
Abstract
Extracellular vesicles (EVs) mediate intercellular communication through transfer of RNA and protein between cells. Thus, understanding how cargo molecules are loaded and delivered by EVs is of central importance for elucidating the biological roles of EVs and developing EV-based therapeutics. While some motifs modulating the loading of biomolecular cargo into EVs have been elucidated, the general rules governing cargo loading and delivery remain poorly understood. To investigate how general biophysical properties impact loading and delivery of RNA by EVs, we developed a platform for actively loading engineered cargo RNAs into EVs. In our system, the MS2 bacteriophage coat protein was fused to EV-associated proteins, and the cognate MS2 stem loop was engineered into cargo RNAs. Using this Targeted and Modular EV Loading (TAMEL) approach, we identified a configuration that substantially enhanced cargo RNA loading (up to 6-fold) into EVs. When applied to vesicles expressing the vesicular stomatitis virus glycoprotein (VSVG) – gesicles – we observed a 40-fold enrichment in cargo RNA loading. While active loading of mRNA-length (>1.5 kb) cargo molecules was possible, active loading was much more efficient for smaller (~0.5 kb) RNA molecules. We next leveraged the TAMEL platform to elucidate the limiting steps in EV-mediated delivery of mRNA and protein to prostate cancer cells, as a model system. Overall, most cargo was rapidly degraded in recipient cells, despite high EV-loading efficiencies and substantial EV uptake by recipient cells. While gesicles were efficiently internalized via a VSVG-mediated mechanism, most cargo molecules were rapidly degraded. Thus, in this model system, inefficient endosomal fusion or escape likely represents a limiting barrier to EV-mediated transfer. Altogether, the TAMEL platform enabled a comparative analysis elucidating a key opportunity for enhancing EV-mediated delivery to prostate cancer cells, and this technology should be of general utility for investigations and applications of EV-mediated transfer in other systems.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Since the discovery that extracellular vesicles (EVs), including exosomes and microvesicles, can transfer functional RNA and protein between cells (Citation1–Citation3), there has been much interest in understanding the rules that govern the loading of biomolecular cargo into EVs and subsequent EV-mediated delivery of such cargo to recipient cells. While miRNA loading and functional delivery is now relatively well-studied (Citation1, Citation4–Citation10), mRNA and protein delivery has been less thoroughly explored. Open questions thus include: Why are EVs enriched in small RNAs compared to EV-producing cells (Citation1, Citation11) (Citation12)? How does RNA-loading efficiency depend on the size of the RNA molecule? What factors impact the degree to which RNA and protein cargo are delivered to the cytoplasm of recipient cells?
EVs are generally enriched in small RNAs, when compared to the RNA pools in the cells from which EVs are derived, but our understanding of this phenomenon is incomplete (Citation1, Citation11–Citation13). One potential explanation for this observation is the existence of sorting mechanisms that actively incorporate small RNAs into EVs. For example, Villarroya-Beltri et al. discovered sequence motifs that are highly enriched in EV-associated miRNAs, compared to cell-associated miRNAs, and determined that sumoylated hnRNAPA2B1 binds to these “EXOmotifs” to mediate loading of bound RNAs into EVs (Citation8). Annexin A2 was also shown to play a role in loading a subset of miRNAs into EVs, though no sequence dependence was observed (Citation14). A process for actively loading miRNAs into EVs could bias the EV RNA population towards smaller RNAs. However, there seem to be active processes for loading some mRNAs into EVs as well. Bolukbasi et al. discovered sequence motifs that lead to enhanced loading of mRNAs into EVs, via a mechanism that involves sequence-specific interactions between mRNA and specific miRNA (Citation15). EVs are also enriched in other small RNAs, including yRNAs, tRNAs and mRNA fragments (Citation13), although no active loading mechanism has yet been identified for these cargo molecules. Altogether, these observations suggest that while some specific active loading mechanisms may exist, it is possible that RNA size directly impacts EV-loading efficiency as well, although this hypothesis has not been directly tested. Thus, the general rules governing RNA-loading efficiency remain rather poorly understood.
