
Abstract
The surface protein composition of extracellular vesicles (EVs) is related to the originating cell and may play a role in vesicle function. Knowledge of the protein content of individual EVs is still limited because of the technical challenges to analyse small vesicles. Here, we introduce a novel multiplex bead-based platform to investigate up to 39 different surface markers in one sample. The combination of capture antibody beads with fluorescently labelled detection antibodies allows the analysis of EVs that carry surface markers recognized by both antibodies. This new method enables an easy screening of surface markers on populations of EVs. By combining different capture and detection antibodies, additional information on relative expression levels and potential vesicle subpopulations is gained. We also established a protocol to visualize individual EVs by stimulated emission depletion (STED) microscopy. Thereby, markers on single EVs can be detected by fluorophore-conjugated antibodies. We used the multiplex platform and STED microscopy to show for the first time that NK cell–derived EVs and platelet-derived EVs are devoid of CD9 or CD81, respectively, and that EVs isolated from activated B cells comprise different EV subpopulations. We speculate that, according to our STED data, tetraspanins might not be homogenously distributed but may mostly appear as clusters on EV subpopulations. Finally, we demonstrate that EV mixtures can be separated by magnetic beads and analysed subsequently with the multiplex platform. Both the multiplex bead-based platform and STED microscopy revealed subpopulations of EVs that have been indistinguishable by most analysis tools used so far. We expect that an in-depth view on EV heterogeneity will contribute to our understanding of different EVs and functions.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Extracellular vesicles (EVs) comprise exosomes, microvesicles and apoptotic bodies which are all cell-derived and enclosed by a lipid bilayer (Citation1). Exosomes are released from intact cells after inward budding of multivesicular bodies and fusion with the plasma membrane. They have the same membrane orientation as the originating cell, i.e. displaying extracellular domains on their surface (Citation2, Citation3). Exosomes are secreted by many cell types (Citation4) into diverse body fluids such as blood (Citation5), semen (Citation6), urine (Citation7), saliva (Citation8), breast milk (Citation9), ascites fluid (Citation10) and cerebrospinal fluid (Citation11). In addition to their size, exosomes are characterized by their density, lipid composition, and certain protein markers, such as tetraspanins, Alix and tumour susceptibility gene 101 (Citation1, Citation2). Exosomes have been shown to transport RNA (Citation12–Citation14), proteins, and other cytosolic components [reviewed by Simpson in (Citation15)]. The surface proteins on exosomes can affect the cellular uptake, and the exosome load can impact the physiology of target cells (Citation2).
CD9, CD63, and CD81 are 3 of the most-studied members of the tetraspanin protein family. Tetraspanins contain 4 transmembrane domains that promote associations between tetraspanins and other proteins (Citation16). Tetraspanin proteins are thought to be enriched in exosomes (Citation17) because they mediate exosome secretion as well as protein sorting into exosomes by assembling tetraspanin-enriched microdomains (TEMs) (Citation18). For example, CD63 was shown to be essential for the sorting of a melanosomal protein into exosomes (Citation19), CD9 knockout impairs the exosome secretion by dendritic cells (Citation20) and the transfer of MHC (major histocompatibility complex) class II into exosomes is correlated with its association with CD9 (Citation21). Accordingly, the composition of TEMs and their binding partners are specific for each cell type and related to cellular functions, as reviewed by Levy and Shoham (Citation16). Origin-specific proteins described for cells were also found on the respective exosomes, as reviewed by Théry (Citation3). In general, it is difficult to compare the composition of different EVs described in the literature as the experimental focus and methods often diverge. Moreover, the multitudes of EV populations from different cell types in a donor's sample, e.g. plasma, add another level of complexity that is difficult to resolve. For western blotting or mass spectrometry the bulk sample is analysed. Whether the detected protein was present on all EVs or just on a subpopulation of EVs cannot be discriminated. Single EV analysis, e.g. by electron microscopy, is time-consuming, and multiple pictures must be analysed to provide sufficient statistical rigor (Citation22). In addition, the detection of two markers present on the same vesicle is limited to abundant epitopes, and a systematic analysis of several markers requires several experiments (Citation23). High-resolution flow cytometry might currently be the most promising technique to analyse surface marker distributions on single EVs (Citation24, Citation25).
To discriminate EV subpopulations in one sample more efficiently, we have developed a multiplex bead-based platform that detects up to 39 different surface proteins and enables EV subpopulation identification by staining with different antibodies. Additionally, we established a protocol to visualize single EVs by high-resolution microscopy using stimulated emission depletion (STED). STED bypasses the diffraction limit of light microscopy. The excitation beam is supplemented by a STED beam that de-excites fluorophores by stimulated emission. The combination of these two beams limits fluorescence emission to predefined sample coordinates to increase resolution (Citation26, Citation27). Our data indicate the existence of distinct EV subpopulations and a heterogeneous distribution of tetraspanins on EVs.
Material and methods
Cell isolation, cultivation, and stimulation for EV production
For platelet isolation, fresh whole blood was diluted with an equal volume of Krebs Ringer buffer (100 mM NaCl, 4 mM KCl, 20 mM NaHCO3, 2 mM Na2SO4, 4.7 mM citric acid, 14.2 mM tri-sodium citrate) at pH 7.4 and centrifuged at 190×g for 10 min (Citation28). To deplete leukocytes and erythrocytes, the platelet-rich plasma was centrifuged at 100×g for 20 min. Platelets were pelleted at 1,000×g for 15 min and washed twice with Krebs Ringer buffer; 1 to 9×107 platelets per ml whole blood were isolated and platelet purities ranged from 82 to 99%. After adjusting to 1×109 platelets per ml Krebs Ringer buffer, they were activated with 50 nM calcium ionophore and 10 mM calcium chloride for 30 min at room temperature (Citation29).
Natural killer (NK) cells were isolated from buffy coats using the MACSxpress® NK Cell Isolation Kit and cultivated in TexMACS medium with 5% human AB serum and 500 U/ml IL-2 (Miltenyi Biotec) for 14 days.
Monocytes were isolated from buffy coats after Ficoll gradient by immunomagnetic cell sorting using CD14 MicroBeads (Miltenyi Biotec) with purities of 92–98% and cultivated in RPMI1640 with 2 mM L-glutamine, 50 U/ml penicillin and 50 µg/ml streptomycin for 24 h with viability rates >90%.
B cells were isolated from a buffy coat after Ficoll gradient by immunomagnetic cell sorting using CD19 MicroBeads (Miltenyi Biotec) with a purity of 96%. 2×106 B cells per ml were stimulated in StemMACS HSC Expansion Media XF with 5% vesicle-depleted human AB serum, 1 µg/ml CD40 ligand, 3.6 µl/ml cross-linking antibody, 20 U/ml IL-4, 50 U/ml penicillin and 50 µg/ml streptomycin according to the B cell Expansion Kit (Miltenyi Biotec) for 5 days. On the day of EV harvest, the cell viability rates were >90%.
Isolation of extracellular vesicles
Cell culture supernatants were depleted of cells and large cell debris by centrifugation at 2,000×g for 30 min. Dead cells and cell debris were depleted by centrifugation at 10,000×g for 45 min and larger vesicles by filtration through 0.22 µm. The filtrated supernatants were centrifuged at 108,000×g for 2 h and the pellets were washed with PBS (Citation23). The EV-enriched pellet was resuspended in PBS using 1/500 to 1/50 of the initial cell culture supernatant volume. The vesicle concentration was measured indirectly by BCA Assay (Thermo Fisher Scientific) using BSA as standard.
