
Abstract
Objective
Isolation from human plasma of exosomes that retain functional and morphological integrity for probing their protein, lipid and nucleic acid content is a priority for the future use of exosomes as biomarkers. A method that meets these criteria and can be scaled up for patient monitoring is thus desirable.
Methods
Plasma specimens (1 mL) of patients with acute myeloid leukaemia (AML) or a head and neck squamous cell carcinoma (HNSCC) were differentially centrifuged, ultrafiltered and fractionated by size exclusion chromatography in small disposable columns (mini-SEC). Exosomes were eluted in phosphate-buffered saline and were evaluated by qNano for particle size and counts, morphology by transmission electron microscopy, protein content, molecular profiles by western blots, and for ability to modify functions of immune cells.
Results
Exosomes eluting in fractions #3–5 had a diameter ranging from 50 to 200 nm by qNano, with the fraction #4 containing the bulk of clean, unaggregated exosomes. The exosome elution profiles remained constant for repeated runs of the same plasma. Larger plasma volumes could be fractionated running multiple mini-SEC columns in parallel. Particle concentrations per millilitre of plasma in #4 fractions of AML and HNSCC were comparable and were higher (p<0.003) than those in normal controls. Isolated AML exosomes co-incubated with normal human NK cells inhibited NKG2D expression levels (p<0.004), and HNSCC exosomes suppressed activation (p<0.01) and proliferation of activated T lymphocytes (p<0.03).
Conclusions
Mini-SEC allows for simple and reproducible isolation from human plasma of exosomes retaining structural integrity and functional activity. It enables molecular/functional analysis of the exosome content in serial specimens of human plasma for clinical applications.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Exosomes, a type of extracellular vesicles (EVs) produced by most if not all cells, are characterized by size (30–150 nm in diameter), morphology (spherical, membrane-bound), the endosomal origin and cellular release by fusion of multivesicular bodies (MVBs) with the plasma membrane of the parent cell (Citation1–Citation3). Exosomes present in human body fluids, including plasma, derive from many different cells, and their content may vary depending on current physiological or pathological events (Citation4, Citation5). It has been observed that the exosome content of plasma or other body fluids increases in disease and may subside upon successful treatment or recovery (Citation5, Citation6). Emerging evidence suggesting that exosomes might serve as a benchmark for the presence of a disease has created enormous interest in their properties, including molecular profiles and biological activities (Citation7).
Exosomes are usually isolated by a complex and lengthy procedure which includes differential centrifugation to remove cellular fragments and large vesicles, ultracentrifugation at 100,000×g for 2–3 h and sucrose density gradient centrifugation to recover purified exosomes floating at the density of 1.13–1.19 g/mL (Citation8). Although some modifications to this procedure were introduced (Citation9), high-speed ultracentrifugation has remained the “classical” method for exosome isolation. The method is laborious, time-consuming and, most importantly, is not applicable to high-throughput processing of exosomes in plasma needed for biomarker studies or clinical monitoring. While a variety of other approaches to the isolation of EVs have been available (Citation10), a simple and reproducible procedure that can be readily up-scaled for exosome recovery from numerous human specimens and can be applied to serial monitoring has not been described. By adapting size-exclusion chromatography (SEC), first used in 1983 by Taylor for isolation of plasma membrane fragments from cell culture supernatants (Citation11), to exosome isolation from plasma, we established and standardized a procedure that separates exosomes from the bulk of “contaminating” plasma proteins. The procedure, called mini-SEC because it utilizes small-size ion exchange columns, yields a fraction enriched in biologically active and morphologically intact vesicles with the diameter size of exosomes, which are recovered in the quantity sufficient for molecular profiling of their cargo. Most importantly, mini-SEC can be readily adapted to simultaneous processing of several plasma aliquots using a series of small columns and providing a reliable, simple and inexpensive approach to a large-scale fractionation of human plasma for exosome recovery. Here, the mini-SEC method has been successfully applied to exosome isolation from plasma of patients with a solid tumour [head and neck squamous cell carcinoma (HNSCC)] and a haematological malignancy [acute myeloid leukaemia (AML)]. We describe advantages of this one-step fractionation of plasma obtained from patients with cancer without ultracentrifugation, emphasizing the fact that the recovered exosomes retain their functional as well as morphological integrity.
Materials and methods
Plasma of cancer patients and healthy donors
Venous blood samples were obtained from patients with AML or HNSCC seen in the UPCI Clinics as well as from healthy volunteers. All subjects donating blood specimens for this study signed an informed consent approved by the Institutional Review Board of the University of Pittsburgh (IRB #960279, #0403105 and #0506140).
All blood samples were centrifuged at 1,000×g for 10 min to collect the plasma. Plasma was aliquoted into 2 mL vials and stored frozen for various time periods prior to exosome isolation. Heparinized peripheral blood specimens were obtained from normal donors and were separated on Ficoll-Hypaque gradients (GE Healthcare Bioscience, Pittsburgh, PA, USA) to isolate peripheral blood mononuclear cells (PBMCs). PBMCs were washed in medium and immediately used for experiments.
