for example, to treat immunological disorders, such as graft-versus-host disease (GvHD), Crohn's disease and rheumatoid arthritis (Citation91–Citation95). Although many of these studies have reported beneficial effects, engrafted MSCs were rarely found in corresponding tissues. These observations led to the assumption that, instead of direct cellular effects, secreted factors induce the MSC pro-regenerative and/or immune-modulatory functions (Citation96). Indeed, some recent data suggest that the delivery of viable MSCs to damaged tissues is not required to exert the MSC therapeutic effects (Citation97–Citation102).
Initial evidence that EVs are responsible for the therapeutic MSC effects was presented by the groups of G. Camussi and S.K. Lim and D. de Kleijn in experimental models of acute kidney failure or myocardial infarction, respectively (Citation99,Citation103). By exerting anti-apoptotic activities, human MSC-EVs were as effective as their parental cells in promoting kidney regeneration in severe combined immunodeficiency (SCID) mice with glycerol-induced acute kidney injury (Citation103). Subsequent work confirmed the protective properties of MSC-derived EVs in both acute and chronic renal damage (Citation104–Citation110). With respect to myocardial infarction, supernatants of in vitro expanded MSCs had been successfully used to reduce the myocardial infarction size in mice (Citation100–Citation102). The pro-regenerative activity of supernatants was identified to be enriched in the EV fraction, rather than in the EV-depleted soluble fraction (Citation99). It was confirmed that MSC-EVs exert immune-suppressive effects, by enforcing M2 macrophage polarization and indirectly driving regulatory T cell induction (Citation111). MSC-EVs have also been shown to suppress the activation of NK cells and other peripheral blood leukocytes of healthy donors as well as those of a GvHD patient (Citation112). Based on this therapeutic potential, the first documented clinical MSC-EV administration was performed in 2011. MSC-EVs were administered in escalating doses to a steroid-refractory GvHD patient. MSC-EVs were infused intravenously at intervals of 2 or 3 days during a period of 2 weeks. The MSC-EV administration was well tolerated, and no side effects were observed. Remarkably, during and following MSC-EV therapy, the GvHD-symptoms declined significantly and the patient was stable for more than 4 months following MSC-EV treatment (Citation112).
Many preclinical models have shown the beneficial effects of MSC-EVs. After demonstrating that human liver stem cell–derived EVs accelerated hepatic regeneration in hepatectomized rats (Citation113), MSC-EVs were successfully tested for their capability to alleviate drug-induced liver injury (Citation114,Citation115). Furthermore, MSC-EVs mediated cytoprotective effects on hypoxia-induced pulmonary hypertension reduced Escherichia coli endotoxin-induced acute lung injury and accelerated muscle regeneration in mice (Citation116–Citation118). In rat models, MSC-EVs significantly improved perfusion in hind limb ischaemia, accelerated re-epithelialization following skin burn and enhanced survival of allogeneic skin grafts (Citation111,Citation119) (Citation120).
MSC therapies have additionally emerged as promising approach to treat stroke patients (Citation121–Citation123) and possibly brain injury in newborn infants, for example, following perinatal asphyxia (Citation124,Citation125). In stroke animal models, intravenously transplanted human MSCs, obtained from different sources, promote neuroprotection and peripheral immunomodulation, reducing the central nervous system (CNS) ischaemic levels and ameliorating the stroke-associated neurological deficits (Citation126,Citation127). In an in vitro model for stroke, similar to the myocardial infarction model, MSCs were found to exert therapeutic activity in a paracrine manner by releasing neuroprotective factors which enhanced neurogenesis and angiogenesis, rather than by direct cellular interactions (Citation128). In line with these results, MSC-EV administration has been shown to promote functional recovery and neovascularization following ischaemic occlusion in a rat stroke model (Citation129) and to enhance sciatic nerve regeneration (Citation130). In a murine stroke model, a direct side-by-side comparison of the therapeutic impacts of MSCs and MSC-EVs revealed no detectable difference in the functional outcome measured in three independent behaviour tests at different time points, post experimental stroke induction. In comparison to untreated controls, both treatment strategies significantly improved the functional outcome after stroke induction in a very similar way (Citation131).
In the immature brain, hypoxic-ischaemic encephalopathy (HIE) following birth asphyxia and premature birth represent major problems affecting development at an early age with lifelong personal consequences. Term newborn babies suffering from asphyxia are treated with hypothermia, which improves the outcome in mild to moderate cases of asphyxia but not in severe cases (Citation132). Hypothermia is, however, not available for preterm babies. Thus, additional and regenerative treatment strategies for both patient groups are urgently needed. In addition to pharmacological approaches, such as sildenafil, xenon and erythropoietin treatment (Citation133), new cell-based or cell-derived therapeutic strategies are tested to treat human preterm and term neonates with the developmental brain injuries that frequently result in serious long-term deficiencies (Citation125). In both mouse and sheep models of neonatal ischaemic brain injury, MSC administration was shown to provide a powerful therapeutic option to promote brain regeneration leading to improved neuro-behavioural and neurological outcome (Citation124,Citation134) (Citation135). In the sheep model, improvements of hypoxic damages have also been observed following systemic MSC-EV administration (Citation136).
A variety of additional applications of MSC-EV therapy appear to be feasible, such as in multiple sclerosis (MS) and Alzheimer's disease (AD). MS is an autoimmune demyelinating disease of the CNS. Several studies have shown that MSCs promote neuroprotection, immunomodulation and, eventually, remyelination in different in vitro and in vivo experimental approaches for MS (Citation137). Moreover, this therapeutic activity is mainly mediated by MSC-secreted factors, suggesting the possible involvement of MSC-EVs and their potential use for the treatment of MS. In AD, adipose tissue-derived MSCs were shown to secrete EVs with enzymatically active neprilysin, which is the rate-limiting enzyme of intra-cerebral amyloid beta peptides, the causative molecules for AD (Citation138). Considering the potential of EVs to deliver their contents to targeted organs, including the brain (Citation139–Citation142), it might be possible to efficiently deliver functional neprilysin to the brain using EVs derived from autologous MSCs.
Thus, MSC-EVs seem to mediate beneficial therapeutic effects in a variety of different diseases. Apart from immune modulation, several studies have shown a direct positive effect of the MSC-EVs on angiogenesis (Citation143–Citation146). Although pro- and anti-tumourigenic effects of MSC-EVs have been observed (Citation147–Citation151), no side effects have been reported so far. However, future studies are needed to confirm their clinical safety and potential in this regard. Furthermore, and as discussed in more detail at the end of this section, heterogeneity among independent MSCs and MSC-EV preparations, as well as the EV heterogeneity in obtained MSC-EV samples, need to be addressed.
Other cell sources under investigation for regenerative medicine are endothelial cells and endothelial colony–forming cells (ECFCs), including human umbilical vein endothelial cells (HUVEC) and late outgrowth endothelial cells (OECs) (Citation152–Citation157). ECFCs are non-haematopoietic cells that can be readily expanded ex vivo and have been shown to functionally integrate into newly formed vessels. In addition, haematopoietic progenitors that are capable of differentiating into myeloid and lymphoid cells may exert pro-angiogenic functions (Citation153,Citation158–Citation161). Although their derivatives are often found to reside in close contact to newly formed vessels, they do not integrate into the endothelial network (Citation162–Citation165). It became evident that pro-angiogenic processes are supported either directly via homing and integrating into sites of endothelial damage and tumours, or indirectly by the release of cytokines, growth factors and EVs (Citation166–Citation168). EVs released from ECFCs stimulated neo-angiogenesis in vitro and in vivo and have been shown to enhance recovery in a murine hind limb ischaemia model by promoting revascularization and protecting the kidneys from ischaemia-reperfusion injury (Citation169–Citation171). Furthermore, such EVs have been found to suppress monocyte activation (Citation172). EVs mediating immune-suppressive function have also been harvested from regulatory T cells (Treg) (Citation173–Citation175). In a rat model, it has been shown that Treg-EVs promote prolonged kidney allograft survival (Citation176).
Neural stem cells (NSCs) have been used in the preclinical models of a variety of neurologic and neuro-inflammatory disorders such as MS, spinal cord injury and stroke (Citation177–Citation184). It was initially assumed that transplanted NSCs home to affected sites and, upon expansion and differentiation, directly replace the lost cell types and tissues (Citation185). It became evident, however, that analogously to MSCs also NSCs exert their therapeutic effects in a paracrine and systemic manner rather than by intercalating into sites of lesion (Citation184,Citation186). In this context, NSC-derived EVs are considered to interact with the host's immune system to mediate neuroprotection and immunomodulation (Citation186,Citation187). Neuroprotection and regeneration can also be mediated by EVs released by the resident glia cells of the nervous system. For example, oligodendrocyte-derived EVs enhance the tolerance of target neurons to various forms of cellular stress and activated pro-survival signalling pathways (Citation188,Citation189). In demyelinating diseases, in which axons degenerate due to lack of glial support, such EVs might be of therapeutic value. Furthermore, Schwann cells secrete EVs which enhance axonal regeneration in the peripheral nervous system by substantially increasing neurite outgrowth and axonal elongation in vitro and in vivo (Citation190,Citation191). It will be interesting to evaluate whether Schwann-cell EVs or EVs from other cell sources such as MSCs or NSCs also promote CNS axon regeneration. In general, it should be considered that CNS therapies might be complicated by the fact that the blood brain barrier (BBB) isolates the brain tissue from the periphery. However, several studies indicate that EVs cross the BBB and enter neural cells, at least under certain conditions, such as inflammation (Citation192). Targeting the CNS might be achieved through the systemic or even the intranasal administration of EVs, giving them a potential advantage over many drugs.
Another source of EVs with immunomodulatory molecules is represented by parasitic helminths like trematodes (Citation193,Citation194). In this context, recent studies have shown that the administration of EVs from the nematode Heligmosomoides polygyrus suppresses type 2 innate responses and eosinophilia in a rodent model of allergy (Citation195).
Finally, very recent studies describe the isolation of EVs from induced pluripotent stem cells (iPSCs), their ability to transfer RNAs and proteins into heart cells (Citation196), and their healing abilities in vivo in ischaemic myocardia (Citation197). In addition, iPSCs might be used as a source to raise somatic stem cells in a scaled manner for the large scale EV-production or to obtain cells as an EV source which can hardly be obtained from primary donor material, such as human NSCs. In this context, EVs from iPSC-derived MSCs have already been shown to attenuate limb ischaemia (Citation198). It is tempting to speculate that the combination of iPSC and EV technologies will provide novel therapeutic options in the future.
Although the previous discussion suggests that a variety of cells release EVs with pro-regenerative and immunosuppressive capabilities, all of these EV-releasing cell types represent heterogeneous populations rather than well-defined cell types. Due to this heterogeneity, it must be kept in mind that even apparently homogenous cell types release different EV subtypes. Furthermore, donor-related variability may be responsible for therapeutic differences among comparable EV fractions.
Regarding MSCs themselves, increasing evidence suggests that independent MSC preparations indeed differ in their therapeutic potentials. Accordingly, attempts have been made to identify surrogate markers and establish potency assays for discriminating such subtypes (Citation199–Citation202). Similar to the heterogeneity of MSCs, independent MSC-EV preparations show different immune-modulatory capabilities (Citation112). Thus, it has to be considered that therapeutic potentials vary among EV preparations harvested from independent preparations of the same cell types. In addition, the functionality of harvested EV fractions might largely depend on the method used to enrich EVs. To our knowledge, no investigation has explored whether co-purified non-EV associated molecules affect the activity of obtained samples. Co-purified molecules might be functional neutral, act synergistically or antagonistically. Furthermore, only a proportion of EVs within given supernatants might mediate the desired therapeutic effect, whereas others might be neutral or act in an antagonistic manner. Since the EVs’ therapeutic potential might depend on their quantity, heterogeneity and quality, methods must be validated to enable the appropriate quantification of EVs in given fractions as well as to analyse their function in suitable potency assays. Despite the advantages of using EVs instead of cells for the therapeutic application, it has to be considered that purified EV fractions may be less therapeutically active than corresponding cell products; certain paracrine effectors might get lost or altered during the purification of the EVs or, as a result of their short half-life, EVs might not remain continuously and sufficiently present in EV-treated patients, than they probably would in patients following cellular treatment.
In the future, it will be interesting to compare the immune-suppressive potential of the different EV types and unravel both common and EV-cell type specific mechanisms to promote regeneration or inhibit inflammation, respectively. Furthermore, we need to understand whether the different EV types discussed here act synergistically or, rather, redundantly.
We are at the very beginning of gathering knowledge of the mechanisms mediating the EVs’ therapeutic effects, the so called mode of action. However, a few molecules have been identified that seem to mediate some of these effects. There is increasing evidence that miRNAs are essentially involved in mediating the EVs’ therapeutic activities (Citation203). For example, miRNA-133b seems to be responsible for the MSC-EV mediated functional recovery following ischaemic stroke in a rat model (Citation204) and miR-22 for the MSC-EV mediated anti-apoptotic effects on cardiomyocytes in ischaemic heart diseases (Citation205). In addition, several proteins have been described to control intrinsic versus reactive immune-stimulating features of EVs, for example, CD86, CD40, MHC-I and -II as well as Toll-like receptors (TLRs) (Citation206–Citation209). Compelling evidence also exists that EVs of certain cell types can modulate the purinergic signalling known to control inflammatory processes (Citation210,Citation211): As a consequence of pathologic conditions such as inflammation or ischaemia, multiple cell types release nucleotides including ATP and ADP into their extracellular environment. Extracellular ATP predominantly functions as a signalling molecule to activate purinergic P2 (P2X/P2Y) receptors. Upon activation, purinergic P2 (P2X/P2Y) receptors trigger inflammatory processes, which can be suppressed by the inhibition of the purinergic signalling pathway (Citation212,Citation213). The molecules ectonucleoside triphosphate diphosphohydrolase 1 (CD39) and ecto-5′-nucleotidase (CD73) are required to metabolize extracellular ATP and ADP into adenosine that, in contrast to ATP and ADP, exerts immune-suppressive effects (Citation214,Citation215). Following MSC transplantation in a mouse GvHD model, elevated levels of CD73 expressing EVs were observed. Like CD39 and CD73 expressing tumour-derived EVs, these EVs were found to metabolize extracellular ATP into adenosine and, coupled to this, to inhibit T cell effector functions (Citation210,Citation211) (Citation216). Since CD73 is a well-known cell surface antigen on MSCs (Citation217), the pyrogenic pathway might essentially contribute to the therapeutic activity mediated by MSC-EVs.
Unmodified EVs from MSCs, endothelial progenitors, Tregs, DCs, and NSCs, as well as of many other cell types, hold promising therapeutic potential in regenerative medicine and immune therapy. As in many of the described studies, human EVs proved effective in different animal models, and the therapeutic capability of at least some EV entities seem to be conserved across species.
Modified EVs for targeted drug delivery
EVs are being increasingly explored as systems for therapeutic delivery of different drug types. Recent reviews highlight the most relevant features of using EVs in targeted drug delivery such as their circulation time, bio-distribution, cellular interactions and the different methods for therapeutic cargo loading and administration (Citation218–Citation220). Potential advantages of EV-based drug delivery over the existing synthetic delivery systems (such as liposomes) include decreased immunogenicity and toxicity, increased stability in circulation and tissue, and intrinsic homing abilities (Citation221). Drugs that could particularly benefit from delivery by EVs are small RNA therapeutics, including miRNAs and siRNAs, and anti-inflammatory agents as well as anti-cancer drugs (Citation219). Small RNAs can trigger the inhibition of virtually any gene expression via RNA interference, giving them enormous therapeutic potential. However, cellular entry for such large, hydrophilic and charged molecules is restricted by the plasma membrane. Thus, shuttle carriers are required. Viral and cationic carriers are potentially unsafe because of the uncontrolled integration of viral material or toxicity, respectively (Citation222).