Multiplex bead-based platform
Carboxylated polystyrene beads (Microparticles GmbH) were hard dyed by swelling them in an organic solution of pyrromethene/LD688. For the calculation of dye concentrations in the mixture, beads were dyed with single dyes at varying concentrations beforehand. The emission into fluorescence channels was measured and linear equations correlating mean fluorescence intensities (MFIs) of the different emission channels with staining concentrations were established. The data were combined into a matrix equation to allow calculation of MFIs from the concentrations of a dye mixture. By inverting the matrix, the equation was used to calculate concentrations of dye mixtures for the desired MFIs of bead populations.
Capture antibodies (Supplementary Table I) were reduced at room temperature and incubated with dyed, imide covered beads. The capture antibody beads were washed with PBS + 0.03% Pluronic F68 + 0.05% sodium azide.
Approximately 800 beads per bead population were incubated with EVs isolated from NK cells, B cells, or monocytes in 100 µl or platelet supernatant in 300 µl at 4°C overnight. To adjust the volume of different samples, PAP buffer consisting of PBS with 0.1% Pluronic and 0.09% azide was used. The optimal amount of isolated EVs (typically 8–32 µg) was determined for each EV type by titration of the EV sample. For the analysis after immunomagnetic separation, 100, 70 and 30% of the original protein amount used for separation and 130 µl of the eluted sample were each adjusted to a total volume of 300 µl with PAP buffer. These samples and the 300 µl flow-through were each incubated with a 39-plex bead set.
To remove unbound EVs, the beads were washed in PAP and pelleted at 3,000×g for 5 min. The beads were resuspended in 100 µL PAP and bound EVs were stained with 0.5 µg of each detection antibody. The capture antibodies used are listed in Supplementary Table I. The same antibody clones from the same companies conjugated to allophycocyanin (APC) were used as detection antibodies.
Flow cytometry analysis
For flow cytometry analysis, the MACSQuant Analyzer 10 with the corresponding software (Miltenyi Biotec) was used. For the multiplex bead-based platform, a trigger for the side scatter (SSC) and the forward scatter (FSC) were selected to confine the measurement on the multiplex beads. Single beads were gated to exclude bead doublets and non-bead events. Voltages for the FITC and PE channels were adapted to ensure that each of the differentially labelled bead populations were detectable. The 39 single bead populations were gated to allow the determination of the APC signal intensity on the respective bead population ().
Fig. 1. The multiplex bead-based platform. (a) Workflow of the multiplex platform. Isolated EVs or cell culture supernatant were incubated overnight with up to 39 different bead populations, each coupled to a different capture antibody. The different bead populations are distinguishable by flow cytometry. EVs bound to the beads were detected with single antibodies, e.g. cell-specific markers, or exosome markers such as anti-CD9-APC, anti-CD63-APC, anti-CD81-APC antibodies, or with a cocktail of the latter 3 antibodies. (b–d) Analysis example showing (b) exclusion of doublets and no bead events, (c) discrimination of differently labelled bead populations, and (d) measurement of signal intensities of the single bead populations. Positive bead populations are highlighted in colours. Black events represent beads that did not bind EVs or beads with EVs not detected by the staining cocktail.
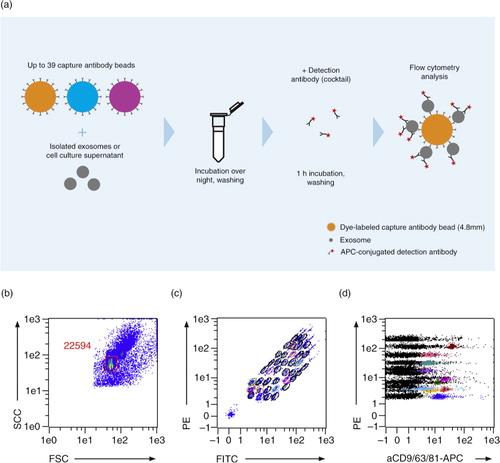
Background signals were determined by analysing beads incubated only with the respective staining antibodies. The background signals were subtracted from the signals obtained for beads incubated with EVs and stained with the respective antibody to receive background corrected signals.
For the NK cell–derived EVs that were isolated from medium with human AB serum, an equivalent volume of medium including human AB serum was used for EV isolation. The signals gained for the isolated AB serum EVs were set as background and were subtracted from the NK cell EV sample signals.
Stimulated Emission Depletion
Standard glass slides (Menzel, Thermo Fisher Scientific) were cleaned in a 1% (w/v) ammonium persulfate solution in sulphuric acid for 1.5 h at room temperature and modified with amino groups by incubation with 2% (w/v) (3-aminopropyl)triethoxysilane (APTES) in ethanol for 1 hour at room temperature. Succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was dissolved in dimethyl sulfoxide and further diluted with PBS containing 2 mM EDTA. After cleaning in ethanol, slides were incubated with 0.5 mg/ml SMCC for 3 hours at room temperature to be activated with maleimide groups. Anti-CD63 (clone H5C6) antibody or a cocktail of anti-CD9 (clone SN4), anti-CD63 and anti-CD81 (clone 5A6) antibodies (Miltenyi Biotec) were reduced by TCEP-HCl for 1 hour at room temperature. The antibodies were incubated for 3 hours at room temperature to bind to the maleimide groups on the activated glass slides.
Antibody-coated slides were washed in PBS with 2 mM EDTA, 0.03% Pluronic F68 and 0.05% sodium azide and stored in the same buffer at 4°C until use. 13 µg B cell EVs, 2 µg monocyte EVs or a mixture of 2.4 µg platelet EVs and 2 µg NK cell EVs in 30 µl PBS/EDTA each were immobilized on slides overnight at 4°C. Slides were washed with PBS and bound EVs were stained with a final concentration of 5 µg/ml of each respective antibody conjugate for 3 hours at 4°C, i.e. anti-CD9-STAR488, anti-CD9-STAR RED, anti-CD81-STAR488, anti-CD81-STAR RED, anti-IgG1-STAR RED, anti-CD42a-STAR RED, and anti-CD19-STAR488 (all Miltenyi Biotec). All incubation steps were performed in a humidified chamber and cover slips were used to prevent the slides from drying. Slides were washed twice in PBS and the stained EVs were preserved with Pro Long Gold antifade reagent (Thermo Fisher Scientific).
For STED analysis a Leica TCS SP8 gSTED microscope with a 100× objective was used. Fiji (ImageJ 2.0.0-rc-38, www.fiji.sc) was used to align the STED images and perform a basic peak detection. After this initial step, R (3.3.0) was used to calculate the full width at half maximum for each spot identified at the respective peak positions (Gaussian fitting on neighbourhood surrounding each peak). Identified spots with intensities below an automatically determined threshold (histogram based) were ignored for further analysis. Using a nearest neighbour approach, colocalized spots were identified.
Immunomagnetic separation of EVs
EVs (25 µg NK cell EVs, 50 µg platelet EVs or a mixture of 12.5 µg NK cell EVs and 25 µg platelet EVs) were incubated with 50 µL CD9-, CD63- or CD81-MicroBeads (Miltenyi Biotec) for 1 hour at room temperature in a MACSmix Tube Rotator in the dark. The mixture was applied to an equilibrated (50 µL equilibration buffer for protein applications, then 3 times 100 µL PBS) µ column placed in a µMACS Separator (Miltenyi Biotec). For the western blot analysis, the µ column was washed 4 times with 200 µL PBS before EVs and beads were co-eluted outside the magnetic field with 100 µL hot (95°C) 1×SDS loading buffer (200 mM Tris, pH 6.8, 0.02% SDS, 0.5% glycerol, 0.005% bromophenol blue). For the multiplex platform analysis, the flow-through of the µ column and 100 µl PAP buffer applied as wash were collected. EVs and beads were co-eluted outside the magnetic field with 100 µL PAP buffer. In both cases, a 5 ml syringe plunger was used for elution.