Although EV-mediated delivery of mRNA and protein products has been reported for several systems, there remains substantial uncertainty as to whether the cargo molecules were delivered to the cytoplasm and to what extent, if any, post-delivery translation of cargo mRNA may have occurred. Mizrak et al. found that overexpressing an exogenous cargo protein (CD-UPRT, a prodrug-converting enzyme) in EV-producing cells was sufficient to load both CD-URPT mRNA and protein into EVs (Citation16). EV-receiving cells treated with the cognate prodrug experienced toxicity, whereas control EV-receiving cells did not. While this observation confirmed that CD-UPRT enzyme was functional in recipient cells, such activity could be mediated from either endosomal or cytoplasmic locations. Whether CD-UPRT mRNA was translated in recipient cells was not investigated, such that it is not clear whether endosomal escape (e.g. via intermembrane fusion between EVs and endosomes) and cytoplasmic delivery occurred in this system. Kanada et al. observed that exosomes and microvesicles delivered a luciferase-RFP fusion protein to recipient cells (Citation17). Delivered protein was observed in punctae within recipient cells, again suggesting accumulation of EV cargo in endosomes. The luciferase-RFP mRNA could be detected in recipient cells 24 h post EV-delivery, and by 48 h this mRNA could no longer be detected – such degradation could have occurred via either lysosomal or cytoplasmic routes. Perhaps, the strongest evidence for cytoplasmic delivery of mRNA or protein EV cargo was reported by Zomer et al. and Ridder et al., who observed that EV-mediated transfer of nuclear-targeted Cre protein could catalyze recombination in recipient cells (Citation18, Citation19). Since Cre mRNA but not protein was detected in EVs, both studies concluded that post-delivery translation of Cre mRNA conferred the observed recombination. Finally, it is possible that the relative rates at which mRNA and protein are loaded into EVs and delivered by EVs to recipient cells depend on both mRNA and protein abundance as well as the cell types serving as EV senders and EV receivers. Resolving these important questions, and elucidating which steps limit EV-mediated delivery in any one system, thus requires a comparative approach.
The above questions and phenomena have proven challenging to investigate quantitatively, in part because we lack tools for modulating the efficiency with which RNA cargo is loaded into EVs. For example, a generalizable method for actively loading RNAs into EVs would enable a direct comparison of loading efficiency between RNAs by testing what can be loaded. Importantly, such a method would facilitate direct comparison within a single EV producer cell type. Furthermore, the ability to tune RNA cargo loading into EVs would enable an evaluation of the effects of RNA cargo abundance on functional cargo delivery to the cytoplasm of recipient cells. To meet these needs, in this study we developed a general platform for actively loading RNA into EVs, which we term the Targeted and Modular EV Loading (TAMEL) platform, and we used this technology to investigate the important open questions posed above.
Methods
Plasmid construction
Human Lamp2b cDNA clone 3543019 was purchased from Open Biosystems (now sold by Dharmacon) and inserted into pcDNA3.1+ Hygro (Clontech). A C-terminal GSG spacer and an HA peptide tag (YPYDVPDYA) were added by PCR to create plasmid pMEH31. The pMS2-GFP plasmid, which encodes the MS2 bacteriophage coat protein tandem dimer (mutant d1FG and V29I (Citation20, Citation21)), was obtained from Addgene (plasmid #27121), deposited by Robert Singer (Citation22). The MS2 dimer was amplified from this plasmid and inserted into pMEH31 with a 7-amino-acid peptide spacer (GASGSGS) downstream of Lamp2b and upstream of HA by PCR and restriction digest to create plasmid pMEH60. Human CD63 cDNA was amplified from HEK293FT cell lysate using F: ATCATCGGTACCATGGCGGTGGAAGGAGGAATG and R: GGCTGGGCTAGCCATCACCTCGTAGCCACTTCTGATACTCTTCAC and substituted in