Exosome isolation from human plasma
Frozen plasma specimens were thawed and centrifuged at 2,000×g for 10 min at 4°C and then at 10,000–14,000×g for 30 min at 4°C. Clarified plasma was passed through 0.22 µm-pore Millipore filter and used for exosome isolation by SEC performed using 1.5 cm×12 cm mini-columns (Bio-Rad, Hercules, CA, USA; Econo-Pac columns) packed with Sepharose 2B (Sigma-Aldrich, St. Louis, MO, USA). The column bed volume is 10 mL. Prior to applying clarified plasma, the column is washed with 20 mL of phosphate-buffered saline (PBS), and a porous frit is placed at the top of the gel to prevent its disturbance during subsequent elution with PBS. Clarified plasma (0.5–1.0 mL) was loaded onto the column and five 1 mL fractions corresponding to the void volume peak were collected. Fractions # 3, #4 and #5 were tested for protein content, morphology by transmission electron microscopy (TEM) and in functional assays. In preparation for western blots, the fractions were concentrated using 300,000 MWCO VivaSpin 500 Centrifugal Concentrators (Sartorius Corp, New York, NY, USA) by centrifugation at 5,000×g for 2–15 min, depending on the content. Supplementary Fig. 1 shows the schema for isolation of plasma exosomes by the mini-SEC method.
Protein measurements
Protein concentrations in isolated exosome fractions were measured using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer's instructions.
Particle size and concentration by resistive pulse sensing
Tunable resistive pulse sensing (RPS) by qNano (Izon, Cambridge, MA, USA) was used to measure the size distribution and concentration of particles in isolated exosome fractions. To prevent protein binding to the pore, an Izon reagent kit was used, and 0.03% Tween/PBS was added to each of the collected SEC fractions. An aliquot of exosomes from each fraction or calibration particles included in the reagent kit (1:1, 200 EV, Izon) were placed in the Nanopore (NP150, A37355, Izon). All samples were measured at 43.3 mm stretch with a voltage of 0.74 V at 2 pressure levels of 4 and 8 mbar. Particles were detected in short pulses of the current (blockades). The calibration particles were measured directly after the experimental sample under identical conditions. The sizes and concentrations of particles were determined using software provided by Izon (version 3.2).
Transmission electron microscopy
TEM of isolated exosomes was performed at the Center for Biologic Imaging at the University of Pittsburgh as previously described (Citation12). Briefly, freshly isolated exosomes were put on a copper grid coated with 0.125% Formvar in chloroform. The grids were stained with 1% (v/v) uranyl acetate in ddH2O, and the exosome samples were examined immediately. A JEM 1011 TEM was used for imaging.
Western blots
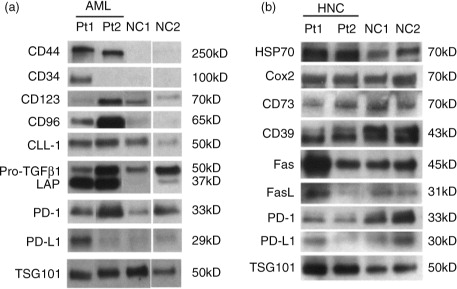
Isolated exosomes were tested for the presence of exosomal markers, CD9, CD63 or TSG101 and other protein markers of interest using western blots as previously described (Citation12). Briefly, exosomes (10 µg protein after concentration of the collected 1 mL fractions by VivaSpin 500 or 50 µL of each unconcentrated fraction) were lysed with Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, USA), separated on 7–15% SDS/PAGE gels and transferred onto the polyvinylidene flouride (PVDF) membrane (Millipore, Billerica, MA, USA) for western blot analysis. Membranes were incubated overnight at 4°C with antibodies specific for the designated antigens and purchased from Abcam, Cambridge, MA, USA: CD9 (1:500, ab97999), CD34 (1:2,000, ab81289), TSG101 (1:500, ab30871), CD96 (1:500, ab56653), Fas (1:1,000, ab1333619), FasL (1:500, ab68338), CD44 (1:1,000, ab41478), HSP70 (1:5,000, ab9920); from Santa Cruz, Dallas, TX, USA: CD63 (1:200, sc-15363), CD39 (1:400, sc-33558), CD73 (1:400, sc-25603), PD-L1 (1:500, sc-19090), COX-2 (1:500, sc-1745CD81); from Thermo Fisher, Pittsburgh, PA, USA: CD123 (1:200, PA5-13582), from R&D, Minneapolis, MN, USA: CLL-1 (1:2,000, AF2946), PD-1(1:500, MAB1086); from Cell Signaling, Danvers, MA, USA: TGF-β1 (1:1,000, #3711). Next, the horse radish peroxidase (HRP)-conjugated secondary antibody (1:3,000–1:5,000, Pierce, Thermo Fisher) was added for 1 h at room temperature (RT), and blots were developed with ECL detection reagents (GE Healthcare Biosciences). Band intensities on exposed films were quantified using Image J software (NIH, USA). Following SDS/PAGE electrophoresis, gels were stained with the Coomassie blue dye (Bio-Rad) for 1 h.
Functional studies with isolated exosomes
NKG2D down-regulation assay
PBMCs obtained from healthy volunteers were co-incubated with isolated exosomes to determine the percentage of NK cells (CD3negCD56+) expressing the natural cytotoxicity receptor NKG2D on the cell surface, as previously described (Citation12). Briefly, PBMCs were co-incubated with exosomes (5 µg protein/mL) collected in the SEC fractions #3, #4 or #5 for 24 h at 37°C. Then, flow cytometry for frequency of NKG2D was performed gating on CD3neg/CD56+ NK cells. The data are expressed as%NKG2D+ cells/total CD3negCD56+. The mean fluorescence intensity (MFI) of NKG2D on NK cells was also recorded. PBMC incubated in medium without exosomes were used as controls. PE-conjugated anti-NKG2D antibody or isotype controls were purchased from Beckman Coulter, Atlanta, GA, USA.