Results
Multiplex bead-based platform
The multiplex platform is based on polystyrene beads with a diameter of 4.8 µm that were labelled with varying amounts of 2 dyes to generate 39 different bead populations distinguishable by flow cytometry. Each bead population is coupled to a different capture antibody that recognizes EVs that bear the respective antigen. EVs bound to beads are detected by an APC-conjugated antibody. The workflow of the assay is depicted in . For a broad detection, captured exosome-enriched EV samples were stained with an antibody cocktail of the commonly used exosome markers anti-CD9-, anti-CD63- and anti-CD81-APC (Citation30). This marker set might not be suitable for all kinds of EVs and a missing marker on the EVs could bias the results. The parallel detection of up to 39 different surface markers results in a characteristic profile of the EVs.
EVs from NK cells and platelets carry surface markers of the originating cells
To investigate the surface marker profile from primary human blood cells, EVs were isolated from stimulated platelets and expanded NK cells. NK cell EVs from 4 donors or platelet EVs from 11 donors were incubated overnight with 39 bead populations, washed and stained with a cocktail of anti-CD9-, anti-CD63- and anti-CD81-APC antibodies.
Besides the signals for the exosome markers CD9, CD63 and CD81, well-established cell markers could be detected. For example, the NK cell markers CD2 (Citation31), CD8 (Citation32) and CD56 (Citation33) were detected on NK cell EVs while the platelet markers CD41b, CD42a and CD61 (Citation34) were detected on platelet EVs ().
Fig. 2. Surface marker profiles of NK and platelet EVs. Background corrected APC median signal intensities after incubation of NK cell–derived EVs from 4 donors (8 µg each) or platelet-derived EVs from 11 donors (32 µg each) with 39 capture antibody bead populations, followed by staining with a cocktail of anti-CD9-, anti-CD63- and anti-CD81-APC antibodies. REA, mIgG1, mIgG2a and mIgG2b indicate isotype control-beads, i.e. beads linked to an antibody which does not bind EVs.
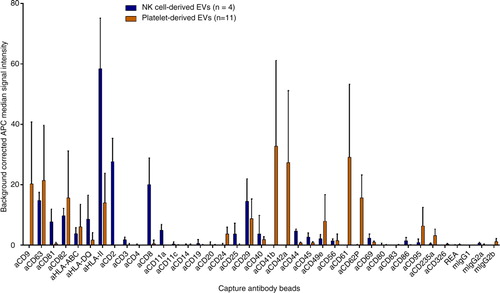
To assess non-specific binding, we incubated multiplex beads with detection antibodies without EVs. The obtained signals were subtracted from the EV sample signals for each bead population to receive background corrected signals. In addition, beads coupled to isotype control antibodies were used to assess potential non-specific binding of EVs to the capture beads. Both negative controls showed low signals (maximal APC median signals of 2.7 and 5.3 for the buffer control and the isotype control-beads, respectively).
The specificity of EV binding was also tested by antibody blocking experiments. Addition of soluble isotype control antibody did not reduce signal intensities. In contrast, using the same excess of CD63 antibody nearly completely abolished the signals of anti-CD63-bead bound EVs, whereas the signals on the anti-CD9 and anti-CD81-beads were not affected (Supplementary Fig. 1a).
Theoretically, different capture antibodies can compete for antigens on an EV population. This could bias the analysis towards larger vesicles, high abundant antigens, or high affinity antibodies. However, only a fraction of the EVs binds to the beads as demonstrated in a control experiment using the supernatant after incubating an EV sample with a 39-plex bead set. The protein profile gained by adding fresh beads to the diluted supernatant was comparable. The signal intensities were lower likely due to the dilution of the sample (Supplementary Fig. 2). In addition, we did not observe competition between capture beads for EVs. EVs that were incubated with a single bead population (data not shown), a bead set consisting of 8 bead populations, or a set of 34 bead populations gave comparable signals (Supplementary Fig. 1b). To minimize potential bias due to the size of the vesicles, we assured a small size range of the EV samples as monitored by Nanoparticle Tracking Analysis (NTA) (Supplementary Fig. 3). In summary, the signal intensity of captured EVs appeared to be independent of the number of beads or the composition of the bead set.
EVs from NK cells and platelets differ in their tetraspanin composition
For a detailed analysis, bead set captured EVs can be split into samples that are each stained with a different APC-conjugated antibody. We hypothesized that swapping capture and detection antibodies would result in different signal intensities. Differing signal intensities could provide information about the relative amount of different markers on an EV population and the proportion of EVs that comprise a given marker. Theoretically, one marker molecule would be sufficient for EV binding to a bead, while more marker molecules would not enhance the EV binding. In contrast, if the marker is used for detection, more marker molecules would ideally increase the signal (a).
Fig. 3. (a, b) Theoretical example of matrix profiles (a) of EVs carrying 10 molecules A and 1 molecule B on each EV or (b) of EVs carrying 10 molecules A and 50% of the EVs comprise also 10 molecules B. Every detected molecule giving a signal of 1 light unit. (c–f) Background corrected APC median signal intensities of anti-CD9-, anti-CD63-, anti-CD81-beads and isotype control-beads after incubation with (c) 16 µg NK cell–derived EVs, (d) 16 µg platelet-derived EVs, (e) a mixture of NK cell–derived EVs and platelet-derived EVs (8 µg each) or (f) 8 µg monocyte-derived EVs, followed by staining with anti-CD9-APC, anti-CD63-APC, or anti-CD81-APC antibodies or with a cocktail of these antibodies. MIgG1 indicates isotype control-beads.
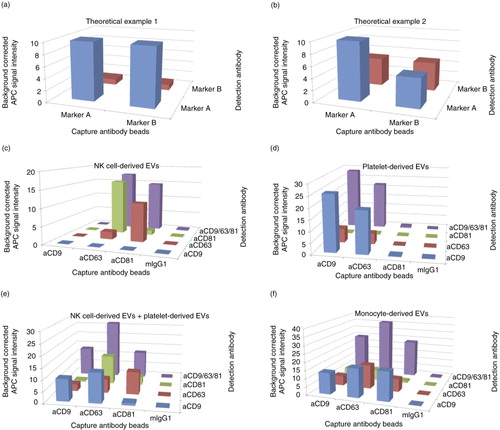
Likewise, the proportion of EVs in a sample comprising a given marker will also affect the signal intensities. For instance, all EVs carry a similar number of marker molecule A but only half of the EVs carry a similar number of marker molecule B (b). Capturing EVs with anti-marker A beads and detecting with anti-marker A antibodies would result in higher signals than capturing A and detecting B. In contrast, by capturing with marker B would give the same signal intensity when detecting with marker A or B. The signal would be as high as capturing with marker A and detecting with marker B but lower than capturing with marker A and detecting with marker A.
In general, lower signal intensities for a bead population indicate less captured EVs likely because of a subpopulation that lack the respective marker.