EVs are natural carriers of RNA molecules and the delivery of their content can lead to functional changes in recipient cells (Citation11–Citation13),Citation192, (Citation223–Citation225). Initially described for their ability to transfer mRNA, an increasing number of studies have affirmed their ability to transfer miRNA into cells. These findings suggest that EVs utilize native mechanisms for cellular internalization and trafficking, and a potential role for EVs for small RNA delivery (Citation226). In order to load EVs with therapeutic small RNA molecules, two encapsulation approaches have been explored: (a) post-loading, that is, after EV isolation (also known as exogenous method), or (b) pre-loading, that is, during EV formation (also called endogenous method) (Citation218,Citation219) (Citation227). Several recent reports have shown functional siRNA delivery into recipient cells using EVs loaded by electroporation (Citation139,Citation141) (Citation228). However, the efficacy of this exogenous method has not been fully demonstrated, and the initially reported loading efficiencies may have been overestimated due to the possible aggregation of siRNAs in the electroporation buffer (Citation229). Other teams have reported that they were unsuccessful in using electroporation to load EVs with miRNA (Citation140). The fact that various sources of EVs have different molecular composition could influence the susceptibility of particular EVs to electroporation (Citation3,Citation230). Therefore, further studies are needed to confirm the feasibility and efficiency of this method for EVs loading.
The endogenous approach exploits the cellular machinery for small RNA loading into EVs after overexpression or the direct transfection of the RNAs of interest into the cells from which the EVs are subsequently derived (Citation12). This method has been successfully used for the packaging of both siRNA and miRNA in EVs. Functional delivery into recipient cells has been shown in several reports (Citation140,Citation231–Citation234). The feasibility of this method, however, likely varies depending on the siRNA or miRNA species, as cells seem to have selective sorting mechanisms for the incorporation of small RNAs into EVs (Citation235,Citation236). Furthermore, as a result of the overexpression or the direct transfection of a particular small RNA in the EV donor cells, other changes to the EV content may occur. Finally, when transfection reagents are being used, consideration should be given as to whether or not any residual or co-released reagents are co-purified during EV isolation. Such impurities could affect the EV behaviour, induce false-positive effects and/or cause toxicity.
The observation that EVs released by tumour cells, in vivo and in vitro, can transport cytotoxic drugs, such as cisplatin in its native form, demonstrated that EVs can transport drugs from one cellular compartment to another (Citation237). Accordingly, EVs are considered promising anti-tumour drug delivery vehicles. They may help to circumvent the mechanisms mediating chemo-resistance following conventional drug application. For instance, tumour acidity represents a very efficient, though non-specific cause of chemo-resistance, inducing the protonation of the drug and consequent neutralization in the extracellular environment (Citation237,Citation238). Drugs transported via EVs may be protected within such acidified microenvironments and, thus, might facilitate the efficient delivery of active drugs into tumour cells in an acidic microenvironment. Notably, microenvironmental acidity has been shown to increase both EV-targeting to the tumour sites and EV-uptake by tumour cells (Citation237,Citation239).
Anti-inflammatory drugs such as curcumin or chemotherapeutic agents (paclitaxel, PTX and doxorubicin, Dox) are under investigation for their suitability for EV-mediated transport. PTX-loaded EVs, released from PTX-treated MSCs in vitro, have been shown to inhibit the proliferation of cultured tumour cells (Citation240). EVs from immature mouse DCs, engineered to express a fusion of lamp2b with alpha-5 integrin-specific peptide for tumour targeting and loaded with Dox by electroporation, were efficiently incorporated in breast cancer cells in vitro. In vivo, they were specifically delivered to implanted breast tumour tissues and suppressed the tumour growth without causing any toxicity (Citation241). A phase II clinical trial has been registered to test the safety and efficacy of tumour cell-derived EVs to treat malignant ascites and pleural effusion (NCT01854866, Table ). 100–1,000-nm-sized EVs were harvested from methotrexate (MTX), Dox, cisplatin or hydroxyl camptothecin-loaded tumour cells following apoptosis induction by ultraviolet light irradiation. In vitro, the drug-loaded EVs were found to be cytotoxic to tumour cells and more effective than direct treatment with the same drug on a dose-per-dose basis (Citation242). Furthermore, MTX-encapsulating EVs were shown to inhibit ascites hepatocarcinoma growth following intravenous or intraperitoneal administration into mice, while cisplatin-loaded as well as cisplatin and PTX co-loaded EVs inhibited ovarian cancer growth without producing strong adverse effects (Citation242).
Curcumin-loaded EVs have already made their way into the clinic. Curcumin is a natural polyphenol with anti-inflammatory properties which, as a hydrophobic substance, interacts with lipid membranes and is poorly soluble in aqueous solutions (Citation243). Upon mixing curcumin with EVs, curcumin was found to bind in quantitative amounts to the EVs released by different cell types. At the therapeutic level, in contrast to their native forms, curcumin-loaded liposomes and free curcumin, curcumin-loaded EVs were found to protect mice from LPS-induced sepsis (Citation244). Upon administration to the brain through intranasal routes, curcumin-loaded EVs protected mice from LPS-induced brain inflammation and from the progression of myelin oligodendrocyte glycoprotein peptide–induced experimental autoimmune encephalomyelitis (Citation245). Furthermore, curcumin-loaded EVs delayed brain tumour growth in the GL26 tumour model (Citation245). Curcumin-loaded EVs were shown to specifically suppress the activation of myeloid cells and to be taken up by the microglial cells, which subsequently become apoptotic (Citation244,Citation245). As curcumin has strong inhibitory effects on the progression of many tumour types, including colorectal carcinoma (Citation243), a phase I clinical trial using curcumin-loaded vesicles (deciphered as plant exosomes) has been registered (NCT01294072, Table ). This study investigates the ability of nanosized plant vesicles (nanovesicles) to deliver curcumin to normal colon tissue and colon tumour cells in patients undergoing surgery for newly diagnosed colon cancer. It is aimed at studying the immune modulation, cellular metabolism and phospholipid profile of normal and malignant colon cells.
Although it has not been demonstrated that nanovesicles harvested from the freshly prepared juice of edible plants are of extracellular origin, obtained nanovesicles provide promising potentials as drug delivery. Nanovesicles harvested from grapefruit juice have been shown recently to deliver short interfering RNAs, DNA expression vectors, proteins and chemotherapeutic agents in different types of cells and animal models (Citation246). Another study explored the effect of unmodified and modified grape-juice-derived nanovesicles on different stem cells and showed that they protected mice intestine from dextran sulphate sodium-induced colitis (Citation247). In addition to the phase 1 clinical trial NCT01294072, there is another ongoing study (NCT01668849), which will evaluate the ability of grape-derived nanovesicles to reduce the incidence of oral mucositis during irradiation and chemotherapy treatment for head and neck tumours.
Similar to plant nanovesicles, non-human EVs are currently tested for their ability to serve as effective drug carrier systems. Animal milk-derived EVs have been shown to act as an effective drug carrier (Citation248). Like plant nanovesicles, bovine milk provides a scalable source for isolating large quantities of EVs and provides a cost-effective and biocompatible material.
Although it is beyond the scope of this position paper, it should be noted that technologies have been developed that allow for the production of ex vivo artificially generated nanovesicles obtained from broken cells, which mimic the structure and physical features of EVs. Such vesicles have been called exosome-mimetic nanovesicles and, depending on the preparation method, can either be enriched for intracellular or for plasma membrane vesicles (Citation249,Citation250).
EVs can be loaded with a range of molecules and serve as drug delivery vesicles, which provide new options in anti-tumour and immune therapy for targeted drug delivery.
EV-based therapeutics: regulatory aspects of pharmaceutical development including categories, safety and manufacturing requirements
Early pharmaceutical development is strongly dependent on results derived from the observations and data generated by basic researchers. If novel approaches proceed towards the translational phases, the strategy of how to address questions and regularly acquire data has to change considerably, in order to focus strongly on the validation and certification of the applied technologies. Upon developing novel therapeutics for humans, issues related to pharmaceutical categorization become essential. The regulatory aspects of manufacturing and application of new therapeutics have to be implemented. Safety aspects must be highlighted from various perspectives (e.g. donor, recipient, product, manufacturing, clinical application, biovigilance). Thus, it may appear that the same information has to be provided repeatedly. In the following section, we summarize the most relevant issues to be addressed at the various levels of the developmental processes to translate EV-based therapeutics into the clinic.
Pharmaceutical category of EV preparations
The definition of biological medicine is relevant for EV-based therapies
The development of human EV-based therapeutics is subject to the regulatory frameworks concerning biological medicinal products in the European Union (EU), United States of America, Australia and Japan (see ). “A biological medicine is a medicine that contains one or more active substances made by or derived from a biological cell. Some of them may be already present in the human body and examples include proteins such as insulin, growth hormone and erythropoietin. The active substances of biological medicines are larger and more complex than those of non-biological medicines. Only living organisms are able to reproduce such complexity. Their complexity as well as the way they are produced may result in a degree of variability in molecules of the same active substance, …” (Citation251). Synonyms for the term “biological medicine” are “biologic drugs, biologicals or biopharmaceuticals” and are differentially used in regulatory documents depending on regional practice (Citation252–Citation254).
Fig. 1. Pharmaceutical categories and a suggested classification of EV-based therapeutics. Chart depicts the Categories of Medicinal Products with respect to their origin (chemical, biological, herbal). Medicinal Products (according to DIRECTIVE 2001/83/EC) include any substance or combination of substances for treating or preventing disease in humans. Any substance or combination of substances which may be administered to humans with a view to making a medical diagnosis or to restoring, correcting or modifying physiological functions in humans is likewise considered a medicinal product. The suggested classification of EV-based therapeutics within the class of biological medicinal products is provided (grey fields). Bold indicates categories from which existing legislation is recommended to be considered for preclinical and clinical development of EV-therapeutics.
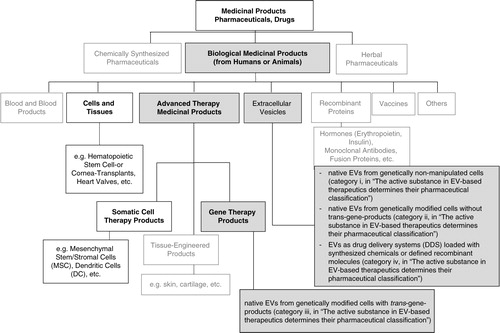
In the EU, detailed guidance on the preclinical development, quality aspects, non-clinical safety requirements and the clinical testing of novel biological medicinal products is provided. As EVs will be considered biological medicinal products, it is anticipated that new rules explicitly regulating EV-based therapies are not needed. Existing European guidance on biological active substances covers the manufacturing and clinical evaluation of novel EV-based therapeutics, in large part (Citation255–Citation262). However, for now, an open question remains about whether special guidelines targeting EV-based therapeutics may be needed. In Australia, the Therapeutic Goods Administration (TGA) office of the government provides rules and guidelines relating to the manufacture and use of therapeutics that are frequently adopted from EU rules. In the United States, EV-based therapies for human use would be also considered biological products, and would be regulated by the Center for Biologics Evaluation and Research (CBER) within the Food and Drug Administration (FDA). Depending on the type of EV-based therapy, pre-existing regulatory guidance may be applicable. For example, EVs used in anti-tumour vaccination may be regulated as therapeutic cancer vaccines for which specific guidance has been issued (Citation263). In all cases, the EVs can be classified as biological medicine (Citation254). depicts the suggested pharmaceutical categorization of EVs, based on the anticipated active substance(s).
In summary, EV-based therapeutics can be defined as biological medicine and belong to the pharmaceutical class of biologicals. Regulatory frameworks for manufacturing and clinical trials exist in Europe, Australia and United States, but special guidelines targeting EV-based therapeutics may be needed.
The active substance in EV-based therapeutics determines their pharmaceutical classification
The regulatory classification of any drug and most biological medicinal products depends on a pharmaceutically active substance, which is not necessarily a defined molecule but in terms of cellular therapeutics can be the cells themselves (Citation254,Citation264). Manufacturers are asked to identify, quantify and characterize the main “substance(s)” of a drug causing a certain pharmacological, immunological or metabolic action being responsible for its biologic effect (i.e. “mode or mechanism of action,” “MoA”). In addition, non-active components needed in the final formulation of a drug (“excipients”) have to be characterized. Whether the MoA of EVs depends on the content of vesicles, the vesicle membranes or a combination of both is currently not known, but these issues have to be addressed during development. It may turn out that, for many therapeutic applications of EV-based therapeutics the specific MoA might not be definable, even if they are confirmed to be effective. Nevertheless, defining or, in early-stage clinical development, anticipating the active substance(s) responsible for the MoA will determine the pharmaceutical control strategy. This includes a panel of quality and potency tests that are linked to a hypothesized MoA. During further clinical development (from clinical trials phase I to III), the goal is to unravel and subsequently verify the MoA in a more detailed manner. As knowledge on the therapeutic substance and the MoA increases, control strategies have to be continuously adapted and refined. These requirements are outlined in the Guidelines on “… the requirements to the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials” (Citation265) and “… on strategies to identify and mitigate risk for first-in-human clinical trials with investigational medicinal products” (Citation266) and (Citation258). It is, therefore, not required that the plethora of open questions associated with biological investigational medicinal products are solved for first-in-man-clinical trials. However, the quality and safety of the investigational new drug must be adequate. At the time of application for product licensing, convincing data regarding the MoA, supported by clinical efficacy and safety, must be provided.
Nevertheless, the definition of the active substance(s) will remain a key question in the preclinical development of EV-based therapeutics. The proposed MoA should be discussed upon registration for a phase I clinical trial in the “Investigational Medicinal Product Dossier” (Citation258). “Details should be provided on the biological activity” (i.e. the specific ability or capacity of a product to achieve a defined biological effect) (Citation258). Ideally, prior to the initiation of phase I clinical studies, the biological activity should be determined using a relevant, reliable and qualified method. The lack of a potency assay (can be tolerated but) should be justified. The rationale for selection of the methods used for the characterization of the therapeutic agent should be provided and their suitability be confirmed. “Tests for quantity, identity and purity are mandatory. A test for biological activity (“potency assay”) should be included unless otherwise justified. Upper limits, taking safety considerations into account, should be set for the impurities.”
Although the specifics will have to be discussed with regulators during the approval process on a case-by-case basis, ISEV suggests categorizing EV-based therapeutics. Specifically, in terms of active substances, at least four different scenarios can be anticipated for EV-based therapeutics ():
i) EV-based therapeutics may be derived from unmodified cells containing native EVs. Then, they are categorized as biological medicine.
ii) EV-based therapeutics may be derived from genetically manipulated cells, but the released EVs do not contain trans-gene products; thus, they are categorized as biological medicine.