CD69 down-regulation on activated T cells
CD4+ T cells were isolated from PBMC by negative selection using AutoMACS as previously described (Citation13). The purity of CD4+ T cells was always >95% as determined by flow cytometry. The CD69 down-regulation assay was performed as described by Muller et al. (Citation14). Briefly, CD4+ T cells were activated with anti-CD3/anti-CD28 beads (Miltenyi, San Diego, CA, USA) at the 1:2 beads to cell ratio and IL-2 (150 U/mL, Peprotech) for 2 h. Exosomes (50 µL aliquots of SEC fraction #4) were added to activated T cells and incubated for 40 h in AIMV medium (Life Technologies, Pittsburgh, PA, USA) at 37°C. The percentages of CD69+ live T cells or MFI for CD69 expression levels were measured by flow cytometry after staining with CD69-FITC (BD Bioscience, San Jose, CA, USA), CD4-PE (Beckman Coulter) and 7-AAD (BD Bioscience). As controls, matching isotype Abs, non-activated T cells+PBS and activated T cells+normal control (NC) exosomes were tested in parallel.
CFSE-based proliferation assay
CD4+ T cells isolated from PBMC of normal donors as described above were labelled with 1.5 µM CSFE (Cell Trace, Life Technologies) in 0.1% BSA in PBS (w/v) for 10 min at 37°C. The staining was quenched with an equal volume of exosome-depleted FBS (Citation13). CFSE-labelled T cells (105/well) were incubated with plate-bound anti-CD3 and anti-CD28 Abs (3 µg/mL; eBioscience, San Diego, CA, USA) for 24 h and following activation, were co-incubated with exosomes (50 µL aliquots of SEC fractions #4) for 4 days at 37°C. Proliferation of T cells was measured on day 5 by flow cytometry, and the data were analysed by Modfit (Verity Software House, Topsham, ME, USA). The percent suppression of proliferation in co-cultures with exosomes was calculated as described by Strauss et al. (Citation15) and compared to control T cells incubated alone.
Data analysis
Data were summarized by descriptive statistics such as means and standard errors (SE) or means and standard deviations (SD) for continuous variables. Statistical analyses were performed using 2-tailed Student's t-test, and the p-value of <0.05 was considered to be statistically significant.
Results
Characterization of exosomes isolated by mini-SEC
Consecutive vesicle-containing fractions #3, #4 and #5 (each fraction=1 mL) were collected by mini-SEC and analysed without concentration by TEM and qNano. For analyses by western blots only, aliquots of the 3 fractions were concentrated using VivaSpin500 cartridges. Usually, 1 mL of plasma is used for exosome isolation. However, it is important to not to overload the column with proteins, and 0.5 ml aliquots of cancer patients’ plasma may be optimal.
To evaluate protein contents of the vesicle-containing fractions, 50 µL aliquots of each fraction (without concentration) were used for SDS/PAGE. Representative gels stained with Coomassie blue are shown in . Western blots of each fraction indicate the presence of exosomes based on positivity for CD9 and TSG101. The SDS gel protein profiles for the exosome-containing fractions isolated from plasma of patients with cancer or NCs are very consistent. We independently tested gel protein profiles of exosomes isolated from plasma of 10 AML and 5 HNSCC patients as well as 5 NCs with results that were identical to those presented in . While the #3 fraction always has the lowest protein levels (Table ), it contains a few vesicles (), which carry TSG101 (). The #4 fraction is largely depleted of the heavy and light immunoglobulin (Ig) chains and other plasma proteins, including albumin and is enriched in exosomes (). While the #5 fraction also contains exosomes, the presence of Igs, albumin and many other plasma proteins () appears to contribute to exosome aggregation (d). Later fractions contained no exosomes (data not shown).
Fig. 1. The protein content of vesicle-containing fractions after mini-SEC of cancer patients’ or normal donors’ plasma. 50 µg aliquots of each fraction (unconcentrated) were separated by PAGE and gels were stained with a Coomassie blue dye. Exosomes were visualized in western blots using anti-CD9- and anti-TSG101-specific Abs. The #3 fraction contains little protein but vesicles are present as shown by TEM in . Fractions #4 and #5 are enriched in exosomes but contain protein bands. Note that the heavy chain of IgG and albumin are the major proteins in fractions # 5, while fractions #3 and #4 are relatively free of IgG and albumin. Shown are representative data from 1 of more than 15 PAGE performed with different column eluates.
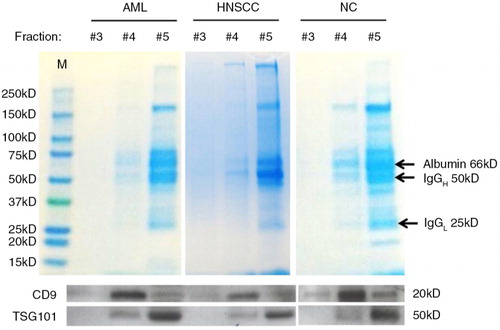
Fig. 2. Transmission electron microscopy (TEM) of vesicles eluted in mini-SEC fractions #3, #4 and #5. Following mini-SEC of (a) AML plasma, (b) HNSCC plasma and (c) NC plasma, TEM was performed. Unconcentrated aliquots of vesicle-containing fractions were placed on grids and negatively stained with uranyl acetate. Note the presence of single exosomes in the size range of 30–100 nm. (d) Exosome aggregates present in #5 fractions of HNSCC and AML plasma are shown. Representative images from 1 of 6 experiments performed are presented. AML=acute myeloid leukaemia; HNSCC=head and neck squamous cell carcinoma; NC=normal control.