Indeed, we could observe differences in EV marker levels on EVs isolated from different cell types. After staining with a cocktail of anti-CD9, anti-CD63, and anti-CD81-APC antibodies, no signals were detected on anti-CD9 beads with NK cell–derived EVs. Additionally, no anti-CD9-APC antibody staining was observed on any bead population (c). The lower signals on anti-CD63-beads stained with the same anti-CD63 antibody or the low anti-CD81-bead/anti-CD81-APC signal most likely indicate blocking effects due to the same antibodies used for capturing and staining.
For platelet-derived EVs, hardly any EVs were detected on anti-CD81-beads or on any bead population after anti-CD81-APC antibody staining (d). To exclude any technical issue with the assay, we analysed monocyte EVs giving signals for all combinations of anti-CD9, anti-CD63 and anti-CD81 beads and stainings (f).
To reveal possible subpopulations of platelet EVs, single stainings were performed (d). Irrespective of the detection antibody, stronger signals were detected on anti-CD9-beads as compared to anti-CD63-beads. This indicates that less EVs were captured by the anti-CD63-beads likely due to a subpopulation of platelet EVs carrying no CD63. The captured CD63 positive subpopulation gave a stronger signal after anti-CD9 staining as compared to the larger population of captured CD9 positive EVs stained by anti-CD63 antibodies (d). Taken together, only a subpopulation of platelet EVs carry CD63 and the level of CD63 per EV within this subpopulation was lower as compared to the level of CD9.
The above examples emphasize one important principle of the platform: The detected signal intensity results from the simultaneous binding of the capture antibody and the detection antibody. If no EVs were captured or no marker could be detected, there would be no signal.
Therefore, besides the identification of surface markers on a given EV population, the platform can also be used to discover the co-existence of two markers on the same vesicle.
As an example, a mixture of two distinct EV populations in one sample was mimicked by mixing NK cell EVs and platelet EVs (Supplementary Fig. 4). After staining with an antibody cocktail of anti-CD9-, anti-CD63- and anti-CD81-APC antibodies, each capture antibody bead population gave a signal indicating that EVs bound on all 3 bead populations (e). Due to the used staining cocktail, CD9−/CD81+ EV populations could not be distinguished from CD9+/CD81− populations. But the single stainings with either anti-CD9 or anti-CD81 antibody resulted in very low CD9/CD81-double-positive signals demonstrating that the markers do not co-exist on the same EV.
Our approach of applying different capture and detection antibody combinations allows to discriminate EV subpopulations in one sample and a relative quantitation of surface markers.
Analysis of surface markers on B cell EVs suggests EV subpopulations
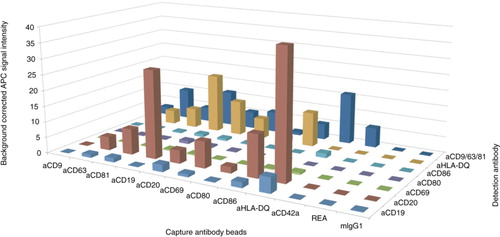
B cells were isolated resulting in high purities (96%, Supplementary Fig. 5a) and activated with CD40 ligand and IL-4. After 5 days, 86 and 98% of the cells were positive for the B cell activation markers CD80 and CD86, respectively (Supplementary Fig. 5b). EVs from activated B cells were incubated with multiplex beads and stained with a cocktail of anti-CD9, anti-CD63 and anti-CD81-APC or with selected single antibodies. For the cocktail staining, weaker signals were detected on the activation marker beads anti-CD80 and anti-CD86 (APC median signal 1.0 and 5.0, respectively) than on the exosome marker bead anti-CD63 (APC median signal 10.0, ). This might be due to a subpopulation of CD63+CD80−CD86− EVs that were likely derived from B cells before activation. Differences in signal intensities could also originate from different antibody affinities, but the weaker signal was detected for both activation markers strengthening the hypothesis of EVs from not-yet activated cells. Furthermore, a strong staining of the antibody conjugates on activated B cells demonstrated antibody functionality (Supplementary Fig. 5b).
The anti-CD42a-beads and the anti-CD9-beads gave signals exclusively after staining with the anti-CD9/63/81-APC cocktail. The signal on the anti-CD42a-beads is a hint of platelets. Control stainings after B cell isolation detected 20% CD42a-positive events if no gates and triggers were set (Supplementary Fig. 6), confirming the presence of platelets within the isolated B cell fraction. We conclude that the signal on the anti-CD9-beads was most likely also due to platelet EVs.
For the B cell markers, signals for anti-CD19-beads were stronger than for anti-CD20-beads (APC median signal 11.3 and 5.4, respectively, ) after the cocktail staining. This might be due to less CD20 positive EVs than CD19 positive EVs. However, the anti-CD20-APC signals were stronger than the anti-CD19-APC signals on all bead populations, suggesting higher levels of CD20 per EV as compared to CD19. Taken together, we found a subpopulation of B cell EVs carrying high levels of CD20.
Fig. 4. Matrix profile of B cell EVs. Background corrected APC median signal intensity of different capture antibody bead populations after incubation with 32 µg EVs from activated B cells, followed by staining with anti-CD19-APC, anti-CD20-APC, anti-CD69-APC, anti-CD80-APC, anti-CD86-APC, anti-HLA-DQ-APC, or a cocktail of anti-CD9-APC, anti-CD63-APC and anti-CD81-APC antibodies. REA and mIgG1 indicate isotype control-beads.
EV visualization by STED microscopy
To analyse differences in protein composition and visualize different EV populations, the analysis on a single EV level would be beneficial. To resolve the small size of EVs, we used a superresolution microscopy technique, called stimulated emission depletion (STED). To overcome bleaching effects on the dyes, we reduced the STED power to 30 or 50% resulting in minor resolution. With the applied settings, we could detect two fluorescence dyes in parallel with a spatial resolution of at least 100 nm.
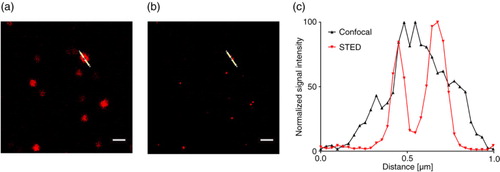
We established a protocol using antibody-coated glass slides to fix EVs and subsequently stained the bound EVs by antibody-fluorophore conjugates optimized for STED microscopy. shows an example of two single EVs located close to each other. While a common fluorescent area appears by confocal microscopy (a), two independent spots can be distinguished by STED demonstrating the higher resolution (b). The risk of incorrectly identifying two neighbouring EVs as a single event is minimized by STED microscopy.
Fig. 5. NK cell–derived EVs bound to an anti-CD63-coated glass slide, stained with anti-CD81-STAR RED, and visualized by (a) confocal microscopy at 100-fold magnitude and (b) STED. (c) Intensity profile of the image section marked with the white line after confocal microscopy (grey) and STED microscopy (red). Scale bars represent 1 µm.
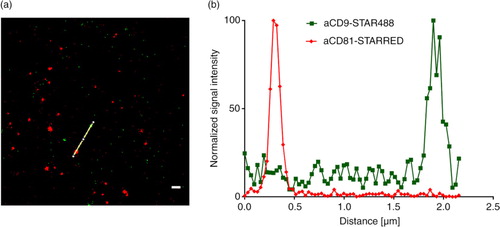
The analysis on the multiplex platform indicated that EVs from NK cells are CD9−CD81+, whereas platelet EVs appeared CD9+CD81−. We analysed a mixture of NK cell EVs and platelet EVs by STED after staining with anti-CD9-STAR488 and anti-CD81-STAR RED. On 4 different image sections, no spots were detected positive for both conjugates (). In control experiments using either NK cell EVs or platelet EVs stained with anti-CD9-STAR488 and anti-CD81-STAR RED, we saw either green (CD9+) spots or red (CD81+) spots, respectively (data not shown). The negligible co-staining demonstrates that single positive EVs can be distinguished from each other.