In these two scenarios, the EVs can be regarded as the active substance which is, due to its overall composition, capable of entering recipient cells and altering them by influencing downstream pathways. Since the EVs’ MoA is defined by the composition of their membranes together with their cargo molecules, it will be challenging – and, indeed, may turn out to be not essential – to decipher these functions from each other.
iii) EV-based therapeutics may be derived from genetically manipulated cells and contain trans-gene products. These are categorized as gene therapy products (GTPs) that belong to an independent sub-category of biologicals (i.e. advanced therapy medicinal products, ATMPs), depending on whether the therapeutic effect is explicitly ascribed to the trans-gene-product or rather to the EVs themselves.
iv) Native EVs may be used as drug-delivery systems for chemical drugs (category: combined biological AND chemical therapeutic, being regarded as biological medicine) or for other molecular components, such as miRNAs or siRNAs (categorized as biological medicine). It would have to be determined whether or not the EVs themselves mediate parts of the therapeutic effects, at least, and therefore, whether they are or are not part of the active substance.
If the whole therapeutic effect could be ascribed to the loaded molecules and not to the EVs, the EVs would be regarded as “excipients.” The regulatory consequences of this distinction are that characterization requirements for the EVs would be reduced. This means that only the safety profile, but not characterization of the MoA, would be required because, as per definition, excipients do not exert a therapeutic action.
Searching for the MoA of EV-based therapeutics is essential and will proceed as an iterative process during clinical translation. The dissection between “active substances” and “excipients” (“claim of action”) is important for the characterization and definition of appropriate strategies to control the quality of EV-based therapeutics. Phase I clinical trials may be permitted, if safety and quality standards are adequately met and a plausible hypothesized MoA is provided.
The importance of legislation on “tissues and cells” and “advanced therapy medicinal products” (ATMPs) for EV-based therapies
EVs derive from complex tissues or cells and may have much in common with their source material with respect to complexity, composition and biological action. Accordingly, the development of EV-based therapeutics will be closely related to tissue- and cell-based products. These products (e.g. haematopoietic stem cell or cornea transplants) are harvested from donors and transplanted without any excessive alterations to fulfil their original function in the graft receiving patients. In the EU, tissue- and cell-based products are regulated by the DIRECTIVE 2004/23/EC (Citation267) and in the DIRECTIVE 2006/17/EC (Citation268). Unlike biological medicinal products, these directives do not demand a definition of the active substance but regulate safety aspects regarding the donation, procurement, testing, traceability, processing, preservation, storage and distribution of the human material to guarantee health protection of both donors and recipients.
Because the biological medicinal products cover a broad range of different pharmaceuticals, a regulatory category termed advanced therapy medicinal products (ATMPs) was introduced in 2007 (). This category covers a subgroup that differs from conventional biological medicinal products with regard to physicochemical, immuno-chemical and biological properties. It includes gene therapy medicinal products, somatic cell therapy medicinal products and tissue-engineered products (Citation254,Citation269–Citation272). Simply put, a prerequisite to call a biological medicinal product an ATMP is that the product has been extensively manipulated (including cell expansion in vitro) or is intended to exert functions that are different from its original functions (e.g. the usage of haematopoietic stem cell transplants for the treatment of myocardial infarction), and contains nucleated, viable cells. If products contain therapeutically active trans-gene constructs derived from genetically engineered cells, they are considered as ATMPs, independent of the presence of any nucleated, viable cell. For the development of EV-based therapeutics ATMP, scientific guidance (Citation273) may be relevant because EVs are produced, in many cases, from human material by a manufacturing process comparable to ATMP production. The medicinal product framework addresses the safety standards for inadvertent microbial and viral contamination and demands GxP standards (GxP=Good Manufacturing/Good Laboratory/Good Distribution/Good Clinical/Good Scientific Practice or GMP/GLP/GDP/GCP/GSP) for the production and quality control of corresponding therapeutics. Furthermore, it regulates the conduct of clinical trials (Citation142).
The conventional pharmacokinetic, non-clinical pharmacology and toxicology studies required for most medicinal products (Citation274) may not be appropriate or meaningful for the development of EV-based therapeutics. Then, in a manner comparable to that for cell-based products, product-specific approaches would have to be applied. In this context, preclinical safety testing and the transition from preclinical to clinical development may use a risk analysis approach, such as is applied to human cell–based products (Citation267) and takes into account their heterogeneity (Citation264).
In the United States, human cells, tissues and cellular and tissue-based products (HCT/Ps) are defined as therapeutics containing or consisting of human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient. Like ATMPs, HCT/Ps are considered distinct from conventional pharmaceuticals and regulated by CBER (or, in some cases, by the Center for Devices and Radiological Health) as biologics. In general, secreted or extracted human products other than semen are not considered as HCT/P. This would include cellular factors such as EVs. However, considerations relevant to the use of human cells, such as the transmission of communicable diseases, would be important for the approval of EV-based therapies derived from human cells.
Compliance with established frameworks is pivotal from a legal point of view and will increase the confidence of different stakeholders (patients, ethical review boards, competent authorities, funding bodies, investors and sponsors for clinical trials).
In addition to the existing guidance on the manufacture of biological medicinal products (biologicals), it is conceivable that safety and quality standards for the EV manufacturing and related clinical trials will emerge as extrapolations from legislation for tissue- or cell-based products. Concerning the preclinical safety testing, a risk analysis approach – as is applicable for ATMPs – may be a helpful tool.
Safety and biological activity evaluation in advance of clinical trials
The demonstration of the safety and efficacy of novel drugs is a challenge for developers and clinical investigators, a difficulty that is not restricted to biopharmaceuticals. Conventional approaches to toxicity testing are usually not considered appropriate for biopharmaceuticals due to unique structural and biological properties that may include species specificity, immunogenicity and (unpredicted) pleiotropic activities (Citation275). These properties apply to tissues and cells, as well as to ATMPs. According to the legislation for tissues and cells, and ATMPs, a panel of minimal criteria to characterize human cell-based medicinal products needs to be considered before use in clinical trials. It has to be addressed whether products are (a) of autologous, allogeneic or xenogeneic origin; (b) extensively or minimally manipulated in vitro and (c) immunologically active or neutral. In addition, (d) the proliferative capacity of cells and (e) the cell or tissue-like organization as well as the dynamic interaction amongst cells with structural components and (f) the intended use have to be defined.
Because it is anticipated that requirements (a–d) and (f) will be relevant for the characterization of sources used to generate EV-based therapeutics, information should be provided by EV-based therapeutic producing manufacturers (Citation276). Of note, due to the facts that many cell products are composed of a heterogeneous mixture of cells, the identification of the cellular component(s) responsible for a proposed biological activity is a big challenge.
Similar issues will, undoubtedly, emerge during the preclinical development of EVs. Nevertheless, the identity and purity of the expected active substance(s) should be shown, and impurities minimized as effectively as possible. As for every biopharmaceutical, the non-active constituents must be characterized and considered for safety evaluation. For the sake of patients’ safety, the therapeutic effects expected should be predicted with standardized potency assays, and the potential for unexpected adverse effects should be explored and minimized during the preclinical development. Three issues have to be considered in advance of first-in-man-clinical trials: (a) Quality aspects (characterization, determination of strength and potency and comparability of material used in non-clinical studies), (b) non-clinical requirements (pharmacodynamics, pharmacokinetics, demonstration of the relevance of animal models for the intended use (if possible), safety pharmacology and toxicology issues and the calculation of the treatment dose for first-in-man) and (c) clinical requirements (general aspects of clinical trial design, such as study population, inclusion/exclusion criteria, first dose, size of cohort, intervals between dosing within same cohort, dose escalation, stopping rules, defining responsibilities for decisions with respect to dosing, route and rate of administration and long-term monitoring of patients including biovigilance) (Citation266). Due to multidimensional complexity, these questions will not be resolved by isolated scientific efforts. Interdisciplinary activities and collaborations between academia and industry will certainly help to accelerate preclinical development and the successful clinical translation. If preclinical research is designed in the context of the subsequent translation into the clinic, adherence to the existing regulatory standards is highly recommended. Uncertainties regarding the extent of the preclinical characterization of EV-based therapeutics prior to clinical trials may be solved with the help of institutional exchange procedures. National agencies as well as the European Medicines Agency (EMA) and the FDA, CBER in the United States offer advice to investigators and clinical trial sponsors at a national or international level, depending on the developmental stage of a new drug. Recently, an EMA-FDA parallel scientific advice program was initiated, which is highly relevant when advancing to phase III multicentre multinational pivotal trials.
Safety standards for tissues and cells and, in special cases, for ATMPs may serve as roadmaps for the characterization of EV-based therapeutics in preclinical and clinical development. Quality aspects, non-clinical and clinical requirements should be considered in advance of phase I clinical trials.
The importance of determining if EV-therapeutics constitute “high risk medicinal products” for clinical trials
Novel pharmaceuticals may pose risks to patients that are considered “high”. A new MoA can result in a classification as a “high risk medicinal product” with the definition including: (a) that only particular knowledge or uncertainties exist with regard to the MoA of the novel substance(s), (b) that the nature of the target, which makes a drug a potent substance, may be unclear and (c) that the relevance of selected animal models for confirming an expected MoA is limited (Citation266). Although all three criteria appear to be currently appropriate for EV-based therapeutics, it should be decided on a case-by-case basis, whether or not EV-based therapeutics would fall under the high-risk definition.
The high-risk definition would substantially augment preclinical safety testing requirements in advance to clinical trials. This may be adjusted by adducing a number of justifications that might mitigate the high-risk aspects of EV-based therapies. While acknowledging that these criteria will need to be addressed on a case-by-case basis, the ISEV proposes: (a) Autologous EVs naturally occur in the human body. (b) EVs are physiologically produced by all cell types, and their production and uptake in target cells is a natural process. (c) Substances contained within EVs are physiological body constituents (unlike fusion proteins, synthetic agonists or kinase inhibitors). (d) Increasing evidence indicates that DCs and (autologous and allogeneic) MSCs show good safety profiles in numerous patients that have previously been treated; therefore, it is plausible that DC-EVs or MSC-EVs will not cause more harm to patients than their respective cells of origin. Finally, (e) despite the huge number of daily transfusion of blood products, there is little published and accessible evidence that allogeneic EVs co-transfused with blood products cause adverse events (Citation277,Citation278). Taken together, these arguments support the assumption that EVs generated from a human cellular source do not carry higher risks than the risks that are carried from the EV-generating tissues or cells and, thus, are not automatically classified as high-risk medicinal products.
In summary, although uncertainties about the MoA, the nature of the targets and the relevance of animal models exist, a number of arguments support the assumption that EV-based therapeutics derived from human tissues and cells do not per se fall under the high-risk definition of investigational new drugs.
Requirements for manufacturing of EV-based pharmaceuticals
Approval of the technical requirements and quality risk management
The manufacturing of EV-based therapeutics will involve the use of living cell systems for the generation of EVs. Minor changes in the tissue culture may have profound impacts on the EV-production process, changing the EVs biological properties and so, in turn, their characteristics (physicochemical, immunochemical and biological). Accordingly, the product characterization of all biological medicines depends on a thorough characterization and control of the manufacturing process (Citation258). The transfer of a therapeutic concept into an investigational product depends on prerequisites such as adequate technical equipment, an established pharmaceutical engineering system with trained personnel and a quality management system. The implementation and maintenance of reliable quality standards according to internationally harmonized proceedings are essential to achieve a required manufacturing license. The production has to be performed in compliance with GxP regulations. GxP standards are being harmonized between regions by “The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH” (Citation261). Since 2009, the ICH has been commissioned to coordinate the pharmaceutical industry and regulatory authorities of EU, Japan and the United States in tripartite discussions on the scientific and technical aspects of drug registration since 1990. In Europe, the harmonized ICH guidelines are published on the official EMA website. Regulatory agencies of ICH members worldwide are continuously monitoring industrial and academic manufacturers by periodical inspections to assess their compliance with to GxP standards.
Donor safety, recipient safety and release criteria for EV-based therapeutics
Physicians and the manufacturers in particular are strictly obliged to comply with donor and recipient safety issues. Donor safety and donor qualification requirements are laid down in blood safety legislation (Citation279), the tissue and cells quality and safety legislations (Citation267) and the DIRECTIVE 2006/17/EC (Citation268). Recipient safety is covered by guidelines from the ICH regarding product safety, efficacy and quality that provide a comprehensive guidance for the development and generation of novel biological medicinal products (Citation261,Citation262). In particular, the quality guideline, “Derivation and Characterization of Cell Substrates used for Production of Biotechnological/Biological Products” (Citation259) and the safety guidelines, “Safety Pharmacology studies for Human Pharmaceuticals” and “Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals” must be considered (Citation260,Citation274). Biopharmaceuticals have to be characterized by a combined approach of testing the active substances (safety pharmacology, pharmacodynamics and toxicology testing) and the final medicinal product, together with a detailed assessment of the pharmaceutical production processes and associated controls. Validated manufacturing processes guarantee the consistent quality of the drug: “The release specifications of the active substance and finished product should be selected on the basis of parameters defined during (non-clinical) characterization studies”. Current studies revealing mechanistic insights into EV biology will determine the future release criteria of EV-based therapeutics. Well-defined release criteria relevant for the respective therapeutic use of EVs should be elaborated by the manufacturer, on the basis of data generated during preclinical characterization (in vitro and in vivo) and process validation. Release criteria will include EV characterization (product definition considering identity/purity/impurities), hypothesized MoA, microbiological control of cell populations, growth media, supplements and materials (such as disposables). Microbial contamination has to be excluded from the final product by testing according to pharmacopoeia (the European Ph.EU and the United States pharmacopoeia, USP). Specific release criteria for each type of modified or unmodified, human allogeneic or autologous, or xenogeneic EVs must be defined according to the intended use of EV-based therapeutics. Due to a broad variety of therapeutic approaches, specific release criteria to suit each scenario are beyond the scope of this position statement. Release criteria and control strategies will be refined in the course of the clinical development.
The manufacturing of EV-based therapeutics requires adequate and appropriate infrastructure and technology, a quality management system and compliance with GxP standards that take into account both donor and recipient safety. Release criteria relevant for the investigational product in its intended use will have to be defined on the basis of preclinical characterization studies.
Requirements to be fulfilled for clinical trials evaluating EV-based therapies
The use of novel drugs in early phase clinical trials requires robust preclinical models indicative for the respective target disease to estimate in vivo response in humans. Limitations in the availability of relevant animal models may result in weak predictability and the resulting misinterpretation of pharmacokinetic results and reduced chances of identifying toxic effects. The predictability of in vivo responses and its easy translation to humans will be reduced if a novel biopharmaceutical displays particular high species specificity. However, current observations that human EVs mediate effects in different animal models suggest reduced species specificities. A low EV species specificity may help to translate results from animal models to humans and may justify the amount of non-clinical safety data.
At least in autologous situations, the assumed immunogenicity risk of EV-based therapies is excluded if the manipulation does not raise additional risks by using allogeneic or xenogeneic factors during EV production. So far, the results of three phase I clinical studies have demonstrated the feasibility and safety of autologous EV-based therapeutics. Table summarizes key information about those studies, together with unpublished EV studies registered at www.ClinicalTrials.gov. In addition to the surveillance regarding the acute toxicity of an EV-based therapeutic medicinal product, it will be necessary to follow up long-term adverse effects such as tumourigenicity and immunogenicity via clinical trial registers and vigilance systems. Table lists potential issues to be defined by each manufacturer of EV-based therapeutics, together with the treating and examining physicians who are designing and executing the intended clinical trials.