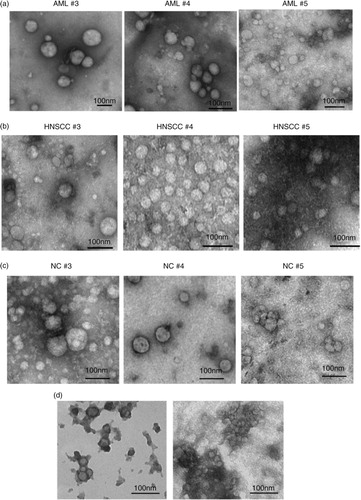
Table I. Exosome protein concentration and particle concentrations in mini-SEC fractions of plasma obtained from patients with AML and HNSCC and NCsa
TEM of the collected fractions confirmed the presence of exosomes in all 3 fractions (). The images of negatively stained exosomes obtained from AML plasma (a), HNSCC plasma (b) and NC plasma (c) were morphologically similar. By TEM, individual exosomes ranged in diameter from 50 to 100 nm, and in fractions #5, exosome aggregates larger than 100 nm were also present (d). The tendency of plasma-derived exosomes to aggregate was greater than that of exosomes in fractions of supernatants from the AML cell line (Kasumi) or HNSCC cell line (PCI-13) isolated by mini-SEC (data not shown).
Results of the qNano analysis of exosomes in vesicle-containing fractions #4 are shown in . The particles present in fractions #4 ranged in size from 50 to 200 nm in diameter, a somewhat larger size and a broader range than that measured in the same fractions by TEM. The differences in diameter observed between TEM () and qNano () measurements could reflect: (a) exosome tendency to shrink during preparation for TEM and/or (b) properties of the NP150 membrane which filters out larger (>200 nm) and smaller (<40 nm) vesicles and thus might not rigorously discriminate between vesicles and vesicle aggregates. In fact, the #5 fractions tended to block the membrane pores, indicating the presence of aggregates larger than 200 nm, as also seen in TEM (d).
Fig. 3. Sizing and counting of particles by qNano. Fractions #4 were isolated from plasma of 2 AML patients, 2 HNSCC patients and 2 NCs by mini-SEC and evaluated by qNano as described in Materials and Methods. AML=acute myeloid leukaemia; HNSCC=head and neck squamous cell carcinoma; NC=normal control.
As shown in and Table , mini-SEC fractions #5 had the highest protein content but contained “contaminating” Igs, albumin as well as other plasma proteins, which likely contributed to aggregation of the vesicles. In contrast, mini-SEC fractions #4 had low content of “contaminating” plasma proteins, were enriched in unclustered morphologically intact exosomes () and, upon concentration by VivaSpin, as described in Materials and Methods, provided sufficient material for western blot analysis of the exosome cargo. The protein contents of #3 fractions were too low for western blot analyses even after VivaSpin concentration. Based on these observations, we routinely collect and use exosomes eluting in #4 fractions for all further studies.
Interestingly, the qNano measurements confirmed that mean diameters of particles (~75–85 nm) recovered in #4 fractions were comparable in the plasma of AML and HNSCC patients and NCs (). Also, the particle concentrations/mL plasma were comparable in #4 fractions of AML and HNSCC plasma (Table ). However, both AML and HNSCC fractions #4 contained significantly higher (p<0.003 and p<0.001) particle concentrations/mL than #4 fractions of NCs (Table ).
Exosome recovery using mini-SEC columns
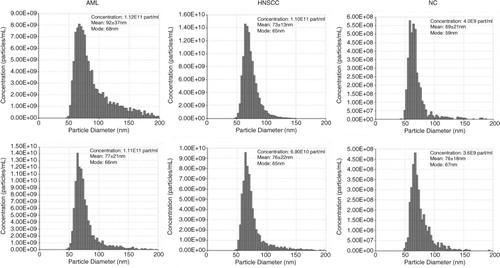
To evaluate exosome recovery in the #4 fractions collected from mini-SEC columns based on total µg protein/mL plasma, the isolation procedure was performed with plasma specimens of 10 newly diagnosed AML patients, 5 HNSCC patients with active untreated disease and 5 NCs (a). The median protein concentration/mL plasma measured in #4 fractions isolated from AML plasma was145 µg (range 30–225 µg), that from HNSCC plasma was 66 µg (range 48–84 µg) and that from NC plasma was 31 µg (23–49 µg). These results indicated that the recovery of exosomes from plasma of patients with cancer was significantly higher than that from plasma of NCs (p<0.02). In addition, b shows that reproducibility of the mini-SEC method was excellent when specimens of the same plasma were eluted using different mini-SEC columns. Protein levels of exosome fractions #4 repeatedly (×3) isolated from plasma aliquots of the same NC and the same HNSCC patient were almost identical.