Fig. 6. STED analysis of EVs bound to an anti-CD63-coated glass slides. (a) A mixture of NK cell EVs and platelet EVs stained with anti-CD9-STAR488 and anti-CD81-STAR RED and (b) the corresponding intensity profile. Scale bar represents 500 nm.
Next, we had a closer look on EVs isolated from B cells that might be contaminated with platelet EVs (). After staining with anti-CD19-STAR488 and anti-CD9-STAR RED, we detected 162 green spots on 3 different image sections (39–84 spots each) and 1,355 (353–619) red spots. In only one of the three image sections, 1% of the spots were positive for both antibodies (a), indicating that the CD19-positive B cell EVs do not carry CD9. In the control experiment, we detected 255 anti-CD9-STAR488 positive spots (72–105 each on 3 different images) and 1,660 spots that were positive for the platelet marker-specific antibody conjugate anti-CD42a-STAR RED (500–617). 8–12% of green spots were also red and 1–2% of the red spots were also green (b), confirming that CD42a+ platelet EVs give rise to the CD9 positive events detected by the multiplex platform.
Fig. 7. STED analysis of B cell EVs bound to anti-CD9/63/81-coated slides and stained with (a) anti-CD19-STAR488 and anti-CD9-STAR RED or (b) stained with anti-CD9-STAR488 and anti-CD42a-STAR RED. Scale bars represent 500 nm.

It is still challenging to visualize the colocalization of markers on the single EV level. The spatial resolution of STED microscopy enables single EV detection, and the data give no indication on false positives due to EV doublets or aggregates. Therefore, co-stainings of single EVs with two different antibodies confirm that there are single EVs carrying two different markers, as suggested by the multiplex data.
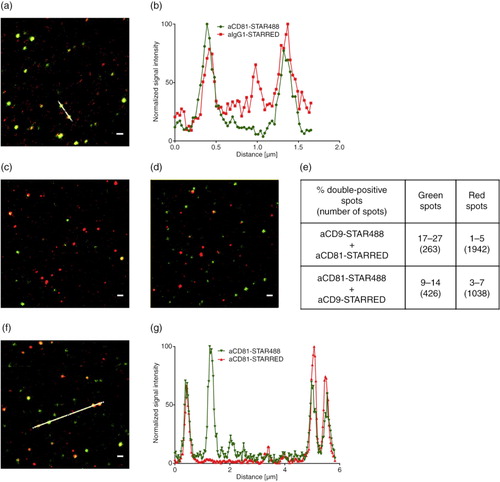
As positive control for the detection of two antibody-fluorophore conjugates on the same EV, we stained monocyte EVs with the humanized anti-CD81-STAR488 and anti-human IgG1-STAR RED as a secondary antibody. In two image sections, 73 and 89% of all detected green fluorescent spots (anti-CD81-STAR488) were also positive for the secondary antibody staining in red (a and b). We detected a higher number of red spots than green spots (118 and 139 versus 177 and 216) resulting in a lower proportion (49 and 57%) of red spots that were double positive. The STAR RED dye appeared brighter or more stable resulting in an improved detection limit for STAR RED. In the control staining of EVs with only the secondary antibody, hardly any signals were detected (data not shown). To evaluate the effect of the 2 dyes, we used monocyte EVs stained with anti-CD9 and anti-CD81 antibodies, each coupled to STAR488 or STAR RED and vice versa. After staining with anti-CD9-STAR488 and anti-CD81-STAR RED, we analysed 3 images and detected a total of 263 green spots (63–113 green spots per image) and 1942 (558–744) red spots (c). After we swapped the dyes (anti-CD81-STAR488/anti-CD9-STAR RED), we detected on 3 images 426 (97–175) green spots and 1,038 (320–364) red spots (d). Comparing the STAR RED or STAR488 positive counts, we see more CD81 positive spots as compared to the respective CD9 staining. The double-positive fractions converge if the brighter STAR RED dye is used for the anti-CD9 antibody (e). The brighter dye somewhat compensates for the lower amounts of CD9 on the EVs or the lower affinity of the antibody. Although we cannot rule out differences in antibody affinity, the results could indicate more CD81 positive EVs or more CD81 epitopes giving brighter stainings as compared to CD9.
Fig. 8. STED analysis of EVs bound to anti-CD63-coated glass slides. (a) Monocyte EVs stained with anti-CD81-STAR488 and anti-IgG1-STAR RED and (b) corresponding intensity profile, (c) monocyte EVs stained with anti-CD9-STAR488 and anti-CD81-STAR RED and (d) monocyte EVs stained with anti-CD81-STAR488 and anti-CD9-STAR RED. (e) Overview of the percentage of double-positive monocyte EVs from 3 images and total number of analysed spots. (f) Monocyte EVs stained with anti-CD81-STAR488 and anti-CD81-STAR RED and (g) corresponding intensity profile. Scale bars represent 500 nm.
As additional control, we stained monocyte EVs with a mixture of the anti-CD81 antibody coupled to either STAR488 or STAR RED to investigate whether the frequency of antigens on one EV is sufficient to enable the binding of 2 antibodies in parallel. We analysed 599 (146–278 each on 3 different images) green spots and 983 (223–404) red spots resulting in only 16–28% green spots that were also red and 7–18% double-positive red spots (f and g). As the proportion of double-positive spots in this control is much lower as compared to the staining using a secondary antibody, the ratio of co-staining results with two different antibodies should be interpreted with care. Co-staining is likely not representative for the proportion of the EVs carrying the two respective epitopes.
Still, our control experiment using a mixture of the same antibody labelled with two different dyes raises the question why the majority of the EVs appeared only single positive. We could rule out the technical limitations of the STED microscope as well as the potential repulsion of the dyes demonstrated by the secondary antibody control staining.
We suggest two possibilities to explain these results. Either EVs carry a limited number of tetraspanins CD9 and CD81 per vesicle or tetraspanin clusters might sterically limit antibody binding and many of these vesicles are stained by a single antibody.
EVs differing in surface markers can be separated using MicroBeads
The separation of EV subpopulations allows further analysis of differences. We used antibody loaded magnetic MicroBeads to isolate EVs according to specific surface markers. As a proof of principle, we mixed NK cell EVs and platelet EVs. By western blot, we detected CD29, CD81 and CD9 in the control sample before separation as well as after anti-CD63 isolation (Supplementary Fig. 7, middle). Obviously, the western blot gave no indication of the two artificial subpopulations. Using the anti-CD9 MicroBeads, only CD29 and CD9 positive EVs (platelet origin) were isolated. Using anti-CD81 MicroBeads, only CD9−CD81+ NK cell EVs were isolated (Supplementary Fig. 7 middle).
To purify the B cell EV sample, we depleted platelet EVs using anti-CD61 MicroBeads. Subsequently, the original B cell EV sample and the isolated fractions were analysed on the multiplex platform. In the CD61 depleted flow-through fraction, the signals for each of the 3 investigated platelet markers (CD41b, CD42a and CD62P) were dramatically lower in comparison to the original sample (Supplementary Fig. 8). The signals for the B cell markers CD19 and CD20 were comparable to the initial sample, indicating high recovery (APC median signal for CD19 of 6.1 before and 5.9 after separation, 2.2 and 1.7, respectively, for CD20; Supplementary Fig. 8). The platelet EVs were conversely detected in the eluate of the column based sorting and appeared CD9 positive in contrast to the flow-through. This confirms again the hypothesis that the signal on anti-CD9 beads derived from platelet EVs in the original B cell preparation ().