Table II. To be defined and established by investigators before (or concomitant to) clinical application of EV-based therapeutics
Representative animal models should be identified and applied in advance of clinical studies investigating EV-based therapeutics whenever possible. Safety, toxicity and immunogenicity need to be monitored in the course of early phase clinical trials. Reliable information concerning efficacy and long-term adverse effects of autologous or allogeneic EVs will be obtained from later phase clinical trials (phase III–IV).
Issues to be considered for the production of EV-based therapeutics
Based on the therapeutic potential of various EV preparations and the relevant regulatory requirements discussed above, we propose to systematically consider the following topics in the context of progression towards clinical trials: (a) Characterization of the EV source, (b) EV isolation, characterization and storage strategies, (c) Pharmaceutical quality control requirements, and (d) in vivo analyses of EVs.
Characterization of the EV source
An important issue in characterizing EVs is the origin of the EV producing tissue or cell source and the relationship between donor and host (i.e. autologous, allogeneic, xenogeneic, cross-kingdom relation). If human cells are the source of the EVs, data on the collection procedure, patient/donor eligibility criteria, health status and medication must be documented. The informed consent of donors and the approval of appropriate ethics committees are mandatory. The screening of the human donors for signs of infection prior to tissue collection is obligatory. For the allogeneic use of EVs, we propose that similar donor inclusion and release criteria be used as for allogeneic donors for blood products and haematopoietic stem cell grafts, respectively (Citation254,Citation255,Citation267–Citation270,Citation280). Clear regulations and national and international guidelines already exist (Citation279,Citation281). In autologous settings, a reduced testing regime according to tissue and cell regulations might be sufficient. Provided that cellular EV sources intended to be “substantially manipulated,” ATMP guidelines should be considered (Citation280). This may include a description of cellular characteristics, such as their cell surface phenotype. Due to the release of apoptotic bodies and subcellular compartments, dying cells may affect the quality of obtained EV fractions. Thus, the viability of cells releasing EVs in vitro should be documented.
It should be taken into account that a disease status may affect cell and subsequent EV characteristics in autologous EV-based therapeutics. A potential impact of tissue compatibility (MHC class I and II class surface antigens) has to be considered in the case of allogeneic EV applications. If HLA-matching turns out to be essential for the success of EV-based therapies, the extent and resolution (low versus high, i.e. 2-digit versus 4-digit resolution) of matching must be defined. Whether or not the matching of classical HLA loci as determined prior to organ transplantation would be sufficient (HLA-A/B/DR or 6/6 matching) has to be clarified. Alternatively, it may turn out to be important to aim for a 10/10 matching (HLA-A/B/C/DR/DQ), as is frequently the practice in haematopoietic stem cell transplantation centres (Citation282). Culture-expanded MSCs are commonly negative for MHC class II, and thus they have been regarded to be immune-privileged cells and have been transplanted without HLA-matching in a number of allogeneic treatments. However, upon stimulation with IFN-γ or upon differentiation, MSCs can severely change their immunological properties and can express significantly more MHC class I and II molecules (Citation108,Citation283) (Citation284). Coupled to that evidence, it has been concluded that allogeneic MSCs may be rejected by the host's immune system, and a discussion has begun about the efficacy of allo-MSC transplantation (Citation285). Currently, knowledge is lacking about the clinical implications of immune response mechanisms, which might be activated following allogeneic MSC-EV administration.
Furthermore, donor-to-donor variabilities have to be considered as comparable cell types generated from varying donors may secrete EVs with different functional properties (Citation112). The variability in EV preparations might be mitigated by using immortalized clonal cell lines that have been stringently tested for stability in their genotype, phenotype and EV production (Citation286). The immortalization of MSCs has been reported to maintain the proliferation rate without compromising the quality or quantity of EVs that they produce (Citation287). In contrast to immortalized cells, which are to our understanding unacceptable for the treatment of patients, such cells might be acceptable for the clinical grade production of human EVs as long as corresponding EVs are free of products of the immortalizing trans-genes. However, since regulations vary from nation to nation and since no directly applicable guidance exists, we recommend discussing projects intending the use of EVs from immortalized cell lines with national regulators at early stages.
If EVs are harvested from cell culture supernatants, culture conditions must be standardized and batch reproducibility has to be ensured (Citation288). It should be considered that cellular densities may affect the amount and quality of EVs (Citation289) and that EVs harvested from early passage supernatants may qualitatively differ from EVs harvested from late passages. The oxygen concentration, the addition of supplementary molecules, for example, cytokines or heparin, and the tissue culture ware or media formulation may significantly affect EV quantity and quality (Citation108,Citation120) (Citation290–Citation296). Of note, growing cells in serum-free media or EV-depleted media as compared to conventional growth media can have profound impacts on the cells themselves, as well as on the quantity and quality of released EVs (Citation289,Citation297) (Citation298).
In addition to EV sources, all reagents used for the EV production must be described regarding their origin. According to the European guidance, protocols intended to culture cells for the clinical application should, if applicable, avoid using animal components such as foetal bovine sera (FBS). Reagents derived from animals may contain xenogeneic infectious agents and/or increase undesirable immunological responses in recipients. Furthermore, in the presence of FBS human EV preparations are contaminated with bovine EVs that could potentially evoke unintended biologic effects (Citation298). Use of FBS is only accepted in compliance with respective stringent USP/EP pharmacopoeia provisions. Non-animal derived reagents of defined composition or human-based supplements are recommended instead (Citation276,Citation299) (Citation300).
In terms of animal milk-derived EVs or plant-derived nanovesicles, only nanovesicles isolated from edible plant sources have been considered for therapeutic application so far. As part of daily nutrition, clinical usage of xenogeneic EVs or nanovesicles derived from the plant kingdom or prokaryotes may raise different concerns than human cell-based medicinal EV products. Of note, some concerns regarding cell-to-cell transmission of the plant toxin trichosanthin via EVs have been raised (Citation301). Consequently, potential risks of toxins and allergen transfers via milk and plant EVs/nanovesicles should be addressed before considering them for clinical studies (Citation302,Citation303).
The characterization of EV-based therapeutics includes the definition of the host-to-donor relationship and the description, whether EVs derive from cultured or primary cells, tissues or fluids. Donor inclusion or exclusion criteria must be defined and donor variability, manufacturing process-related variability and the use of xenogeneic reagents, such as bovine culture supplements, are expected to influence EVs’ therapeutic activity.
EV isolation and storage
Currently, there is no state-of-the-art technology to isolate EVs, for either therapeutic application or basic research (Citation304). A conventional method to enrich for EVs is differential centrifugation, which is restricted by limited rotor sizes to the processing of approximately 400 mL in one run (Citation305). Provided that large-scale production is desired, other methods may be applied. So far, ultrafiltration to concentrate conditioned medium followed by ultracentrifugation into a sucrose cushion (Citation15–Citation17,Citation30), or a polyethylene glycol (PEG) 6000 precipitation method (Citation112) have been used to purify EVs for clinical application. Currently, ultrafiltration and size-exclusion liquid chromatography–based methods appear very promising for EV large-scale preparation (Citation306,Citation307). Regardless of whether any of these methods will be used in the long term, or whether new methods will be developed, the processes need to be standardized with regard to the reproducibility, purity, impurities, and maintenance of EVs’ functional properties. For each desired application, aspects regarding the EVs’ homogeneity or heterogeneity, purity and recovery rate must be considered. Technologies should be evaluated in the context of their scalability within confined processing times and the reproducibility of the product characteristics.
It has been observed that the method used to purify EVs can influence their integrity and impact on their subsequent bio-distribution in vivo (Citation220,Citation306). Thus, EV enrichment methods have to be carefully considered for clinical applications. Methods yielding the highest EV purity will not necessarily be optimal for recovering the therapeutically most effective EV fractions. For example, ultracentrifugation yields high EV purity but in some cases therapeutically ineffective fractions of EV aggregates (S.K. Lim, unpublished data; A. Görgens, M. Bremer and B. Giebel, unpublished data).
With regard to the development of future strategies to prepare clinical grade EVs, we can learn lessons from the production of proteins and monoclonal antibodies for therapeutic use. Monoclonal antibodies have been extensively developed for research, diagnostics, and therapeutic applications, and chromatographic purification based on size, such as size-exclusion chromatography (SEC), and charge, such as ion-exchange chromatography (IEC), are the methods of choice. Biologically active and pure samples with high yield often require a balance between multiple chromatographic steps. Indeed, EVs purified using a combination of SEC and IEC show a potent functional activity (Citation308). EVs can be fractionated by size with 10-nm accuracy using flow field-flow fractionation (FFFF), a one-phase chromatography method that fractionates particles based on differences in their diffusion properties without applying forces equally high to differential centrifugation (Citation309–Citation311). FFFF combined with multiple detectors is able to rapidly characterize and separate EVs and has the potential to facilitate a greater understanding of EV function by subtype, as well as ultimately allow for the label-free isolation of large-scale clinical EVs for the purpose of developing future EV-based diagnostics and therapeutics (Citation312).
Components attached to the EV surface or non-EV-associated co-factors might be lost during purification. Reliable biological read outs (i.e. potency assays) are required to test for the biological activity of obtained samples (see below “Quality control”).
Nowadays, standardized EV storage protocols are being developed for biobanking in which EVs are stored in their natural matrix, that is, body fluid or cell culture supernatant, after minimal processing steps to remove contents that may influence the EV-pool, for example, cells (Citation288). However for therapeutic application, there is a need to store “pure” EVs. For storage of such “pure” EVs, no standardized protocol is currently available. Since EVs interact with their matrix, for example, via electrostatic forces because of their negative zeta potential (negative surface charges) (Citation313), the matrix might not only influence EV isolation but also EV integrity and storage. For example, acidic pH shifts may neutralize fusion and aggregation inhibiting forces, which might result in EV aggregation and fusion and, consequently, to their loss of functionality. Hence, storage conditions may affect the EVs’ stability and so must be standardized and validated. A number of solvents and buffers are used for EV storage; these ranging from water and sodium chloride to PBS, TRIS-HCl, HEPES and glycerol. Importantly, glycerol and DMSO have been shown to fully or partially lyse EVs (Citation314). To conserve the EVs functional and physical properties, we recommend the storage of EVs in isotonic buffers to prevent pH shifts during storage as well as during freezing and thawing procedures. Although many groups use PBS, it has to be considered that even small amounts of calcium ions, probably included in obtained EV fractions, can be sufficient to result in the formation of nano- and microsized calcium phosphate aggregates in PBS or other phosphate containing buffers, which can interfere with EV quantification based on single particle detection (Citation315).
Storage vials can also affect the quality of EVs obtained, as EVs might unexpectedly and irreversibly bind to certain materials. Thus, vials should be used that do not affect the concentration or integrity of stored EVs. Other critical parameters are the freezing and thawing procedures, and the storage temperature. Apparently, EV integrity is more stable at −80°C and −20°C than at −190°C or at 4°C or higher temperatures (Citation313,Citation314) (Citation316). Due to the lack of sufficient studies assessing EV products after increased storing times, we recommend validation of the assumed storage conditions by conducting stability studies, including functional analyses in potency assays (Citation314). The results of such analyses will facilitate defining provisional shelf-life times of obtained and stored EV products.
Upon setting up purification and storage conditions for the clinical grade production of EVs, care should be taken that all procedures, reagents and plastic and glassware used during EV isolation and storage are certified for the production of human therapeutics. Alternatively, qualification efforts for non-certified material and procedures might be required. Adequate in-process controls during EV production should be implemented. It is recommended that these issues be discussed with national regulatory officials at the earliest possible stages of translational projects.
Currently, no standardized procedure is available for the isolation as well as for the storage of EVs. The impact of the EV matrix and applied technologies, reagents and storage containers as well as of storage times must be investigated for each intended of EV-based product. Tailor-made protocols need to be developed.
Quality control
As an essential prerequisite for the release of EV preparations for clinical application, quality release criteria have to be defined, which should include physicochemical, molecular and functional parameters. As required for each pharmaceutical, the biological activity of EVs used as therapeutics must be tested in qualified biological assays, termed “potency assays”.
Molecular and physical EV characterization
Since there are currently no “gold-standards” for EV isolation, purification and analysis, ISEV recently defined some minimal requirements to define EVs (Citation304). Because technologies to perform robust and reproducible single EV-based quantitative and qualitative analyses are very scarce, currently, analyses of EV content are mostly based on bulk analyses (e.g. the absence or presence of a certain proteins and/or total protein concentration). Single EV-based analysis platforms are used for semi-quantitative concentration analyses and for determining average-size distributions. Under the minimal requirements for the definition of EVs, EV characteristics need to be approved at the protein level, and analyses of additional cargo molecules such as lipids, metabolites and RNAs might be helpful (Citation304).
Established technologies to determine (average) EV sizes and concentrations are electron microscopy (EM), dynamic light scattering (DLS) (Citation317), nanoparticle tracking analysis (NTA) (Citation313,Citation318), resistive pulse sensing (Citation319) and fluorescence correlation spectroscopy (Citation306). Although NTA is most frequently used, all these technologies have their specific pitfalls (Citation320). Currently, a number of other technical platforms are developed or optimized for EV analyses. A special focus is directed towards flow cytometry, which enables the multiparameter analysis of single particles. For now, conventional flow cytometers used in daily routine analysis fail to analyse individual particles smaller than 300–500 nm. In contrast, groups that have optimized the set-up of configurable flow cytometers for the measurement of nanosized particles were already able to analyse viruses and EVs at a single particle resolution (Citation321–Citation323). It can be expected that novel generations of optimized flow cytometers will appear on the market, which in the near future will allow multiparameter EV analyses at the single particle level in a daily routine set-up.
In terms of molecular markers, the presence of at least three different marker proteins or lipids that are specifically enriched in EV fractions should be analysed in a semi-quantitative manner. Apart from the common EV markers, such as the tetraspanins CD9, CD63 and CD81, and components of the late endosomal machinery, for example, Tsg101 and Alix, the ganglioside GM1 has been described as a marker of endosome-derived EVs, that is, exosomes (Citation324). Notably, in the two phase I and the phase II tumour vaccination trials, the concentration of MHC II molecules per millilitre EV suspension was used for defining the applied DC-EV treatment doses (Citation15,Citation16) (Citation18). In addition to qualified EV markers, markers indicative of impurities need to be defined and analysed, for example, mitochondria and the Golgi apparatus marker for intracellular residuals, and serum albumin for the remaining extracellular components. Minimal criteria, which might include specific proteins, lipids or RNAs, depending on the intended therapeutic use, should be defined and tested with regard to the proposed function of given EV fractions (Citation304). Western blotting is currently the most commonly used method to analyse the presence of EV marker proteins. However, other technologies are feasible such as mass spectroscopy that, if desired, would allow for a more complex and comparative characterization of EVs’ constituents including proteins, lipids and metabolites (Citation325).
In addition to basic physical and molecular characterization, sterile EV preparations for pharmaceutical use must be tested for the absence of detectable viral and microbiological contaminants, including Mycoplasma, and must not contain endotoxins above defined levels (Citation326). Quality control procedures are laid down in the regulations for blood products (Citation269) as well as for ATMPs (Citation280) and relevant pharmacopoeia chapters (EP 2.6.1, EP 2.6.14, EP 2.6.27).