Fig. 4. A. Comparisons of the protein content in mini-SEC fractions # 4 isolated from plasma of cancer patients or NCs. (a) Exosome protein levels (in µg/mL plasma) in fractions #4 obtained from plasma of AML or HNSCC patients and NCs. Note higher protein levels in in exosome fractions of cancer patients relative to those of NCs. (b) Reproducibility of mini-SEC: plasma specimens (1 mL) obtained from an NC) and a HNSCC patient were repeatedly chromatographed (×3) using different mini-SEC columns. Protein levels measured in #4 fractions of repeated samples are not significantly different. AML=acute myeloid leukaemia; HNSCC=head and neck squamous cell carcinoma; NC =normal control.
Exosome western blot profiles
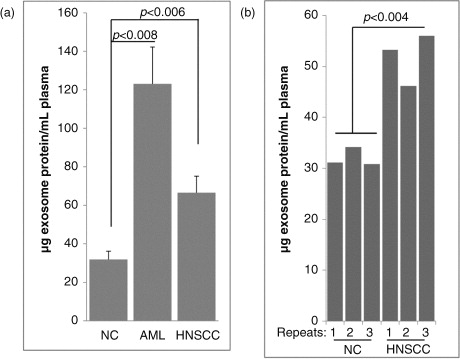
In examining western blot profiles of exosomes recovered by mini-SEC in #4 fractions, we were specifically interested in the proteins that were previously shown by us to mediate biological activities of exosomes upon their co-incubation with immune cells (Citation15, Citation16). Prior to western blots, exosomes present in #4 fractions were routinely concentrated by Vivaspin500, so that we could deliver 10 µg protein/lane for blotting. Exosomes in the #4 fractions isolated from NC plasma served as controls. The VivaSpin membrane allows for the removal of materials with the molecular weight<300 kD, and it concentrates exosomes while removing contaminating proteins associated with exosomes. In this step, exosome recovery is usually 80–90% based on qNano measurements, and protein recovery is 40–50% of the input levels. The exosome enrichment is about 2-fold. The blots in show that plasma-derived exosomes from cancer patients carry the cargo of molecules such as the TGF-β1-associated pro-peptide and latency-associated protein (LAP), PD-1, PD-L1, COX-2, FasL or CD39/CD73 ectoenzymes that are well known to alter functions of immune cells (Citation15–Citation17). Plasma-derived exosomes in patients with AML also carry the leukaemia blast-relevant proteins (CD34, CD44, CD96, CD123 and CLL-1). Exosomes isolated from plasma specimens of NCs have a different protein profile from that seen in exosomes of cancer patients (a and b). Protein profiles of exosomes present in the fraction #4 isolated from cancer patients’ plasma were distinct for each patient.
Fig. 5. Western blot profiles of exosome proteins in fraction #4. Following mini-SEC, western blots were performed as described in Materials and Methods using concentrated aliquots of fractions #4 isolated from plasma specimens of (a) 2 patients with AML and 2 different NCs and (b) 2 patients with HNSCC and 2 different NCs. All lanes were loaded with 10 µg exosomal proteins. TSG101 was used as an exosome marker and a “loading control.” Immunoblotted proteins and their molecular weights are listed. AML=acute myeloid leukaemia; HNSCC=head and neck squamous cell carcinoma; NC=normal control; SEC=size exclusion chromatography.
Isolated exosomes alter functions of normal immune cells
We have previously shown that AML plasma-derived exosomes inhibited cytotoxic activity of NK cells (Citation17). To assess biologic activity of exosomes isolated by mini-SEC from plasma of cancer patients or NCs, we co-incubated fractionated exosomes with in vitro activated immune cells (NK cells or CD4+ T cells) obtained from the peripheral blood of NCs. Following co-incubation, changes in expression levels of functionally relevant surface molecules on lymphocytes or changes in lymphocyte proliferation were measured. The data in a show that exosomes in AML fractions #4 down-regulated expression levels (MFI) of NKG2D on CD3negCD56+ NK cells and significantly reduced the frequency of NKG2D+ NK cells. In contrast, exosomes in fraction #5 were only minimally active. Fraction #5 of plasma obtained from NCs did not alter NKG2D expression levels on NK cells.
Fig. 6. Biological activity of exosomes in #4 fractions tested in vitro using normal human lymphocytes. Exosomes were co-incubated with PBMC (a) or with isolated and in vitro activated T cells (b) under conditions described in Materials and Methods. (a) AML exosomes in fractions #3 and #4, but not in fraction #5 down-regulated NKG2D expression levels (MFI) in activated NK cells and reduced the frequency of NKG2D+ in these cells (see table) as determined by flow cytometry with the gate set on CD3-CD56+ NK cells. The MFI data are representative from 1 of 3 experiments performed with exosomes isolated from different AML patients. (b) HNSCC exosomes in fractions #4 down-regulated expression of CD69 (percentages of expression are shown) in activated human CD4+ T cells (upper panels) and suppressed proliferation of T cells activated via the TcR (lower panels). Data shown are representative for different HNSCC patients as shown in the tables. AML=acute myeloid leukaemia; HNSCC=head and neck squamous cell carcinoma; NC=normal control; PBMC=peripheral blood mononuclear cell.