Immunoaffinity isolation allows the physical separation of EV subpopulations from a sample for further analysis.
Discussion
Several studies described various exosome populations that are characterized by their surface protein profile and related to the type and status of the originating cell [reviewed in (Citation15, Citation35)]. EV proteins are thought to affect intercellular communication via exosomes (Citation4). Exosomes can carry antigenic material and peptide-MHC-complexes. Tumour antigen-harbouring exosomes incubated with dendritic cells were able to induce antitumor immune responses of T cells that lead to tumour rejection (Citation10, Citation36). In addition to these immune activating effects, proteins on tumour exosomes are also able to promote immune escape (Citation37, Citation38). Depending on their protein content, exosomes could be used for immunization against cancer, to improve immunotherapy or as biomarkers (Citation39, Citation40). The term exosome is defined according to the biogenesis and after secretion; there is no unambiguous marker to discriminate exosomes from other EVs (Citation41). We therefore prefer the term “extracellular vesicles”.
As exosomes adopt cell-specific markers during their biogenesis, it would be advantageous to analyse EV subpopulations in a similar manner to well-established cell typing methods, e.g. by flow cytometry. The small size of exosomes requires technical modifications to flow cytometers to reach resolutions sufficient to reliably analyse 100 nm vesicles (Citation24, Citation25) (Citation42). Fluorescent signals are also limited by the small size of the vesicles. In contrast to multicolour flow cytometry used for cell analysis, only few bright fluorescent dyes such as PE or APC can be used for microvesicle analysis (Citation25). Therefore, the routine analysis of a broader range of surface markers would require several independent flow cytometric measurements.
To enable an easy and fast screening of multiple EV surface proteins, we combined 39 different capture antibody beads to analyse a plurality of markers in parallel by standard flow cytometry. Jorgensen and colleagues described a protein microarray-based technique that detects EVs bound to antibody-coated microarray slides (Citation43). The methods mainly differ in the detection platform, i.e. the capture antibodies are bound to glass slides or beads. Accordingly, a microarray scanner or a flow cytometer is used to acquire the signals. However, we suggest an additional dimension for the analysis by using single detection antibodies. Single staining allows distinguishing subpopulations that do not carry a certain marker and EVs with a low level of the marker, as demonstrated by our analysis of EVs from NK cells, platelets and B cells.
In addition to the characterization of EVs by this screening platform, we aimed to visualize the marker presence on single EVs. Transmission electron microscopy is a traditional technique that combines high resolution of up to 1 nm to determine EV size with information about the morphology (Citation22). Further characterization with immunogold-labelled antibodies is time-consuming and the steric hindrance of gold particles must be taken into account due to the small size of exosomes (Citation23). Whereas electron microscopy takes advantage of the high resolution to resolve morphological properties, STED microscopy enables the detection of multiple markers to resolve biochemical properties.
We adapted STED microscopy to analyse single EVs with a resolution of at least 100 nm. Because this resolution limit is in the range of the exosome sizes, EVs carrying different surface markers could be distinguished and the presence of two different markers on the same EV could be confirmed. However, due to the small size of the spots, we set a signal intensity threshold and we might underestimate the number of detected spots. In addition, we cannot determine the proportion of double-positive EVs. Monocyte EVs stained with a mixture of the same anti-CD81 antibody coupled to either one of two dyes revealed that most EVs were stained with only one of the dyes. A control experiment using a secondary antibody demonstrated that colocalized antibodies on EVs can be detected. In addition, the number of protein molecules per exosome had been estimated in a range of 10–30 (Citation44, Citation45) and tetraspanins are among the most prominent exosome surface markers (Citation18, Citation46). Therefore, we would rule out limiting amounts of tetraspanins per EV to explain the low proportion of double-positive EVs. Instead, we speculate that tetraspanins might mainly be present as clusters and not evenly distributed on EVs. Zuidscherwoude and colleagues demonstrated by STED microscopy of plasma membranes that most tetraspanin clusters are composed of one tetraspanin type (Citation47). Interestingly, these clusters appeared quite large even under STED resolution, with diameters well above 100 nm. Exosomes most likely adopt tetraspanin clusters from the plasma membrane because homodimers of CD9 and CD81 were observed to be assembled already from newly synthesized proteins in the Golgi (Citation48). We consider that exosomes adopt these clusters or parts of them. Assuming 4 nm diameter for a tetraspanin molecule (Citation49), a small cluster of, e.g. 6 tetraspanin molecules would give a cluster diameter of about 12 nm. Such an area could be occupied by a single bound antibody.
Smaller clusters would only allow binding of a single antibody per cluster. Larger clusters or more than one cluster per EV would give rise to the proportion of double-positive stained EVs. Interestingly, for the few antibody pairs that recognized different epitopes, we also saw a minor portion of double-positive EVs in the STED pictures. We suggest the following model: Tetraspanins build homoclusters instead of being evenly distributed over the surface of an exosome and most of the exosomes comprise only a subset of tetraspanins.
Accordingly, sandwich assays, such as our multiplex platform, give information on a proportion of EVs carrying at least two accessible epitopes. Even for our STED analysis, we miss EVs that might lack the epitope required for binding to the antibody-coated glass slide.
We suggest that the surface of single EVs and the population of EVs are more heterogeneous than assumed so far.
Marker-specific isolation or depletion of EVs by immunoaffinity isolation enables further analysis of heterogeneous EV mixtures. It has already been demonstrated that EVs from various sources can be isolated using suitable antibody-coated magnetic beads (Citation50–Citation55). According to the protein profiles of EVs isolated by immunoaffinity in comparison to ultracentrifugation and gradient density separation, immunoaffinity might be the “most effective method to isolate exosomes” (Citation54).
It is known that TEMs are cell-specific (Citation16) and are transferred to exosomes (Citation17). Since they interact with each other and other proteins that are important for cell communication and cell activation (Citation17, Citation18), the presence of tetraspanins on distinct exosome populations might be related to different exosome functions. In line with this, we found that the tetraspanins CD63 and CD81 but hardly any CD9 could be detected on NK cell–derived EVs. As another example, platelet-derived EVs were positive for CD63 and CD9, but CD81 was not detectable. In consistency with these findings, NK cell–derived exosomes are described as CD63+ (Citation56), but according to exocarta.org (Citation57), the existence of CD9 or CD81 was not yet investigated. Platelet exosomes were shown to be positive for CD9 (Citation58) and CD63 (Citation28), but CD81 could not so far be detected (Citation28, Citation58).
EVs isolated from activated B cells likely comprise subpopulations. The results obtained from combinations of several capture and detection antibodies indicate the existence of different EV populations: platelet-derived EVs, EVs from activated B cells, EVs from non-activated B cells, CD19dimCD20− EVs and CD19dimCD20high EVs. Consistent with our results, several studies demonstrated that B cell exosomes are enriched in MHC class II (Citation51, Citation59–Citation62). Two published studies used beads coated with an anti-MHC class II antibody to detect CD19, CD20, CD63, CD80, CD81 and CD86 on B cell exosomes (Citation51, Citation60). Clayton and colleagues detected CD20 on B cell exosomes but did not investigate CD19, whereas Admyre and colleagues detected CD19 but did not stain for CD20 (Citation51, Citation60). In another study, Oksvold and colleagues analysed the CD19 and CD20 levels of exosomes from four B cell lines by western blotting (Citation62). These 3 studies confirm that both markers are present on B cell exosomes but do not give a hint on potential B cell EV subpopulations as we have suggested.