If plant nanovesicles (which might represent EVs) or non-human milk EVs are used, the above molecular quality criteria are not applicable. In fact, to date, limited information is available regarding the RNA and protein contents of plant-derived nanovesicles. Although some EV-associated mammalian proteins contain homologues in plants, their association with plant nanovesicles must be demonstrated. Although a good selection of antibodies directed against various Arabidopsis proteins is available, the study of other plant species may suffer from the lack of specific membrane markers and associated antibodies. However, the lack of molecular markers might be of negligible consequence, because most plant nanovesicles that have been considered for the therapeutic application, so far, have been exclusively isolated from edible plant sources. As these are part of our daily nutrition, we speculate that their clinical use may raise concerns different from those regarding the clinical application of human EVs. We recommend using the same tools and methods to analyse plant nanovesicles’ or animal milk EVs’ size distribution and quantity.
A conclusive enumeration of quality release criteria for plant or non-human nanovesicles/EVs is beyond the scope of this article. In any of the nanovesicle/EV characterization approaches, special attention has to be paid to standardization of pre-analytical conditions (Citation288).
In vitro potency assays for biological EV characterization
Due to a current lack of practical technologies to analyse EVs at the single vesicle level, the heterogeneity of EV fractions cannot be comprehensively addressed, even if they are harvested from apparently homogeneous cell sources. In attempts to unravel their biological functions, including their MoA, many studies focus on EV-associated small RNAs, such as miRNAs, and are neglecting the potential roles of proteins and lipids. Efforts to link EVs’ therapeutic effects largely to individual small RNAs may result in overlooking promising findings in different models, in which immediate therapeutic responses are more likely to be mediated by proteins or lipids than by RNAs (Citation327), or in which protein and/or lipids potentiate the biological activity of (small) RNAs. Due to the multiplicity of the active constituents, EVs are understood to act in a complex manner. Thus, it will take a considerable time to unravel the MoA of specific EVs. As therapeutic activities cannot be proposed solely by molecular profiling for pharmaceutical characterization, biologic assays need to be defined that allow the prediction of EVs’ functional properties, at best in a quantifiable manner. After their qualification, such assays can be used as potency assays that should reflect aspects of the proposed/hypothesized MoA of EV fractions being tested. For example, based on the assumption that MSC-EVs exert immune-suppressive functions in vivo, T cell proliferation assays can be applied to assess the EVs’ immune modulatory properties ex vivo (Citation108,Citation112) (Citation314). Functional end-points which qualify product release criteria need to be defined together with molecular and physical quality control data. These criteria have to be met by all manufactured products for therapeutic use. Efficacy and potency end-points will need to be evaluated during both preclinical and clinical testing and tailored for each individual EV therapy. Each type of therapeutic application may require its own specific potency assay.
Gold standards for EV quantification, and molecular and physical EV characterization are still lacking. Qualified in vitro potency assays are required to predict the intended therapeutic potential of EV fractions aimed to be used as EV-based therapeutics.
In vivo analyses of EVs
Provided relevant animal models are available for an intended use of EVs, they will help answer a variety of different questions before starting a clinical trial. EV dose-escalation studies should be performed in such models, together with toxicity assays. It has to be determined whether or not administered EVs cause side effects, for example, general toxicity, immunogenicity, immunotoxicity or tumourigenicity. In addition to safety issues, the route of administration must be defined by answering the following questions: Are the EV fractions intended to be applied systemically or topically into affected tissues, and will they be administered once or repetitively? Does the route of injection affect the relative distribution (Citation220) and the amount of EVs detected systemically? Is topical administration more effective than systemic administration? Even if a clear local distribution can be observed, it must be considered that the observed distribution does not necessarily directly correlate with therapeutic effect; effects might be indirect. Provided that labelled EVs will be used to study their bio-distribution, it must be considered that labelling, either by dyes or by genetic engineering, may affect the function of resulting EV fractions. Coupled to biodistribution experiments, half-life times and the degradation of administered EVs might be studied (Citation220,Citation328). For all animal models, the size of the groups should be sufficiently large to allow meaningful scientific interpretation of the resulting data; appropriate controls should be included. Any exclusion of controls should be justified. If in vivo potency assays are utilized to demonstrate specific therapeutic effects for an intended use of EVs, the relevance of the selected animal model must be discussed. For the translation of EVs into clinic, it will be supportive to generate convincing data in appropriate animal models mimicking clinically relevant disease conditions, whenever available.
In summary, identifying appropriate preclinical in vivo models to study the potential of EV-based therapeutics might be challenging. Nevertheless, definitive data from in vivo safety and potency assays will support the translation process for EV-based therapeutics into the clinic.
Conclusion
Although EVs were administered to humans already in the early 2000s for the treatment of cancer patients and then, more recently, for a GvHD patient, no recommended standard techniques have been established for the clinical grade production and quality control of EV-based therapeutics so far. Coupled with the fact that several manufacturing and safety considerations need to be addressed and appropriate quality controls have to be implemented and validated, it remains a challenge to set up platforms for the EVs’ clinical grade production that fulfils all necessary criteria for the successful approval of subsequent EV-based clinical trials. However, as a consequence of the substantial progress made in the EV research field, improved and standardized protocols for EVs isolation and storage, as well as improved methods, techniques and criteria for quality analyses of EV-based therapeutics, should be available soon. We hope that this position article and the various critical points highlighted for their importance to be considered for the translation of EVs into the clinic, will help to successfully advance, in a timely manner, from the preclinical setting towards clinical evaluation of EV-based therapeutics.
Conflict of interest and funding
The authors have not received any funding or benefits from industry to conduct this study. The authors acknowledge the european COST action for supporting the European Network on Microvesicles and Exosomes in Health and Disease (ME-HaD, BM1202, www.cost.eu/COST_Actions/BMBS/Actions/BM1202) who funded publication of this work.
References
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200: 373–83.
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, etal. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015; 4 27066,.
- Kim DK, Lee J, Kim SR, Choi DS, Yoon YJ, Kim JH, etal. EVpedia: a community web portal for extracellular vesicles research. Bioinformatics. 2015; 31: 933–9.
- Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. 1983; 97: 329–39.
- Pan BT, Johnstone RM. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell. 1983; 33: 967–78.
- Pan BT, Teng K, Wu C, Adam M, Johnstone RM. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol. 1985; 101: 942–8.
- Crawford N. The presence of contractile proteins in platelet microparticles isolated from human and animal platelet-free plasma. Br J Haematol. 1971; 21: 53–69.
- Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles. 2013; 2 20360, doi: http://dx.doi.org/10.3402/jev.v2i0.20360.
- Anderson HC. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J Cell Biol. 1969; 41: 59–72.
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, etal. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996; 183: 1161–72.
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654–9.
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, etal. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10: 1470–6.
- Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, etal. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006; 20: 847–856.
- Zitvogel L, Regnault A, Lozier A, Wolfers J, Flament C, Tenza D, etal. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med. 1998; 4: 594–600.
- Escudier B, Dorval T, Chaput N, Andre F, Caby MP, Novault S, etal. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: results of thefirst phase I clinical trial. J Transl Med. 2005; 3: 10.
- Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM, etal. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005; 3: 9.
- Lamparski HG, Metha-Damani A, Yao JY, Patel S, Hsu DH, Ruegg C, etal. Production and characterization of clinical grade exosomes derived from dendritic cells. J Immunol Methods. 2002; 270: 211–26.
- Viaud S, Ploix S, Lapierre V, Thery C, Commere PH, Tramalloni D, etal. Updated technology to produce highly immunogenic dendritic cell-derived exosomes of clinical grade: a critical role of interferon-gamma. J Immunother. 2011; 34: 65–75.
- Chaput N, Schartz NE, Andre F, Taieb J, Novault S, Bonnaventure P, etal. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004; 172: 2137–46.
- Segura E, Nicco C, Lombard B, Veron P, Raposo G, Batteux F, etal. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood. 2005; 106: 216–23.
- Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, etal. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007; 56: 641–8.
- Besse B, Charrier M, Lapierre V, Dansin E, Lantz O, Planchard D, etal. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology. 2016; 5:in press doi: http://dx.doi.org/10.1080/2162402X.2015.1071008.
- Gehrmann U, Naslund TI, Hiltbrunner S, Larssen P, Gabrielsson S. Harnessing the exosome-induced immune response for cancer immunotherapy. Semin Cancer Biol. 2014; 28: 58–67.
- Gehrmann U, Hiltbrunner S, Georgoudaki AM, Karlsson MC, Naslund TI, Gabrielsson S. Synergistic induction of adaptive antitumor immunity by codelivery of antigen with alpha-galactosylceramide on exosomes. Cancer Res. 2013; 73: 3865–76.
- Naslund TI, Gehrmann U, Qazi KR, Karlsson MC, Gabrielsson S. Dendritic cell-derived exosomes need to activate both T and B cells to induce antitumor immunity. J Immunol. 2013; 190: 2712–9.
- Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, etal. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001; 7: 297–303.
- Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, etal. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002; 360: 295–305.
- Altevogt P, Bretz NP, Ridinger J, Utikal J, Umansky V. Novel insights into exosome-induced, tumor-associated inflammation and immunomodulation. Semin Cancer Biol. 2014; 28: 51–7.
- Adams M, Navabi H, Croston D, Coleman S, Tabi Z, Clayton A, etal. The rationale for combined chemo/immunotherapy using a Toll-like receptor 3 (TLR3) agonist and tumour-derived exosomes in advanced ovarian cancer. Vaccine. 2005; 23: 2374–8.
- Dai S, Wei D, Wu Z, Zhou X, Wei X, Huang H, etal. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol Ther. 2008; 16: 782–90.
- Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, etal. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. 2012; 189: 2833–42.
- Bellier B, Dalba C, Clerc B, Desjardins D, Drury R, Cosset FL, etal. DNA vaccines encoding retrovirus-based virus-like particles induce efficient immune responses without adjuvant. Vaccine. 2006; 24: 2643–55.
- Desjardins D, Huret C, Dalba C, Kreppel F, Kochanek S, Cosset FL, etal. Recombinant retrovirus-like particle forming DNA vaccines in prime-boost immunization and their use for hepatitis C virus vaccine development. J Gene Med. 2009; 11: 313–25.
- Lescaille G, Pitoiset F, Macedo R, Baillou C, Huret C, Klatzmann D, etal. Efficacy of DNA vaccines forming e7 recombinant retroviral virus-like particles for the treatment of human papillomavirus-induced cancers. Human Gene Ther. 2013; 24: 533–44.
- Sedlik C, Vigneron J, Torrieri-Dramard L, Pitoiset F, Denizeau J, Chesneau C, etal. Different immunogenicity butsimilar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. J Extracell Vesicles. 2014; 3 24646, doi: http://dx.doi.org/10.3402/jev.v3.24646 [PubMed Central Full Text].
- Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, etal. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008; 68: 1228–35.
- Hartman ZC, Wei J, Glass OK, Guo H, Lei G, Yang XY, etal. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011; 29: 9361–7.
- Rountree RB, Mandl SJ, Nachtwey JM, Dalpozzo K, Do L, Lombardo JR, etal. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011; 71: 5235–44.
- Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010; 64: 163–84.
- Meckes DG Jr., Raab-Traub N. Microvesicles and viral infection. J Virol. 2011; 85: 12844–54.
- Marcilla A, Martin-Jaular L, Trelis M, de Menezes-Neto A, Osuna A, Bernal D, etal. Extracellular vesicles in parasitic diseases. J Extracell Vesicles. 2014; 3 25040, doi: http://dx.doi.org/10.3402/jev.v3.25040 [PubMed Central Full Text].
- Rodrigues ML, Nosanchuk JD, Schrank A, Vainstein MH, Casadevall A, Nimrichter L. Vesicular transport systems in fungi. Future Microbiol. 2011; 6: 1371–81.
- Schorey JS, Bhatnagar S. Exosome function: from tumor immunology to pathogen biology. Traffic. 2008; 9: 871–81.
- Kruh-Garcia NA, Wolfe LM, Chaisson LH, Worodria WO, Nahid P, Schorey JS, etal. Detection of Mycobacterium tuberculosis peptides in the exosomes of patients with active and latent M. tuberculosis infection using MRM-MS. PLoS One. 2014; 9: e103811.
- Kim JH, Lee J, Park J, Gho YS. Gram-negative and Gram-positive bacterial extracellular vesicles. Semin Cell Dev Biol. 2015; 40: 97–104.
- Oliveira DL, Freire-de-Lima CG, Nosanchuk JD, Casadevall A, Rodrigues ML, Nimrichter L. Extracellular vesicles from Cryptococcus neoformans modulate macrophage functions. Infect Immun. 2010; 78: 1601–9.
- Martin-Jaular L, Nakayasu ES, Ferrer M, Almeida IC, Del Portillo HA. Exosomes from Plasmodium yoelii-infected reticulocytes protect mice from lethal infections. PLoS One. 2011; 6: e26588.
- Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood. 2007; 110: 3234–44.
- Singh PP, Smith VL, Karakousis PC, Schorey JS. Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J Immunol. 2012; 189: 777–85.
- Cheng Y, Schorey JS. Exosomes carrying mycobacterial antigens can protect mice against Mycobacterium tuberculosis infection. Eur J Immunol. 2013; 43: 3279–90.
- Aline F, Bout D, Amigorena S, Roingeard P, Dimier-Poisson I. Toxoplasma gondii antigen-pulsed-dendritic cell-derived exosomes induce a protective immune response against T. gondii infection. Infect Immun. 2004; 72: 4127–37.
- Beauvillain C, Ruiz S, Guiton R, Bout D, Dimier-Poisson I. A vaccine based on exosomes secreted by a dendritic cell line confers protection against T. gondii infection in syngeneic and allogeneic mice. Microbes Infect. 2007; 9: 1614–22.
- Beauvillain C, Juste MO, Dion S, Pierre J, Dimier-Poisson I. Exosomes are an effective vaccine against congenital toxoplasmosis in mice. Vaccine. 2009; 27: 1750–7.
- Roy K, Hamilton DJ, Munson GP, Fleckenstein JM. Outer membrane vesicles induce immune responses to virulence proteins and protect against colonization by enterotoxigenic Escherichia coli. Clin Vaccine Immunol. 2011; 18: 1803–8.
- Kim OY, Hong BS, Park KS, Yoon YJ, Choi SJ, Lee WH, etal. Immunization with Escherichia coli outer membrane vesicles protects bacteria-induced lethality via Th1 and Th17 cell responses. J Immunol. 2013; 190: 4092–102.
- Bottero D, Gaillard ME, Errea A, Moreno G, Zurita E, Pianciola L, etal. Outer membrane vesicles derived from Bordetella parapertussis as an acellular vaccine against Bordetella parapertussis and Bordetella pertussis infection. Vaccine. 2013; 31: 5262–8.
- Alaniz RC, Deatherage BL, Lara JC, Cookson BT. Membrane vesicles are immunogenic facsimiles of Salmonella typhimurium that potently activate dendritic cells, prime B and T cell responses, and stimulate protective immunity in vivo. J Immunol. 2007; 179: 7692–701.
- Dalseg R, Wedege E, Holst J, Haugen IL, Hoiby EA, Haneberg B. Outer membrane vesicles from group B meningococci are strongly immunogenic when given intranasally to mice. Vaccine. 1999; 17: 2336–45.
- Haneberg B, Dalseg R, Wedege E, Hoiby EA, Haugen IL, Oftung F, etal. Intranasal administration of a meningococcal outer membrane vesicle vaccine induces persistent local mucosal antibodies and serum antibodies with strong bactericidal activity in humans. Infect Immun. 1998; 66: 1334–41.