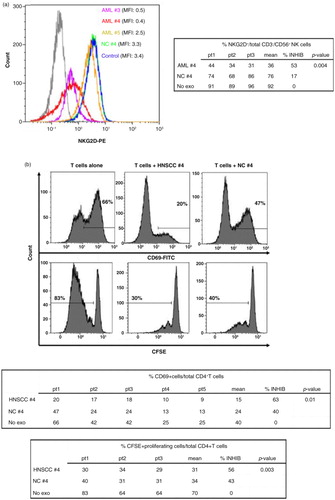
Exosomes isolated from plasma of HNSCC patients were previously shown to induce death and dysfunction of activated peripheral blood T cells (Citation18, Citation19). Therefore, we tested mini-SEC isolated exosomes of HNSCC patients in fractions #4 for the ability to down-regulate expression levels of CD69, an activation marker, which is up-regulated on the surface of in vitro activated CD4+ T cells. As shown in b (top), #4 fraction exosomes down-regulated expression levels of CD69 on the surface of activated T cells (p<0.01). Similarly, these exosomes also inhibited proliferation of activated T cells in culture (p<0.03, b, bottom).
Discussion
Plasma and other body fluids contain a variety of EVs produced by many different tissue cells and hematopoietic cells. Various methods are available for EV isolation, including polyethylene glycol-mediated precipitation, microfluidics, immunoaffinity capture (Citation20–Citation22) and many proprietary commercially available procedures. It remains unclear whether these various technologies isolate vesicles with the same or different properties. Most of these methods were developed by utilizing EVs in supernatants of cultured cell lines [reviewed in Taylor and Shah (Citation10)]. Increasingly, these methods are being applied to the isolation of EVs from plasma/serum of patients with various diseases. However, the use of plasma introduces a new level of complexity to nanovesicle isolation and especially to exosome separation from larger EVs or ubiquitous plasma components. Unlike supernatants of cultured cell lines, which are a source of EVs originating from 1 cell type, plasma/serum is a complex mixture of EVs produced by many different cells at levels that may be individually regulated. Among these EVs of different types and sizes are exosomes, and their isolation from body fluids remains a considerable challenge.
First, the definition of exosomes as spherical, membrane-bound vesicles ranging in size from 30 to 150 nm is arbitrary. It is possible that MVBs upon fusion with the parent cell membrane (Citation23) release exosomes with a much broader size range. Alternatively, exosomes could fuse upon their release to form vesicles with a larger diameter. While these derivative vesicles might have properties similar to exosomes, separation methods based on size alone would tend to eliminate them. Second, plasma-derived exosomes are “contaminated” with high-abundance proteins that in proteomic studies are likely to mask genuine exosome-associated proteins. Thus, not only isolation but also concomitant purification of exosomes is desirable. Third, recent insights into the role of exosomes as potential prognostic biomarkers in human disease Citation17, (Citation24–Citation26) impose the criteria for reliable recovery and consistency in biological activity of isolated exosomes. Finally, to satisfy the above criteria and implement exosome isolation at the clinical monitoring level, it is necessary to have available a simple, high-throughput and inexpensive method that can ensure isolation of exosomes in the quantity and form suitable for RNA, DNA and protein analyses.
The mini-SEC method we have adapted and used appears to meet most of the criteria listed above. In addition, unlike lengthy and cumbersome ultracentrifugation, it accomplishes exosome isolation in 30 min. Others have also reported on the use of SEC to isolate vesicles from human plasma or platelet concentrates and commented on advantages of SEC over other isolation methods (Citation27–Citation29). Aside from a risk of protein complex formation, aggregation, and vesicle losses, methods relying on ultracentrifugation and density gradient exosome separation may lead to a loss of biological functions. Hence, consistency in recovery of vesicles that are structurally intact and functionally active is an important advantage of mini-SEC-based isolation.
Eluted from the column in PBS as non-aggregated vesicles and co-incubated with normal human NK or T cells, cancer plasma-derived exosomes significantly and reproducibly down-regulated functions of these target cells. Similarly isolated exosomes from plasma of NCs did not mediate these effects. The mini-SEC eluted exosomes from plasma of cancer patients carry molecules which suppress functions or induce apoptosis of activated immune cells (Citation12, Citation14) (Citation16–Citation18, Citation30–Citation32) . Based on observations that protein levels of exosomes isolated from cancer patients’ plasma are significantly elevated (Citation5, Citation6) (Citation14), it has been assumed that tumour cells are the major source of these exosomes. Studies indicate that cultured human tumour cells produce large quantities of exosomes which carry immuno-suppressive proteins and inhibit functions of immune cells (Citation16). Tumour-derived exosomes can either directly deliver inhibitory signals to target cells or indirectly mediate suppression by promoting expansion of regulatory T cells (Treg) or myeloid-derived suppressor cells (MDSC) (Citation33–Citation35). These observations provide a strong rationale for considering plasma-derived exosomes in cancer as contributors to immune suppression, which may be responsible, at least in part, for tumour escape from the host immune system (Citation36). It has been suggested that exosomes interacting with lymphocytes deliver signals upon a direct contact with the cell surface rather than internalization (Citation37). The integrity of isolated exosomes and their purity may be of key importance for surface receptor-mediated information transfer. The aggregate formation or an excess of non-exosomal “contaminants” on the exosome membrane presumably could interfere with their signalling functions. Therefore, the ability of mini-SEC purified exosomes from plasma of cancer patients to alter functions of activated NK cells or T cells is an important advantage of this isolation method.