We could assign the CD9+ EVs in our B cell EV preparation to platelet-derived EVs. Oksvold and colleagues did not detect CD9 on any of the 5 investigated B cell line exosomes (Citation62). Therefore, the CD9− B cell EVs, in addition to platelet and NK cell EVs, might be the third type of EVs with very low levels of one of the three so-called exosome markers. Among all tetraspanins, CD9 and CD81 are most structurally related (Citation63) and compete with each other for association with the other tetraspanin proteins (Citation64). On the other hand, CD9 and CD81 are described to preferentially homodimerize in comparison to cross-linking with other tetraspanins (Citation48). Since CD81 has been described to be able to compensate CD9 loss (Citation65), the question arises whether CD9 and CD81 are exchangeable. If the presence of both markers is not necessary, one would assume CD9 is preferentially present on platelet EVs as observed here because CD9 is associated with CD41/CD61 on platelets (Citation63). Similarly, CD81 would be expected on B cell EVs because CD81 was shown to be required for the normal expression of CD19 (Citation66). In addition, CD81 seems to be enriched in B cell exosomes (Citation17) and CD9 is expressed only during B cell development (Citation63). In general, the presence of various proteins is critical for EV function. For example, surface proteins on exosomes and recipient cells were shown to be essential for exosome uptake because the uptake was inhibited by proteinase K treatment (Citation67). Exosomes from APC were observed to activate naïve T cells in absence of APC if they carry MHC class I/peptide complexes, intercellular adhesion molecule I (CD54) and the co-stimulatory molecule B7 (Citation68). In addition, disease-associated antigens were observed to be transferred between cells via exosomes. The main co-receptor CCR5 for macrophage-tropic human immunodeficiency virus (HIV)-1 is transferred via microparticles to CCR5 negative peripheral blood mononuclear cells leading to infection of tissues without endogenous CCR5 expression (Citation69). Similarly, aggressive glioma cells have been shown to transfer the oncogenic form of the epidermal growth factor receptor (EGFRvIII) to receptor-negative cancer cells by microvesicles (Citation70). Melanoma derived, MET-containing exosomes appeared to educate bone marrow progenitor cells towards metastasis (Citation71). Exosomes might “adopt the homing pattern of the parental cell of origin” (Citation72) and targeted delivery could be important for the development of vaccines or therapies. These examples demonstrate the need to investigate potential subpopulations of EVs not only with regard to their origin, but also with the focus on potential functional effects.
Taken together, we demonstrate here that immunoaffinity isolation is a fast and easy tool to physically separate EV subpopulations. It can be combined with the multiplex bead-based platform to screen the initial sample and the separated fractions for EV subpopulations. Visualization of single EVs and their surface marker profile by STED microscopy gives insight into EV heterogeneity.
The novel methods introduced here can change the way we look at the origins and functions of the plethora of EV populations.
Conflict of interest and funding
Nina Koliha, Yvonne Wiencek, Ute Heider, Susanne Krauthäuser, Ian Johnston, Andreas Bosio and Stefan Wild are employed at Miltenyi Biotec GmbH. A patent application has been submitted including the concept of matrix protein profiles: A method for analysing markers on the surface of vesicles, submission number 3496895, application number EP15167673.1
Supplementary Material
Download PDF (1.5 MB)Acknowledgements
We thank Falk Nimmerjahn and Andreas Baur (Friedrich-Alexander-University Erlangen-Nuremberg) for discussions. We gratefully acknowledge Eva Bergschneider, Markus Granzin, Sabine Müller, Ursula Bissa, and Sabine Schmachtenberg (Miltenyi Biotec) for their technical assistance. We thank Martin Wiemann and Christian Schechtmann (IBE R&D GmbH) for performing NTA.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- EL Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013; 12: 347–57.
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev. 2009; 9: 581–93.
- Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev. 2002; 2: 569–79.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200: 373–83.
- Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005; 17: 879–87.
- Aalberts M, van Dissel-Emiliani FM, van Adrichem NP, van Wijnen M, Wauben MH, Stout TA, etal. Identification of distinct populations of prostasomes that differentially express prostate stem cell antigen, annexin A1, and GLIPR2 in humans. Biol Reprod. 2012; 86: 82.
- Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004; 101: 13368–73.
- Ogawa Y, Miura Y, Harazono A, Kanai-Azuma M, Akimoto Y, Kawakami H, etal. Proteomic analysis of two types of exosomes in human whole saliva. Biol Pharmaceut Bull. 2011; 34: 13–23.
- Admyre C, Johansson SM, Qazi KR, Filen JJ, Lahesmaa R, Norman M, etal. Exosomes with immune modulatory features are present in human breast milk. J Immunol. 2007; 179: 1969–78.
- Andre F, Schartz NE, Movassagh M, Flament C, Pautier P, Morice P, etal. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002; 360: 295–305.
- Street JM, Barran PE, Mackay CL, Weidt S, Balmforth C, Walsh TS, etal. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med. 2012; 10: 5.
- Lasser C, Alikhani VS, Ekstrom K, Eldh M, Paredes PT, Bossios A, etal. Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J Transl Med. 2011; 9: 9.
- Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, etal. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Comm. 2011; 2: 282.
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654–9.
- Simpson RJ, Lim JW, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics. 2009; 6: 267–83.
- Levy S, Shoham T. The tetraspanin web modulates immune-signalling complexes. Nat Rev. 2005; 5: 136–48.
- Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998; 273: 20121–7.
- Andreu Z, Yanez-Mo M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. 2014; 5: 442.
- van Niel G, Charrin S, Simoes S, Romao M, Rochin L, Saftig P, etal. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev Cell. 2011; 21: 708–21.
- Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol. 2010; 190: 1079–91.
- Buschow SI, Nolte-‘t Hoen EN, van Niel G, Pols MS, ten Broeke T, Lauwen M, etal. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic. 2009; 10: 1528–42.
- van der Pol E, Coumans FA, Grootemaat AE, Gardiner C, Sargent IL, Harrison P, etal. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J Thromb Haemost. 2014; 12: 1182–92.
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. Chapter 3: Unit 3.22.
- Kormelink TG, Arkesteijn GJ, Nauwelaers FA, van den Engh G, Nolte-‘t Hoen EN, Wauben MH. Prerequisites for the analysis and sorting of extracellular vesicle subpopulations by high-resolution flow cytometry. Cytometry A. 2015
- van der Vlist EJ, Nolte-‘t Hoen EN, Stoorvogel W, Arkesteijn GJ, Wauben MH. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protocol. 2012; 7: 1311–26.
- Vicidomini G, Schonle A, Ta H, Han KY, Moneron G, Eggeling C, etal. STED nanoscopy with time-gated detection: theoretical and experimental aspects. PLoS One. 2013; 8: e54421.
- Willig KI, Rizzoli SO, Westphal V, Jahn R, Hell SW. STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. Nature. 2006; 440: 935–9.
- Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999; 94: 3791–9.
- Leong HS, Podor TJ, Manocha B, Lewis JD. Validation of flow cytometric detection of platelet microparticles and liposomes by atomic force microscopy. J Thromb Hemostasis. 2011; 9: 2466–76.
- Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014; 3: :24641, doi: http://dx.doi.org/10.3402/jev.v3.24641.