- McConnell MJ, Rumbo C, Bou G, Pachon J. Outer membrane vesicles as an acellular vaccine against Acinetobacter baumannii. Vaccine. 2011; 29: 5705–10.
- Saunders NB, Shoemaker DR, Brandt BL, Moran EE, Larsen T, Zollinger WD. Immunogenicity of intranasally administered meningococcal native outer membrane vesicles in mice. Infect Immun. 1999; 67: 113–9.
- Olaya-Abril A, Prados-Rosales R, McConnell MJ, Martin-Pena R, Gonzalez-Reyes JA, Jimenez-Munguia I, etal. Characterization of protective extracellular membrane-derived vesicles produced by Streptococcus pneumoniae. J Proteomics. 2014; 106: 46–60.
- Holst J, Martin D, Arnold R, Huergo CC, Oster P, O'Hallahan J, etal. Properties and clinical performance of vaccines containing outer membrane vesicles from Neisseria meningitidis. Vaccine. 2009; 27(Suppl 2): B3–12.
- Carter NJ. Multicomponent meningococcal serogroup B vaccine (4CMenB; Bexsero((R))): a review of its use in primary and booster vaccination. BioDrugs. 2013; 27: 263–74.
- Kim OY, Choi SJ, Jang SC, Park KS, Kim SR, Choi JP, etal. Bacterial protoplast-derived nanovesicles as vaccine delivery system against bacterial infection. Nano Lett. 2015; 15: 266–74.
- Meeusen EN, Walker J, Peters A, Pastoret PP, Jungersen G. Current status of veterinary vaccines. Clin Microbiol Rev. 2007; 20: 489–510.
- Zhu L, Song H, Zhang X, Xia X, Sun H. Inhibition of porcine reproductive and respiratory syndrome virus infection by recombinant adenovirus- and/or exosome-delivered the artificial microRNAs targeting sialoadhesin and CD163 receptors. Virol J. 2014; 11: 225.
- Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, etal. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008; 10: 619–24.
- Gentili M, Kowal J, Tkach M, Satoh T, Lahaye X, Conrad C, etal. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 2015; 349: 1232–6.
- Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968; 6: 230–47.
- Friedenstein AJ, Deriglasova UF, Kulagina NN, Panasuk AF, Rudakowa SF, Luria EA, etal. Precursors for fibroblasts in different populations of hematopoietic cells as detected by the in vitro colony assay method. Exp Hematol. 1974; 2: 83–92.
- Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, etal. Multilineage potential of adult human mesenchymal stem cells. Science. 1999; 284: 143–7.
- Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012; 10: 709–16.
- Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, etal. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. 2013; 19: 35–42.
- Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, etal. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001; 7: 211–28.
- Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. 2000; 109: 235–42.
- Mareschi K, Biasin E, Piacibello W, Aglietta M, Madon E, Fagioli F. Isolation of human mesenchymal stem cells: bone marrow versus umbilical cord blood. Haematologica. 2001; 86: 1099–100.
- Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008; 2: 313–9.
- Kogler G, Sensken S, Airey JA, Trapp T, Muschen M, Feldhahn N, etal. A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med. 2004; 200: 123–35.
- Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, etal. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002; 418: 41–9.
- Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, etal. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002; 99: 3838–43.
- Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol. 2012; 12: 383–96.
- Di Ianni M, Del Papa B, De Ioanni M, Moretti L, Bonifacio E, Cecchini D, etal. Mesenchymal cells recruit and regulate T regulatory cells. Exp Hematol. 2008; 36: 309–18.
- Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, etal. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006; 107: 367–72.
- Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004; 103: 4619–21.
- Selmani Z, Naji A, Zidi I, Favier B, Gaiffe E, Obert L, etal. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells. 2008; 26: 212–22.
- Casiraghi F, Azzollini N, Cassis P, Imberti B, Morigi M, Cugini D, etal. Pretransplant infusion of mesenchymal stem cells prolongs the survival of a semiallogeneic heart transplant through the generation of regulatory T cells. J Immunol. 2008; 181: 3933–46.
- Adutler-Lieber S, Ben-Mordechai T, Naftali-Shani N, Asher E, Loberman D, Raanani E, etal. Human macrophage regulation via interaction with cardiac adipose tissue-derived mesenchymal stromal cells. J Cardiovasc Pharmacol Ther. 2013; 18: 78–86.
- Heldring N, Mager I, Wood MJ, Le Blanc K, Andaloussi SE. Therapeutic potential of multipotent mesenchymal stromal cells and their extracellular vesicles. Hum Gene Ther. 2015; 26: 506–17.
- Prockop DJ, Kota DJ, Bazhanov N, Reger RL. Evolving paradigms for repair of tissues by adult stem/progenitor cells (MSCs). J Cell Mol Med. 2010; 14: 2190–9.
- Te Boome LC, Mansilla C, van der Wagen LE, Lindemans CA, Petersen EJ, Spierings E, etal. Biomarker profiling of steroid-resistant acute GVHD in patients after infusion of mesenchymal stromal cells. Leukemia. 2015; 29: 1839–46.
- Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, etal. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008; 371: 1579–86.
- Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, etal. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004; 363: 1439–41.
- MacDonald GI, Augello A, De Bari C. Role of mesenchymal stem cells in reestablishing immunologic tolerance in autoimmune rheumatic diseases. Arthritis Rheum. 2011; 63: 2547–57.
- Dalal J, Gandy K, Domen J. Role of mesenchymal stem cell therapy in Crohn's disease. Pediatr Res. 2012; 71(4 Pt 2): 445–51.
- Tolar J, Le Blanc K, Keating A, Blazar BR. Concise review: hitting the right spot with mesenchymal stromal cells. Stem Cells. 2010; 28: 1446–55.
- Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, etal. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009; 5: 54–63.
- Zanotti L, Sarukhan A, Dander E, Castor M, Cibella J, Soldani C, etal. Encapsulated mesenchymal stem cells for in vivo immunomodulation. Leukemia. 2013; 27: 500–3.
- Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, etal. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010; 4: 214–22.
- Timmers L, Lim SK, Arslan F, Armstrong JS, Hoefer IE, Doevendans PA, etal. Reduction of myocardial infarct size by human mesenchymal stem cell conditioned medium. Stem cell Res. 2007; 1: 129–37.
- Gnecchi M, He H, Liang OD, Melo LG, Morello F, Mu H, etal. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005; 11: 367–8.
- Gnecchi M, He H, Noiseux N, Liang OD, Zhang L, Morello F, etal. Evidence supporting paracrine hypothesis for Akt-modified mesenchymal stem cell-mediated cardiac protection and functional improvement. FASEB J. 2006; 20: 661–9.
- Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, etal. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009; 20: 1053–67.
- Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, etal. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant. 2011; 26: 1474–83.
- Bruno S, Grange C, Collino F, Deregibus MC, Cantaluppi V, Biancone L, etal. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One. 2012; 7: e33115.
- He J, Wang Y, Sun S, Yu M, Wang C, Pei X, etal. Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology. 2012; 17: 493–500.
- Zhou Y, Xu H, Xu W, Wang B, Wu H, Tao Y, etal. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res Ther. 2013; 4: 34.
- Kilpinen L, Impola U, Sankkila L, Ritamo I, Aatonen M, Kilpinen S, etal. Extracellular membrane vesicles from umbilical cord blood-derived MSC protect against ischemic acute kidney injury, a feature that is lost after inflammatory conditioning. J Extracell Vesicles. 2013; 2 21927, doi: http://dx.doi.org/10.3402/jev.v2i0.21927 [PubMed Central Full Text].
- Reis LA, Borges FT, Simoes MJ, Borges AA, Sinigaglia-Coimbra R, Schor N. Bone marrow-derived mesenchymal stem cells repaired but did not prevent gentamicin-induced acute kidney injury through paracrine effects in rats. PLoS One. 2012; 7: e44092.
- Zou X, Zhang G, Cheng Z, Yin D, Du T, Ju G, etal. Microvesicles derived from human Wharton's Jelly mesenchymal stromal cells ameliorate renal ischemia-reperfusion injury in rats by suppressing CX3CL1. Stem Cell Res Ther. 2014; 5: 40.
- Zhang B, Yin Y, Lai RC, Tan SS, Choo AB, Lim SK. Mesenchymal stem cell secretes immunologically active exosomes. Stem Cells Dev. 2014; 23: 1233–44.
- Kordelas L, Rebmann V, Ludwig AK, Radtke S, Ruesing J, Doeppner TR, etal. MSC-derived exosomes: a novel tool to treat therapy-refractory graft-versus-host disease. Leukemia. 2014; 28: 970–3.
- Herrera MB, Fonsato V, Gatti S, Deregibus MC, Sordi A, Cantarella D, etal. Human liver stem cell-derived microvesicles accelerate hepatic regeneration in hepatectomized rats. J Cell Mol Med. 2010; 14: 1605–18.
- Tan CY, Lai RC, Wong W, Dan YY, Lim SK, Ho HK. Mesenchymal stem cell-derived exosomes promote hepatic regeneration in drug-induced liver injury models. Stem Cell Res Ther. 2014; 5: 76.
- Li T, Yan Y, Wang B, Qian H, Zhang X, Shen L, etal. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013; 22: 845–54.
- Lee C, Mitsialis SA, Aslam M, Vitali SH, Vergadi E, Konstantinou G, etal. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation. 2012; 126: 2601–11.
- Zhu YG, Feng XM, Abbott J, Fang XH, Hao Q, Monsel A, etal. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells. 2014; 32: 116–25.
- Nakamura Y, Miyaki S, Ishitobi H, Matsuyama S, Nakasa T, Kamei N, etal. Mesenchymal-stem-cell-derived exosomes accelerate skeletal muscle regeneration. FEBS Lett. 2015; 589: 1257–65.
- Zhang B, Wang M, Gong A, Zhang X, Wu X, Zhu Y, etal. HucMSC-exosome mediated-Wnt4 signaling is required for cutaneous wound healing. Stem Cells. 2015; 33: 2158–68.
- Zhang HC, Liu XB, Huang S, Bi XY, Wang HX, Xie LX, etal. Microvesicles derived from human umbilical cord mesenchymal stem cells stimulated by hypoxia promote angiogenesis both in vitro and in vivo. Stem Cells Dev. 2012; 21: 3289–97.
- Gutierrez-Fernandez M, Rodriguez-Frutos B, Ramos-Cejudo J, Teresa Vallejo-Cremades M, Fuentes B, Cerdan S, etal. Effects of intravenous administration of allogenic bone marrow- and adipose tissue-derived mesenchymal stem cells on functional recovery and brain repair markers in experimental ischemic stroke. Stem Cell Res Ther. 2013; 4: 11.
- Hess DC, Hill WD. Cell therapy for ischaemic stroke. Cell Prolif. 2011; 44(Suppl 1): 1–8.
- Mendez-Otero R, de Freitas GR, Andre C, de Mendonca ML, Friedrich M, Oliveira-Filho J. Potential roles of bone marrow stem cells in stroke therapy. Regen Med. 2007; 2: 417–23.
- Jellema RK, Wolfs TG, Lima Passos V, Zwanenburg A, Ophelders DR, Kuypers E, etal. Mesenchymal stem cells induce T-cell tolerance and protect the preterm brain after global hypoxia-ischemia. PLoS One. 2013; 8: e73031.
- Gortner L, Felderhoff-Muser U, Monz D, Bieback K, Kluter H, Jellema R, etal. Regenerative therapies in neonatology: clinical perspectives. Klin Pädiatr. 2012; 224: 233–40.
- Chen J, Shehadah A, Pal A, Zacharek A, Cui X, Cui Y, etal. Neuroprotective effect of human placenta-derived cell treatment of stroke in rats. Cell Transplant. 2013; 22: 871–9.
- Cheng Q, Zhang Z, Zhang S, Yang H, Zhang X, Pan J, etal. Human umbilical cord mesenchymal stem cells protect against ischemic brain injury in mouse by regulating peripheral immunoinflammation. Brain Res. 2015; 1594: 293–304.
- Hsieh JY, Wang HW, Chang SJ, Liao KH, Lee IH, Lin WS, etal. Mesenchymal stem cells from human umbilical cord express preferentially secreted factors related to neuroprotection, neurogenesis, and angiogenesis. PLoS One. 2013; 8: e72604.
- Xin H, Li Y, Cui Y, Yang JJ, Zhang ZG, Chopp M. Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab. 2013; 33: 1711–5.
- Raisi A, Azizi S, Delirezh N, Heshmatian B, Farshid AA, Amini K. The mesenchymal stem cell-derived microvesicles enhance sciatic nerve regeneration in rat: a novel approach in peripheral nerve cell therapy. J Trauma Acute Care Surg. 2014; 76: 991–7.
- Doeppner TR, Herz J, Gorgens A, Schlechter J, Ludwig AK, Radtke S, etal. Extracellular vesicles improve post-stroke neuroregeneration and prevent postischemic immunosuppression. Stem Cells Transl Med. 2015; 4: 1131–43.
- Jacobs SE, Berg M, Hunt R, Tarnow-Mordi WO, Inder TE, Davis PG. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev. 2013; 1: CD003311. [PubMed Abstract].
- Buonocore G, Perrone S, Turrisi G, Kramer BW, Balduini W. New pharmacological approaches in infants with hypoxic-ischemic encephalopathy. Curr Pharm Des. 2012; 18: 3086–100.
- Donega V, Nijboer CH, van Tilborg G, Dijkhuizen RM, Kavelaars A, Heijnen CJ. Intranasally administered mesenchymal stem cells promote a regenerative niche for repair of neonatal ischemic brain injury. Exp Neurol. 2014; 261: 53–64.
- van Velthoven CT, Kavelaars A, van Bel F, Heijnen CJ. Repeated mesenchymal stem cell treatment after neonatal hypoxia-ischemia has distinct effects on formation and maturation of new neurons and oligodendrocytes leading to restoration of damage, corticospinal motor tract activity, and sensorimotor function. J Neurosci. 2010; 30: 9603–11.
- Ophelders DR, Wolfs TG, Jellema RK, Zwanenburg A, Ludwig AK, Radtke S, etal. Mesenchymal stromal cell-derived extracellular vesicles protect the fetal brain after hypoxia-ischemia. (in preparation).
- Rivera FJ, Aigner L. Adult mesenchymal stem cell therapy for myelin repair in multiple sclerosis. Biol Res. 2012; 45: 257–68.
- Katsuda T, Tsuchiya R, Kosaka N, Yoshioka Y, Takagaki K, Oki K, etal. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci Rep. 2013; 3: 1197.
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011; 29: 341–5.
- Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, etal. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013; 21: 185–91.
- Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, etal. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord. 2014; 29: 1476–85.
- El-Andaloussi S, Lee Y, Lakhal-Littleton S, Li J, Seow Y, Gardiner C, etal. Exosome-mediated delivery of siRNA in vitro and in vivo. Nat Protoc. 2012; 7: 2112–26.
- Zhang B, Wu X, Zhang X, Sun Y, Yan Y, Shi H, etal. Human umbilical cord mesenchymal stem cell exosomes enhance angiogenesis through the Wnt4/beta-catenin pathway. Stem Cells Transl Med. 2015; 4: 513–22.
- Bian S, Zhang L, Duan L, Wang X, Min Y, Yu H. Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J Mol Med. 2014; 92: 387–97.
- Salomon C, Ryan J, Sobrevia L, Kobayashi M, Ashman K, Mitchell M, etal. Exosomal signaling during hypoxia mediates microvascular endothelial cell migration and vasculogenesis. PLoS One. 2013; 8: e68451.