Mini-SEC columns are now commercially available, and the advantages of these commercially available, pre-packed chromatography columns have been discussed by Welton et al. (Citation27). The disadvantage is the considerable cost of these columns. In fact, similar, equally well-performing and much less costly homemade mini-SEC columns can be successfully used for the isolation of exosomes from human plasma, as shown here. The homemade mini-columns are inexpensive, disposable, and can be set up in a row of 5 or 10 to be run simultaneously when large quantities of exosomes from the same plasma are required. Exosomes collected in the early fractions can be concentrated by using VivaSpin 500 concentrators. This step provides an additional advantage of removing proteins that are not integral components of the exosome membrane.
The recovery of purified exosomes in the #4 fraction was reliably achieved in the same repeatedly separated aliquots of the same or different plasma samples obtained from NCs or cancer patients. Purified plasma exosomes consistently eluted in the #4 fraction as morphologically intact but heterogeneous in size vesicles. Because plasma exosomes originate from various cells which might release vesicles of different sizes, the heterogeneity in size of isolated exosomes (30–150 nm) is not surprising. The precise sizing of isolated exosomes by qNano is complicated by their propensity for aggregation, especially in the #5 fraction which is enriched in plasma proteins after mini-SEC. A decision to harvest only the fraction #4 after mini-SEC was undertaken based on evidence that it contains largely non-aggregated morphologically intact vesicles relatively free of “contaminating” plasma proteins as confirmed by TEM and protein electrophoresis.
By qNano, the particle concentration per millilitre of plasma was routinely higher in #4 exosome fractions obtained from cancer patients than from NCs. How these data in recovered #4 fractions relate to the initial concentration of vesicles in patients’ plasma could not be determined. We were consistently unable to measure EVs in clarified plasma before mini-SEC by qNano, even upon dilutions in of 0.05%Tween. Therefore, we cannot comment on the particle content of the unfractionated plasma and the exosome recovery after mini-SEC using qNano.
Recently, Balaj and colleagues reported a new method for EV purification, which utilizes ultrafiltration followed by EV capture on heparin-affinity beads (Citation31). EVs are captured on heparin conjugated to agarose or to magnetic beads and are eluted from the beads in 2.15 M NaCl. While this method is reported to perform well for RNA extraction from EVs and preserves their ability to be internalized by target cells, it does not discriminate between exosomes and other types of EVs. Also, the process of capture and elution of EV from magnetic beads is more time- and effort-demanding than mini-SEC, which segregates exosomes into vesicles based on their diameter and surface properties (i.e. tendency to aggregate) in a single step. Others have attempted to compare 3 commercially available methods for exosome isolation from supernatants of cultured cells, using a model liposome system as a “gold standard” and tunable-RPS (qNano) for exosome characterization (Citation38). This study concluded that “larger particles present in most purified exosome samples represent co-purified contaminating non-exosome debris .…” Our study supports this conclusion, with a caveat that a one-step, inexpensive and simple mini-SEC can partially remove and fractionate this debris yielding fractions enriched in plasma-derived exosomes which are morphologically intact, largely depleted of contaminating immunoglobulins and able to mediate intercellular communication.
Authors' contributions
C-SH, SF and LM performed the experimental work and analysed the results. TLW and MB conceived of the idea of this study, guided its performance and interpreted the data. TLW wrote the manuscript.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Supplementary figure
Download PDF (223.8 KB)Acknowledgements
This work was supported in part by the NIH grant R01-CA16862 to TLW and CCSG grant P30 CA047904 and by the University of Pittsburgh-Essen-Partnership-Program to SF. Support for leukemia research was also provided by The Plum Running Mustangs The authors acknowledge the expert assistance of the staff in the Cell Imaging Core directed by Dr. Simon Watkins, also supported in part by CCSG grant P30 CA047904.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015; 25: 364–72. doi: http://dx.doi.org/10.1016/j.tcb.2015.01.004.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200: 373–83. doi: http://dx.doi.org/10.1083/jcb.201211138.
- Graner MW, Alzate O, Dechkovskaia AM, Keene JD, Sampson JH, Mitchell DA, etal. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009; 23: 1541–57. doi: http://dx.doi.org/10.1096/fj.08-122184.
- Brinton LT, Sloane HS, Kester M, Kelly KA. Formation and role of exosomes in cancer. Cell Mol Life Sci. 2015; 72: 659–71. doi: http://dx.doi.org/10.1007/s00018-014-1764-3.
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, etal. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012; 18: 883–91. doi: http://dx.doi.org/10.1038/nm.2753.
- Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. 2011; 96: 1302–9. doi: http://dx.doi.org/10.3324/haematol.2010.039743.
- De Toro J, Herschlik L, Waldner C, Mongini C. Emerging roles of exosomes in normal and pathological conditions: new insights for diagnosis and therapeutic applications. Front Immunol. 2015; 6: 203. doi: http://dx.doi.org/10.3389/fimmu.2015.00203.
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3:Unit 3.22. doi: http://dx.doi.org/10.1002/0471143030.cb0322s30.
- Montecalvo A, Larregina AT, Morelli AE. Methods of purification of CTL-derived exosomes. Methods Mol Biol. 2014; 1186: 87–102. doi: http://dx.doi.org/10.1007/978-1-4939-1158-5_7.
- Taylor DD, Shah S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods. 2015; 87: 3–10. doi: http://dx.doi.org/10.1016/j.ymeth.2015.02.019.
- Taylor DD, Chou IN, Black PH. Isolation of plasma membrane fragments from cultured murine melanoma cells. Biochem Biophys Res Commun. 1983; 113: 470–6.