- Ida H, Anderson P. Activation-induced NK cell death triggered by CD2 stimulation. Eur J Immunol. 1998; 28: 1292–300.
- Addison EG, North J, Bakhsh I, Marden C, Haq S, Al-Sarraj S, etal. Ligation of CD8alpha on human natural killer cells prevents activation-induced apoptosis and enhances cytolytic activity. Immunology. 2005; 116: 354–61.
- Caligiuri MA. Human natural killer cells. Blood. 2008; 112: 461–9.
- Schmitz G, Rothe G, Ruf A, Barlage S, Tschope D, Clemetson KJ, etal. European working group on clinical cell analysis: consensus protocol for the flow cytometric characterisation of platelet function. Thromb Haemost. 1998; 79: 885–96.
- Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev. 2014; 14: 195–208.
- Wolfers J, Lozier A, Raposo G, Regnault A, Thery C, Masurier C, etal. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001; 7: 297–303.
- Huber V, Fais S, Iero M, Lugini L, Canese P, Squarcina P, etal. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology. 2005; 128: 1796–804.
- Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E, etal. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood. 2009; 113: 1957–66.
- Revenfeld AL, Baek R, Nielsen MH, Stensballe A, Varming K, Jorgensen M. Diagnostic and prognostic potential of extracellular vesicles in peripheral blood. Clin Ther. 2014; 36: 830–46.
- Pant S, Hilton H, Burczynski ME. The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol. 2012; 83: 1484–94.
- Gould SJ, Raposo G. As we wait: coping with an imperfect nomenclature for extracellular vesicles. J Extracell Vesicles. 2013; 2: :20389, doi: http://dx.doi.org/10.3402/jev.v2i0.20389.
- van der Pol E, Coumans F, Varga Z, Krumrey M, Nieuwland R. Innovation in detection of microparticles and exosomes. J Thromb Haemost. 2013; 11(Suppl 1): 36–45.
- Jorgensen M, Baek R, Pedersen S, Sondergaard EK, Kristensen SR, Varming K. Extracellular Vesicle (EV) Array: microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. J Extracell Vesicles. 2013; 2: :20920, doi: http://dx.doi.org/10.3402/jev.v2i0.20920.
- Estelles A, Sperinde J, Roulon T, Aguilar B, Bonner C, LePecq JB, etal. Exosome nanovesicles displaying G protein-coupled receptors for drug discovery. Int J Nanomedicine. 2007; 2: 751–60.
- Higginbotham JN, Demory Beckler M, Gephart JD, Franklin JL, Bogatcheva G, Kremers GJ, etal. Amphiregulin exosomes increase cancer cell invasion. Curr Biol. 2011; 21: 779–86.
- Simpson RJ, Jensen SS, Lim JW. Proteomic profiling of exosomes: current perspectives. Proteomics. 2008; 8: 4083–99.
- Zuidscherwoude M, Gottfert F, Dunlock VM, Figdor CG, van den Bogaart G, Spriel AB. The tetraspanin web revisited by super-resolution microscopy. Sci Rep. 2015; 5: 12201.
- Kovalenko OV, Yang X, Kolesnikova TV, Hemler ME. Evidence for specific tetraspanin homodimers: inhibition of palmitoylation makes cysteine residues available for cross-linking. Biochem J. 2004; 377: 407–17.
- Min G, Wang H, Sun TT, Kong XP. Structural basis for tetraspanin functions as revealed by the cryo-EM structure of uroplakin complexes at 6-A resolution. J Cell Biol. 2006; 173: 975–83.
- Coren LV, Shatzer T, Ott DE. CD45 immunoaffinity depletion of vesicles from Jurkat T cells demonstrates that exosomes contain CD45: no evidence for a distinct exosome/HIV-1 budding pathway. Retrovirology. 2008; 5: 64.
- Clayton A, Court J, Navabi H, Adams M, Mason MD, Hobot JA, etal. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J Immunol Meth. 2001; 247: 163–74.
- Koga K, Matsumoto K, Akiyoshi T, Kubo M, Yamanaka N, Tasaki A, etal. Purification, characterization and biological significance of tumor-derived exosomes. Anticancer Res. 2005; 25: 3703–7.
- Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008; 110: 13–21.
- Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, etal. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012; 56: 293–304.
- Gieseler F, Gamperl H, Theophil F, Stenzel I, Quecke T, Ungefroren H, etal. Using annexin V-coated magnetic beads to capture active tissue factor-bearing microparticles from body fluids. Cell Biol Int. 2014; 38: 277–81.
- Lugini L, Cecchetti S, Huber V, Luciani F, Macchia G, Spadaro F, etal. Immune surveillance properties of human NK cell-derived exosomes. J Immunol. 2012; 189: 2833–42.
- Simpson RJ, Kalra H, Mathivanan S. ExoCarta as a resource for exosomal research. J Extracell Vesicles. 2012; 1: :18374, doi: http://dx.doi.org/10.3402/jev.v1i0.18374.
- Aatonen MT, Ohman T, Nyman TA, Laitinen S, Gronholm M, Siljander PR. Isolation and characterization of platelet-derived extracellular vesicles. J Extracell Vesicles. 2014; 3: :24692, doi: http://dx.doi.org/10.3402/jev.v3.24692.
- Arita S, Baba E, Shibata Y, Niiro H, Shimoda S, Isobe T, etal. B cell activation regulates exosomal HLA production. Eur J Immunol. 2008; 38: 1423–34.
- Admyre C, Bohle B, Johansson SM, Focke-Tejkl M, Valenta R, Scheynius A, etal. B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce TH2-like cytokines. J Allergy Clin Immunol. 2007; 120: 1418–24.
- Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. EMBO J. 2007; 26: 4263–72.
- Oksvold MP, Kullmann A, Forfang L, Kierulf B, Li M, Brech A, etal. Expression of B-cell surface antigens in subpopulations of exosomes released from B-cell lymphoma cells. Clin Ther. 2014; 36: 847–62.e1.
- Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997; 11: 428–42.
- Rubinstein E, Le Naour F, Lagaudriere-Gesbert C, Billard M, Conjeaud H, Boucheix C. CD9, CD63, CD81, and CD82 are components of a surface tetraspan network connected to HLA-DR and VLA integrins. Eur J Immunol. 1996; 26: 2657–65.
- Parthasarathy V, Martin F, Higginbottom A, Murray H, Moseley GW, Read RC, etal. Distinct roles for tetraspanins CD9, CD63 and CD81 in the formation of multinucleated giant cells. Immunology. 2009; 127: 237–48.
- Shoham T, Rajapaksa R, Boucheix C, Rubinstein E, Poe JC, Tedder TF, etal. The tetraspanin CD81 regulates the expression of CD19 during B cell development in a postendoplasmic reticulum compartment. J Immunol. 2003; 171: 4062–72.
- Escrevente C, Keller S, Altevogt P, Costa J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Canc. 2011; 11: 108.
- Hwang I, Shen X, Sprent J., Direct stimulation of naive T. cells by membrane vesicles from antigen-presenting cells: distinct roles for CD54 and B7 molecules. Proc Natl Acad Sci U S A. 2003; 100: 6670–5.
- Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J, etal. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. 2000; 6: 769–75.
- Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, etal. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008; 10: 619–24.
- Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, etal. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012; 18: 883–91.
- Wiklander OP, Nordin JZ, O'Loughlin A, Gustafsson Y, Corso G, Mager I, etal. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J Extracell Vesicles. 2015; 4: 26316,. doi: http://dx.doi.org/10.3402/jev.v4.26316.