- Chen J, Liu Z, Hong MM, Zhang H, Chen C, Xiao M, etal. Proangiogenic compositions of microvesicles derived from human umbilical cord mesenchymal stem cells. PLoS One. 2014; 9: e115316.
- Zhu W, Huang L, Li Y, Zhang X, Gu J, Yan Y, etal. Exosomes derived from human bone marrow mesenchymal stem cells promote tumor growth in vivo. Cancer Lett. 2012; 315: 28–37.
- Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, etal. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013; 123: 1542–55.
- Bruno S, Collino F, Iavello A, Camussi G. Effects of mesenchymal stromal cell-derived extracellular vesicles on tumor growth. Front Immunol. 2014; 5: 382.
- Katakowski M, Buller B, Zheng X, Lu Y, Rogers T, Osobamiro O, etal. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013; 335: 201–4.
- Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi RU, etal. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci Signal. 2014; 7: ra63.
- Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, etal. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004; 104: 2752–60.
- Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, etal. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. 2007; 109: 1801–9.
- Yoon CH, Hur J, Park KW, Kim JH, Lee CS, Oh IY, etal. Synergistic neovascularization by mixed transplantation of early endothelial progenitor cells and late outgrowth endothelial cells: the role of angiogenic cytokines and matrix metalloproteinases. Circulation. 2005; 112: 1618–27.
- Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, etal. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004; 24: 288–93.
- Denecke B, Horsch LD, Radtke S, Fischer JC, Horn PA, Giebel B. Human endothelial colony-forming cells expanded with an improved protocol are a useful endothelial cell source for scaffold-based tissue engineering. J Tissue Eng Regen Med. 2015; 9: E84–97.
- Reinisch A, Hofmann NA, Obenauf AC, Kashofer K, Rohde E, Schallmoser K, etal. Humanized large-scale expanded endothelial colony-forming cells function in vitro and in vivo. Blood. 2009; 113: 6716–25.
- Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, etal. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003; 348: 593–600.
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, etal. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997; 275: 964–7.
- Ito H, Rovira II, Bloom ML, Takeda K, Ferrans VJ, Quyyumi AA, etal. Endothelial progenitor cells as putative targets for angiostatin. Cancer Res. 1999; 59: 5875–7.
- Gulati R, Jevremovic D, Peterson TE, Chatterjee S, Shah V, Vile RG, etal. Diverse origin and function of cells with endothelial phenotype obtained from adult human blood. Circ Res. 2003; 93: 1023–5.
- Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005; 106: 1525–31.
- Zentilin L, Tafuro S, Zacchigna S, Arsic N, Pattarini L, Sinigaglia M, etal. Bone marrow mononuclear cells are recruited to the sites of VEGF-induced neovascularization but are not incorporated into the newly formed vessels. Blood. 2006; 107: 3546–54.
- Rohde E, Malischnik C, Thaler D, Maierhofer T, Linkesch W, Lanzer G, etal. Blood monocytes mimic endothelial progenitor cells. Stem Cells. 2006; 24: 357–67.
- Rohde E, Bartmann C, Schallmoser K, Reinisch A, Lanzer G, Linkesch W, etal. Immune cells mimic the morphology of endothelial progenitor colonies in vitro. Stem Cells. 2007; 25: 1746–52.
- Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, etal. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ Res. 2011; 109: 724–8.
- Sheldon H, Heikamp E, Turley H, Dragovic R, Thomas P, Oon CE, etal. New mechanism for Notch signaling to endothelium at a distance by Delta-like 4 incorporation into exosomes. Blood. 2010; 116: 2385–94.
- van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM, etal. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood. 2013; 121: 3997–4006, S1–15.
- Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, etal. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007; 110: 2440–8.
- Ranghino A, Cantaluppi V, Grange C, Vitillo L, Fop F, Biancone L, etal. Endothelial progenitor cell-derived microvesicles improve neovascularization in a murine model of hindlimb ischemia. Int J Immunopathol Pharmacol. 2012; 25: 75–85.
- Cantaluppi V, Gatti S, Medica D, Figliolini F, Bruno S, Deregibus MC, etal. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012; 82: 412–27.
- Njock MS, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, etal. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood. 2015; 125: 3202–12.
- Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, etal. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. 2013; 43: 2430–40.
- Xie Y, Zhang X, Zhao T, Li W, Xiang J. Natural CD8(+)25(+) regulatory T cell-secreted exosomes capable of suppressing cytotoxic T lymphocyte-mediated immunity against B16 melanoma. Biochem Biophys Res Commun. 2013; 438: 152–5.
- Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, etal. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity. 2014; 41: 89–103.
- Yu X, Huang C, Song B, Xiao Y, Fang M, Feng J, etal. CD4+CD25+ regulatory T cells-derived exosomes prolonged kidney allograft survival in a rat model. Cell Immunol. 2013; 285: 62–8.
- Pluchino S, Quattrini A, Brambilla E, Gritti A, Salani G, Dina G, etal. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature. 2003; 422: 688–94.
- Pluchino S, Zanotti L, Rossi B, Brambilla E, Ottoboni L, Salani G, etal. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature. 2005; 436: 266–71.
- Pluchino S, Zanotti L, Brambilla E, Rovere-Querini P, Capobianco A, Alfaro-Cervello C, etal. Immune regulatory neural stem/precursor cells protect from central nervous system autoimmunity by restraining dendritic cell function. PLoS One. 2009; 4: e5959.
- Pluchino S, Gritti A, Blezer E, Amadio S, Brambilla E, Borsellino G, etal. Human neural stem cells ameliorate autoimmune encephalomyelitis in non-human primates. Ann Neurol. 2009; 66: 343–54.
- Ziv Y, Avidan H, Pluchino S, Martino G, Schwartz M. Synergy between immune cells and adult neural stem/progenitor cells promotes functional recovery from spinal cord injury. Proc Natl Acad Sci USA. 2006; 103: 13174–9.
- Cusimano M, Biziato D, Brambilla E, Donega M, Alfaro-Cervello C, Snider S, etal. Transplanted neural stem/precursor cells instruct phagocytes and reduce secondary tissue damage in the injured spinal cord. Brain. 2012; 135(Pt 2): 447–60.
- Bacigaluppi M, Pluchino S, Peruzzotti-Jametti L, Kilic E, Kilic U, Salani G, etal. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain. 2009; 132(Pt 8): 2239–51.
- Döppner TR, Ewert TA, Tonges L, Herz J, Zechariah A, ElAli A, etal. Transduction of neural precursor cells with TAT-heat shock protein 70 chaperone: therapeutic potential against ischemic stroke after intrastriatal and systemic transplantation. Stem Cells. 2012; 30: 1297–310.
- Rossi F, Cattaneo E. Opinion: neural stem cell therapy for neurological diseases: dreams and reality. Nat Rev Neurosci. 2002; 3: 401–9.
- Pluchino S, Cossetti C. How stem cells speak with host immune cells in inflammatory brain diseases. Glia. 2013; 61: 1379–401.
- Cossetti C, Iraci N, Mercer TR, Leonardi T, Alpi E, Drago D, etal. Extracellular vesicles from neural stem cells transfer IFN-gamma via Ifngr1 to activate Stat1 signaling in target cells. Mol Cell. 2014; 56: 193–204.
- Frühbeis C, Frohlich D, Kuo WP, Amphornrat J, Thilemann S, Saab AS, etal. Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication. PLoS Biol. 2013; 11: e1001604.
- Fröhlich D, Kuo WP, Fruhbeis C, Sun JJ, Zehendner CM, Luhmann HJ, etal. Multifaceted effects of oligodendroglial exosomes on neurons: impact on neuronal firing rate, signal transduction and gene regulation. Philos Trans R Soc Lond B Biol Sci. 2014; 369 20130510, http://dx.doi.org/10.1098/rstb.2013.0510.
- Lopez-Verrilli MA, Court FA. Transfer of vesicles from schwann cells to axons: a novel mechanism of communication in the peripheral nervous system. Front Physiol. 2012; 3: 205.
- Lopez-Verrilli MA, Picou F, Court FA. Schwann cell-derived exosomes enhance axonal regeneration in the peripheral nervous system. Glia. 2013; 61: 1795–806.
- Ridder K, Keller S, Dams M, Rupp AK, Schlaudraff J, Turco DD, etal. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol. 2014; 12: e1001874.
- Dalton JP, Robinson MW, Mulcahy G, O'Neill SM, Donnelly S. Immunomodulatory molecules of Fasciola hepatica: candidates for both vaccine and immunotherapeutic development. Vet Parasitol. 2013; 195: 272–85.
- Marcilla A, Trelis M, Cortes A, Sotillo J, Cantalapiedra F, Minguez MT, etal. Extracellular vesicles from parasitic helminths contain specific excretory/secretory proteins and are internalized in intestinal host cells. PLoS One. 2012; 7: e45974.
- Buck AH, Coakley G, Simbari F, McSorley HJ, Quintana JF, Le Bihan T, etal. Exosomes secreted by nematode parasites transfer small RNAs to mammalian cells and modulate innate immunity. Nat Commun. 2014; 5: 5488.
- Bobis-Wozowicz S, Kmiotek K, Sekula M, Kedracka-Krok S, Kamycka E, Adamiak M, etal. Human induced pluripotent stem cell-derived microvesicles transmit rnas and proteins to recipient mature heart cells modulating cell fate and behavior. Stem Cells. 2015; 33: 2748–61.
- Wang Y, Zhang L, Li Y, Chen L, Wang X, Guo W, etal. Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int J Cardiol. 2015; 192: 61–9.
- Hu GW, Li Q, Niu X, Hu B, Liu J, Zhou SM, etal. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res Ther. 2015; 6: 10.
- Samsonraj RM, Rai B, Sathiyanathan P, Puan KJ, Rotzschke O, Hui JH, etal. Establishing criteria for human mesenchymal stem cell potency. Stem Cells. 2015; 33: 1878–91.
- Yang Z, Dong P, Fu X, Li Q, Ma S, Wu D, etal. CD49f acts as an inflammation sensor to regulate differentiation, adhesion, and migration of human mesenchymal stem cells. Stem Cells. 2015; 33: 2798–810.
- Kaltz N, Ringe J, Holzwarth C, Charbord P, Niemeyer M, Jacobs VR, etal. Novel markers of mesenchymal stem cells defined by genome-wide gene expression analysis of stromal cells from different sources. Exp Cell Res. 2010; 316: 2609–17.
- Buhring HJ, Treml S, Cerabona F, de Zwart P, Kanz L, Sobiesiak M. Phenotypic characterization of distinct human bone marrow-derived MSC subsets. Ann N Y Acad Sci. 2009; 1176: 124–34.
- Collino F, Bruno S, Incarnato D, Dettori D, Neri F, Provero P, etal. AKI recovery induced by mesenchymal stromal cell-derived extracellular vesicles carrying MicroRNAs. J Am Soc Nephrol. 2015; 26: 2349–60.
- Xin H, Li Y, Liu Z, Wang X, Shang X, Cui Y, etal. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells. 2013; 31: 2737–46.
- Feng Y, Huang W, Wani M, Yu X, Ashraf M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS One. 2014; 9: e88685.
- Walker JD, Maier CL, Pober JS. Cytomegalovirus-infected human endothelial cells can stimulate allogeneic CD4+ memory T cells by releasing antigenic exosomes. J Immunol. 2009; 182: 1548–59.
- Damo M, Wilson DS, Simeoni E, Hubbell JA. TLR-3 stimulation improves anti-tumor immunity elicited by dendritic cell exosome-based vaccines in a murine model of melanoma. Sci Rep. 2015; 5: 17622.
- Giri PK, Kruh NA, Dobos KM, Schorey JS. Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis-infected and culture filtrate protein-treated macrophages. Proteomics. 2010; 10: 3190–202.
- Van Niel G, Mallegol J, Bevilacqua C, Candalh C, Brugiere S, Tomaskovic-Crook E, etal. Intestinal epithelial exosomes carry MHC class II/peptides able to inform the immune system in mice. Gut. 2003; 52: 1690–7.
- Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. 2011; 187: 676–83.
- Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, etal. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol. 2014; 177: 531–43.
- Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. 2012; 367: 2322–33.
- Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014; 509: 310–7.
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, etal. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007; 204: 1257–65.
- Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC. CD39 and control of cellular immune responses. Purinergic Signal. 2007; 3: 171–80.
- Amarnath S, Foley JE, Farthing DE, Gress RE, Laurence A, Eckhaus MA, etal. Bone marrow-derived mesenchymal stromal cells harness purinergenic signaling to tolerize human Th1 cells in vivo. Stem Cells. 2015; 33: 1200–12.
- Nery AA, Nascimento IC, Glaser T, Bassaneze V, Krieger JE, Ulrich H. Human mesenchymal stem cells: from immunophenotyping by flow cytometry to clinical applications. Cytometry A. 2013; 83: 48–61.
- van der Meel R, Fens MH, Vader P, van Solinge WW, Eniola-Adefeso O, Schiffelers RM. Extracellular vesicles as drug delivery systems: lessons from the liposome field. J Control Release. 2014; 195: 72–85.
- Johnsen KB, Gudbergsson JM, Skov MN, Pilgaard L, Moos T, Duroux M. A comprehensive overview of exosomes as drug delivery vehicles - endogenous nanocarriers for targeted cancer therapy. Biochim Biophys Acta. 2014; 1846: 75–87.
- Wiklander OP, Nordin JZ, O'Loughlin A, Gustafsson Y, Corso G, Mager I, etal. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015; 4 26316, doi: http://dx.doi.org/10.3402/jev.v4.26316.
- El Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013; 12: 347–57.
- Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013; 12: 967–77.
- Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, etal. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci USA. 2010; 107: 6328–33.
- Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan ML, Karlsson JM, etal. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012; 119: 756–66.
- Zomer A, Maynard C, Verweij FJ, Kamermans A, Schafer R, Beerling E, etal. In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015; 161: 1046–57.
- Marcus ME, Leonard JN. FedExosomes: engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals. 2013; 6: 659–80.
- Vader P, Kooijmans SA, Stremersch S, Raemdonck K. New considerations in the preparation of nucleic acid-loaded extracellular vesicles. Ther Deliv. 2014; 5: 105–7.
- Wahlgren J, De LKT, Brisslert M, Vaziri Sani F, Telemo E, Sunnerhagen P, etal. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012; 40: e130.
- Kooijmans SA, Stremersch S, Braeckmans K, de Smedt SC, Hendrix A, Wood MJ, etal. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J Control Release. 2013; 172: 229–38.
- Kalra H, Simpson RJ, Ji H, Aikawa E, Altevogt P, Askenase P, etal. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012; 10: e1001450.
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010; 285: 17442–52.
- Zhang Y, Liu D, Chen X, Li J, Li L, Bian Z, etal. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol Cell. 2010; 39: 133–44.
- Zhang Y, Li L, Yu J, Zhu D, Zhang Y, Li X, etal. Microvesicle-mediated delivery of transforming growth factor beta1 siRNA for the suppression of tumor growth in mice. Biomaterials. 2014; 35: 4390–400.
- Zhou Y, Xiong M, Fang L, Jiang L, Wen P, Dai C, etal. miR-21-containing microvesicles from injured tubular epithelial cells promote tubular phenotype transition by targeting PTEN protein. Am J Pathol. 2013; 183: 1183–96.
- Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, etal. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013; 4: 2980.