- Hong CS, Muller L, Boyiadzis M, Whiteside TL. Isolation and characterization of CD34+ blast-derived exosomes in acute myeloid leukemia. PLoS One. 2014; 9: e103310. doi: http://dx.doi.org/10.1371/journal.pone.0103310.
- Saze Z, Schuler PJ, Hong CS, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood. 2013; 122: 9–18. doi: http://dx.doi.org/10.1182/blood-2013-02-482406.
- Muller L, Hong CS, Stolz DB, Watkins SC, Whiteside TL. Isolation of biologically-active exosomes from human plasma. J Immunol Methods. 2014; 411: 55–65. doi: http://dx.doi.org/10.1016/j.jim.2014.06.007.
- Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol. 2009; 182: 1469–80.
- Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol. 2009; 183: 3720–30. doi: http://dx.doi.org/10.4049/jimmunol.0900970.
- Hong CS, Muller L, Whiteside TL, Boyiadzis M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front Immunol. 2014; 5: 160. doi: http://dx.doi.org/10.3389/fimmu.2014.00160.
- Kim JW, Wieckowski E, Taylor DD, Reichert TE, Watkins S, Whiteside TL. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin Cancer Res. 2005; 11: 1010–20.
- Whiteside TL. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem Soc Trans. 2013; 41: 245–51. doi: http://dx.doi.org/10.1042/BST20120265.
- Taylor DD, Zacharias W, Gercel-Taylor C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol Biol. 2011; 728: 235–46. doi: http://dx.doi.org/10.1007/978-1-61779-068-3_15.
- Chen C, Skog J, Hsu CH, Lessard RT, Balaj L, Wurdinger T, etal. Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab Chip. 2010; 10: 505–11. doi: http://dx.doi.org/10.1039/b916199f.
- Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, etal. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012; 56: 293–304. doi: http://dx.doi.org/10.1016/j.ymeth.2012.01.002.
- Kowal J, Tkach M, Thery C. Biogenesis and secretion of exosomes. Curr Opin Cell Biol. 2014; 29: 116–25. doi: http://dx.doi.org/10.1016/j.ceb.2014.05.004.
- Martins VR, Dias MS, Hainaut P. Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Curr Opin Oncol. 2013; 25: 66–75. doi: http://dx.doi.org/10.1097/CCO.0b013e32835b7c81.
- Muller L, Muller-Haegele S, Mitsuhashi M, Gooding W, Okada H, Whiteside TL. Exosomes isolated from plasma of glioma patients enrolled in a vaccination trial reflect antitumor immune activity and might predict survival. Oncoimmunology. 2015; 4: e1008347. doi: http://dx.doi.org/10.1080/2162402X.2015.1008347.
- Khan S, Aspe JR, Asumen MG, Almaguel F, Odumosu O, Acevedo-Martinez S, etal. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br J Cancer. 2009; 100: 1073–86. doi: http://dx.doi.org/10.1038/sj.bjc.6604978.
- Welton JL, Webber JP, Botos LA, Jones M, Clayton A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J Extracell Vesicles. 2015; 4: 27269. doi: http://dx.doi.org/10.3402/jev.v4.27269.
- Boing AN, van der Pol E, Grootemaat AE, Coumans FA, Sturk A, Nieuwland R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J Extracell Vesicles. 2014; 3: 23430. doi: http://dx.doi.org/10.3402/jev.v3.23430.
- de Menezes-Neto A, Saez MJ, Lozano-Ramos I, Segui-Barber J, Martin-Jaular L, Ullate JM, etal. Size-exclusion chromatography as a stand-alone methodology identifies novel markers in mass spectrometry analyses of plasma-derived vesicles from healthy individuals. J Extracell Vesicles. 2015; 4: 27378. doi: http://dx.doi.org/10.3402/jev.v4.27378.
- Bergmann C, Strauss L, Wieckowski E, Czystowska M, Albers A, Wang Y, etal. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head Neck. 2009; 31: 371–80. doi: http://dx.doi.org/10.1002/hed.20968.
- Balaj L, Atai NA, Chen W, Mu D, Tannous BA, Breakefield XO, etal. Heparin affinity purification of extracellular vesicles. Sci Rep. 2015; 5: 10266. doi: http://dx.doi.org/10.1038/srep10266.
- Andreola G, Rivoltini L, Castelli C, Huber V, Perego P, Deho P, etal. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002; 195: 1303–16.
- Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010; 70: 9621–30. doi: http://dx.doi.org/10.1158/0008-5472.CAN-10-1722.
- Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One. 2010; 5: e11469. doi: http://dx.doi.org/10.1371/journal.pone.0011469.
- Mrizak D, Martin N, Barjon C, Jimenez-Pailhes AS, Mustapha R, Niki T, etal. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J Natl Cancer Inst. 2015; 107: 363. doi: http://dx.doi.org/10.1093/jnci/dju363.
- Whiteside TL. Immune responses to cancer: are they potential biomarkers of prognosis?. Front Oncol. 2013; 3: 107. doi: http://dx.doi.org/10.3389/fonc.2013.00107.
- Hwang I, Ki D. Receptor-mediated T cell absorption of antigen presenting cell-derived molecules. Front Biosci (Landmark Ed). 2011; 16: 411–21.
- Lane RE, Korbie D, Anderson W, Vaidyanathan R, Trau M. Analysis of exosome purification methods using a model liposome system and tunable-resistive pulse sensing. Sci Rep. 2015; 5: 7639. doi: http://dx.doi.org/10.1038/srep07639.