- Koppers-Lalic D, Hackenberg M, Bijnsdorp IV, van Eijndhoven MA, Sadek P, Sie D, etal. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 2014; 8: 1649–58.
- Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, etal. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS One. 2014; 9: e88193.
- De Milito A, Fais S. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol. 2005; 1: 779–86.
- Parolini I, Federici C, Raggi C, Lugini L, Palleschi S, De Milito A, etal. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J Biol Chem. 2009; 284: 34211–22.
- Pascucci L, Cocce V, Bonomi A, Ami D, Ceccarelli P, Ciusani E, etal. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: a new approach for drug delivery. J Control Release. 2014; 192: 262–70.
- Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, etal. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014; 35: 2383–90.
- Tang K, Zhang Y, Zhang H, Xu P, Liu J, Ma J, etal. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun. 2012; 3: 1282.
- Bandyopadhyay D. Farmer to pharmacist: curcumin as an anti-invasive and antimetastatic agent for the treatment of cancer. Front Chem. 2014; 2: 113.
- Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, etal. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010; 18: 1606–14.
- Zhuang X, Xiang X, Grizzle W, Sun D, Zhang S, Axtell RC, etal. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. 2011; 19: 1769–79.
- Wang Q, Zhuang X, Mu J, Deng ZB, Jiang H, Zhang L, etal. Delivery of therapeutic agents by nanoparticles made of grapefruit-derived lipids. Nat Commun. 2013; 4: 1867.
- Ju S, Mu J, Dokland T, Zhuang X, Wang Q, Jiang H, etal. Grape exosome-like nanoparticles induce intestinal stem cells and protect mice from DSS-induced colitis. Mol Ther. 2013; 21: 1345–57.
- Munagala R, Aqil F, Jeyabalan J, Gupta RC. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2015; 371: 48–61.
- Jang SC, Gho YS. Could bioengineered exosome- mimetic nanovesicles be an efficient strategy for the delivery of chemotherapeutics?. Nanomedicine. 2014; 9: 177–80.
- Jang SC, Kim OY, Yoon CM, Choi DS, Roh TY, Park J, etal. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano. 2013; 7: 7698–710.
- EMA/837805/2011. Questions and answers on biosimilar medicines. (similar biological medicinal products)..
- Ilic N, Savic S, Siegel E, Atkinson K, Tasic L. Examination of the regulatory frameworks applicable to biologic drugs (including stem cells and their progeny) in Europe, the U.S., and Australia: part II--a method of software documentary analysis. Stem Cells Transl Med. 2012; 1: 909–20.
- Ilic N, Savic S, Siegel E, Atkinson K, Tasic L. Examination of the regulatory frameworks applicable to biologic drugs (including stem cells and their progeny) in Europe, the U.S., and Australia: part I--a method of manual documentary analysis. Stem Cells Transl Med. 2012; 1: 898–908.
- DIRECTIVE2001/83/EC. On the Community code relating to medicinal products for human use.
- European_Union. Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use; Annex 2, Manufacture of Biological Active Substances and Medicinal Products for Human Use.
- GMPguidelines. [cited 20 Oct 2015]. Available from: http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm.
- ClinicalTrialsGuidelines. [cited 20 Oct 2015]. Available from: http://ec.europa.eu/health/documents/eudralex/vol-10/index_en.htm.
- EMA/CHMP/BWP/534898/2008. Guideline on the requirements for quality documentation concerning biological investigational medicinal products in clinical trials.
- DocumentQ5D. Derivation and characterization of cell substrates used for production of biotechnological/biological products.
- DocumentS6R1. Preclinical safety evaluation of biotechnology-derived pharmaceuticals.
- ICH. [cited 20 Oct 2015]. Available from: http://www.ich.org/products/guidelines.html.
- DocumentQ7. Good Manufacturing Practice (GMP) Guide for Active Pharmaceutical Ingredients.
- BiologicsBloodVaccines. [cited 20 Oct 2015]. Available from: http://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/UCM278673.pdf.
- EMA/CAT/CPWP/686637/2011. Guideline on the risk-based approach according to annex I, part IV of Directive 2001/83/EC applied to Advanced therapy medicinal products.
- EMA/CHMP/QWP/185401/2004. Guideline on the requirements to the chemical and pharmaceutical quality documentation concerning investigational medicinal products in clinical trials..
- EMEA/CHMP/SWP/28367/07. Guideline on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials with Investigational Medicinal Product.
- DIRECTIVE2004/23/EC. Setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells..
- DIRECTIVE2006/17/EC. Implementing Directive 2004/23/EC of the European Parliament and of the Council as regards certain technical requirements for the donation, procurement and testing of human tissues and cells..
- DIRECTIVE2002/98/EC. Setting standards of quality and safety for the collection, testing, processing, storage and distribution of human blood and blood components and amending Directive 2001/83/EC..
- REGULATION/EC/726/2004. Laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency..
- EMA/CAT/600280/2010rev.1. Revision of the CAT Reflection Paper on ATMP classification..
- REGULATION/EC/1394/2007. On advanced therapy medicinal products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004..
- Ruiss R, Jochum S, Mocikat R, Hammerschmidt W, Zeidler R. EBV-gp350 confers B-cell tropism to tailored exosomes and is a neo-antigen in normal and malignant B cells--a new option for the treatment of B-CLL. PLoS One. 2011; 6: e25294.
- ICHS7A. Safety pharmacology studies for human pharmaceuticals.
- ICH/harmonized/TripartiteGuideline. Preclinical safety evaluation of biotechnology-derived pharmaceuticals’, S6[R1], page 3..
- EMEA/CHMP/410869/2006. Guideline on human cell-based medicinal products.
- Danesh A, Inglis HC, Jackman RP, Wu S, Deng X, Muench MO, etal. Exosomes from red blood cell units bind to monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood. 2014; 123: 687–96.
- Kriebardis A, Antonelou M, Stamoulis K, Papassideri I. Cell-derived microparticles in stored blood products: innocent-bystanders or effective mediators of post-transfusion reactions?. Blood Transfus. 2012; 10(Suppl 2): s25–38.
- Eichler H, Schrezenmeier H, Schallmoser K, Strunk D, Nystedt J, Kaartinen T, etal. Donor selection and release criteria of cellular therapy products. Vox Sang. 2013; 104: 67–91.
- REGULATION/EC/1394/2007. Regulation on advanced therapy medicinal products..
- Document32004L0023. [cited 20 Oct 2015]. Available from: http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:32004L0023.
- Petersdorf EW. The major histocompatibility complex: a model for understanding graft-versus-host disease. Blood. 2013; 122: 1863–72.
- Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003; 31: 890–6.
- Hemeda H, Jakob M, Ludwig AK, Giebel B, Lang S, Brandau S. Interferon-gamma and tumor necrosis factor-alpha differentially affect cytokine expression and migration properties of mesenchymal stem cells. Stem Cells Dev. 2010; 19: 693–706.
- Ankrum JA, Ong JF, Karp JM. Mesenchymal stem cells: immune evasive, not immune privileged. Nat Biotechnol. 2014; 32: 252–60.
- Katsuda T, Ochiya T. Molecular signatures of mesenchymal stem cell-derived extracellular vesicle-mediated tissue repair. Stem Cell ResTher. 2015; 6: 212.
- Chen TS, Arslan F, Yin Y, Tan SS, Lai RC, Choo AB, etal. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J Transl Med. 2011; 9: 47.
- Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J, etal. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013; 2 20360, doi: http://dx.doi.org/10.3402/jev.v2i0.20360 [PubMed Central Full Text].
- Li J, Lee Y, Johansson HJ, Mager I, Vader P, Nordin JZ, etal. Serum-free culture alters the quantity and protein composition of neuroblastoma-derived extracellular vesicles. J Extracell Vesicles. 2015; 4 26883, doi: http://dx.doi.org/10.3402/jev.v4.26883.
- Belting M, Christianson HC. Role of exosomes and microvesicles in hypoxia-associated tumour development and cardiovascular disease. J Intern Med. 2015; 278: 251–63.
- Eldh M, Ekstrom K, Valadi H, Sjostrand M, Olsson B, Jernas M, etal. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS One. 2010; 5: e15353.
- Ekstrom K, Omar O, Graneli C, Wang X, Vazirisani F, Thomsen P. Monocyte exosomes stimulate the osteogenic gene expression of mesenchymal stem cells. PLoS One. 2013; 8: e75227.
- Kato T, Miyaki S, Ishitobi H, Nakamura Y, Nakasa T, Lotz MK, etal. Exosomes from IL-1beta stimulated synovial fibroblasts induce osteoarthritic changes in articular chondrocytes. Arthritis Res Ther. 2014; 16: R163.
- Saunderson SC, Schuberth PC, Dunn AC, Miller L, Hock BD, MacKay PA, etal. Induction of exosome release in primary B cells stimulated via CD40 and the IL-4 receptor. J Immunol. 2008; 180: 8146–52.
- Atai NA, Balaj L, van Veen H, Breakefield XO, Jarzyna PA, Van Noorden CJ, etal. Heparin blocks transfer of extracellular vesicles between donor and recipient cells. J Neurooncol. 2013; 115: 343–51.
- Jeppesen DK, Hvam ML, Primdahl-Bengtson B, Boysen AT, Whitehead B, Dyrskjot L, etal. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J Extracell Vesicles. 2014; 3 25011, doi: http://dx.doi.org/10.3402/jev.v3.25011.
- Eitan E, Zhang S, Witwer KW, Mattson MP. Extracellular vesicle-depleted fetal bovine and human sera have reduced capacity to support cell growth. J Extracell Vesicles. 2015; 4 26373, doi: http://dx.doi.org/10.3402/jev.v4.26373.
- Shelke GV, Lasser C, Gho YS, Lotvall J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J Extracell Vesicles. 2014; 3 24783, doi: http://dx.doi.org/10.3402/jev.v3.24783 [PubMed Central Full Text].
- EMEA/CHMP/457920/2012. Guideline on the use of bovine serum in the manufacture of human biological medicinal products.
- EMEA/410/01Rev3. Note for guidance on minimising the risk of transmitting animal spongiform encephalopathy agents via human and veterinary medicinal products (2011/C 73/01).
- Abrami L, Brandi L, Moayeri M, Brown MJ, Krantz BA, Leppla SH, etal. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013; 5: 986–96.
- Pieters BC, Arntz OJ, Bennink MB, Broeren MG, van Caam AP, Koenders MI, etal. Commercial cow milk contains physically stable extracellular vesicles expressing immunoregulatory TGF-beta. PLoS One. 2015; 10: e0121123.
- Wolf T, Baier SR, Zempleni J. The intestinal transport of bovine milk exosomes is mediated by endocytosis in human colon carcinoma caco-2 cells and rat small intestinal IEC-6 cells. J Nutr. 2015; 145: 2201–6.
- Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, etal. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014; 3 26913, doi: http://dx.doi.org/10.3402/jev.v3.26913 [PubMed Central Full Text].
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3:Unit 3 22.
- Nordin JZ, Lee Y, Vader P, Mager I, Johansson HJ, Heusermann W, etal. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine. 2015; 11: 879–83.
- Vishnubhatla I, Corteling R, Stevanato L, Hicks C, Sinden J. The development of stem cell-derived exosomes as a cell-free regenerative medicine. J Circ Biomark. 2014. 3:2, doi: http://dx.doi.org/10.5772/58597.
- Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, etal. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012; 151: 1542–56.
- Kang D, Oh S, Ahn SM, Lee BH, Moon MH. Proteomic analysis of exosomes from human neural stem cells by flow field-flow fractionation and nanoflow liquid chromatography-tandem mass spectrometry. J Proteome Res. 2008; 7: 3475–80.
- van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012; 64: 676–705.
- Sitar S, Kejzar A, Pahovnik D, Kogej K, Tusek-Znidaric M, Lenassi M, etal. Size characterization and quantification of exosomes by asymmetrical-flow field-flow fractionation. Anal Chem. 2015; 87: 9225–33.
- Petersen KE, Manangon E, Hood JL, Wickline SA, Fernandez DP, Johnson WP, etal. A review of exosome separation techniques and characterization of B16-F10 mouse melanoma exosomes with AF4-UV-MALS-DLS-TEM. Anal Bioanal Chem. 2014; 406: 7855–66.
- Sokolova V, Ludwig AK, Hornung S, Rotan O, Horn PA, Epple M, etal. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids Surf B Biointerfaces. 2011; 87: 146–50.
- Lorincz AM, Timar CI, Marosvari KA, Veres DS, Otrokocsi L, Kittel A, etal. Effect of storage on physical and functional properties of extracellular vesicles derived from neutrophilic granulocytes. J Extracell Vesicles. 2014; 3 25465, doi: http://dx.doi.org/10.3402/jev.v3.25465.
- Larson MC, Luthi MR, Hogg N, Hillery CA. Calcium-phosphate microprecipitates mimic microparticles when examined with flow cytometry. Cytometry A. 2013; 83: 242–50.
- Kalra H, Adda CG, Liem M, Ang CS, Mechler A, Simpson RJ, etal. Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013; 13: 3354–64.
- Hood JL, Pan H, Lanza GM, Wickline SA. Consortium for Translational Research in Advanced I, Nanomedicine. Paracrine induction of endothelium by tumor exosomes. Lab Invest. 2009; 89: 1317–28.
- Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJ, Hole P, etal. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomedicine. 2011; 7: 780–8.
- Coumans FA, van der Pol E, Boing AN, Hajji N, Sturk G, van Leeuwen TG, etal. Reproducible extracellular vesicle size and concentration determination with tunable resistive pulse sensing. J Extracell Vesicles. 2014; 3 25922, doi: http://dx.doi.org/10.3402/jev.v3.25922 [PubMed Central Full Text].
- Maas SL, de Vrij J, van der Vlist EJ, Geragousian B, van Bloois L, Mastrobattista E, etal. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. J Control Release. 2015; 200: 87–96.
- Steen HB. Flow cytometer for measurement of the light scattering of viral and other submicroscopic particles. Cytometry A. 2004; 57: 94–9.
- Hercher M, Mueller W, Shapiro HM. Detection and discrimination of individual viruses by flow cytometry. J Histochem Cytochem. 1979; 27: 350–2.
- Nolte-‘t Hoen EN, van der Vlist EJ, Aalberts M, Mertens HC, Bosch BJ, Bartelink W, etal. Quantitative and qualitative flow cytometric analysis of nanosized cell-derived membrane vesicles. Nanomedicine. 2012; 8: 712–20.
- Tan SS, Yin Y, Lee T, Lai RC, Yeo RW, Zhang B, etal. Therapeutic MSC exosomes are derived from lipid raft microdomains in the plasma membrane. J Extracell Vesicles. 2013; 2 22614, doi: http://dx.doi.org/10.3402/jev.v2i0.22614 [PubMed Central Full Text].
- Pocsfalvi G, Stanly C, Vilasi A, Fiume I, Capasso G, Turiak L, etal. Mass spectrometry of extracellular vesicles. Mass Spectrom Rev. 2015; 35: 3–21.
- Quah BJ, O'Neill HC. Mycoplasma contaminants present in exosome preparations induce polyclonal B cell responses. J Leukoc Biol. 2007; 82: 1070–82.
- Brouwers JF, Aalberts M, Jansen JW, van Niel G, Wauben MH, Stout TA, etal. Distinct lipid compositions of two types of human prostasomes. Proteomics. 2013; 13: 1660–6.
- Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015; 199: 145–55.