place of Lamp2b; CD63 was inserted with or without a C-terminal fusion to the MS2 dimer (+ MS2: pMEH343, −MS2: pMEH371). The same method was used for human Hspa8 (isoform 1), obtained via amplification from HEK293FT cDNA using F: ATCATTGGTACCATGTCCAAGGGACCTGCAG, and R: ACCAACGCTAGCATCAACCTCTTCAATGGTGGGCCCTG(+MS2: pMEH361, -MS2: pMEH365). This method was also used for vesicular stomatitis virus glycoprotein (VSVG, Clontech) +MS2: pMEH381, −MS2: pMEH380. A plasmid encoding tandem dTomato was purchased from Addgene (plasmid 18917), deposited by Scott Sternson (Citation23). A sequence encoding the dTomato monomer was amplified and inserted with (pMEH275) or without (pMEH373) an N-terminal SV40 nuclear localization sequence (NLS) (Citation24) into pGIPZ (obtained from Northwestern High Throughput Analysis Core Facility) in place of GFP by PCR and restriction digest. Single MS2 loops in the 3′ UTR were added by PCR to pMEH275 to create pMEH294 (high-affinity MS2 loop) and pMEH295 (wild-type MS2 loop). DNA sequences encoding tandem repeats of MS2 loops were synthesized (ThermoFisher Scientific) and inserted downstream of dTomato into pMEH275 by restriction digest to create pMEH280 (3× high-affinity MS2 loops) and pMEH303 (6× high-affinity MS2 loops). Plasmid pXX30 containing Cas9 was a generous gift from Erik Sontheimer (UMass). Cas9 was amplified from this plasmid and inserted in place of dTomato with 3 high-affinity MS2 loops (pMEH305). The gene encoding mTFP1 was synthesized (ThermoFisher Scientific) and inserted into pGIPZ in place of GFP to create pMEH286 (used as a transfection control plasmid in EV production experiments, below). Plasmid maps are included as supplementary information.
Cell culture and transfection
HEK293FT cells (ThermoFisher Scientific) and PC-3 cells (gifted by Chung Lee, Northwestern University) were maintained at 37°C in 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS, 1% penicillin–streptomycin and 4 mM l-glutamine. Sublines generated from these cells (see Cell Line Generation section) were cultured in the same way. HEK293FT cells and sublines were plated at ~106 cells/mL in 15 cm dishes (20 mL medium). After 6–8 h, when cells were ~70–80% confluent and well attached to the plate, 30 µg of DNA was transfected using the CaCl2–HEPES-buffered saline (HeBS) method. For EV production, cells were transfected with 10 µg of plasmid encoding Lamp2b, Hspa8 or CD63 fusion protein, 15 µg of pcDNA 3.1+ Hygro and 5 µg of pMEH 286 as a transfection control. Transfection efficiencies were estimated by visualization of mTFP1 fluorescence and EVs were harvested so long as visual transfection efficiencies exceeded ~60% (typical efficiencies were ~60–80%). For gesicle production, cells were transfected with 15 µg of pMEH380 or pMEH381, 10 µg of pcDNA 3.1+ Hygro and 5 µg of pMEH286 as a transfection control. Transfection efficiency was evaluated as with EV production. And 12–16 h after transfection, medium was changed to EV-depleted medium (see EV Isolation and Characterization section).
Cell line generation
To package lentiviral vectors, HEK293FT cells were plated at ~ 8×105 cells/mL in 10 cm dishes (8 mL). Six to eight hours later, when cells were ~60–70% confluent and well attached to the plate, cells were transfected with 3 µg of pMD2G, 8 µg of pspax (gifted by William Miller, Northwestern University), 10 µg of viral vector (pGIPZ backbones) and 1 µg of pDsRedExpress2 (Clontech). Twelve to fourteen hours later, medium was changed. Twenty-eight hours after the medium change, conditioned medium was harvested, cleared of cells by centrifugation at 500 g for 2 min and filtered through a 0.4-µm pore filter (VWR, Radnor, PA, USA). Cleared supernatant was then concentrated by ultracentrifugation at 24,200 rpm using an SW41 Ti rotor in an L-80 Optima XP ultracentrifuge for 90 min. The pellet was harvested and added to HEK293FT cells (for dTomato and Cas9 viruses), transduced at an estimated multiplicity of infection of 3. All cell lines generated in this study are described in Table .
Table I. Cell lines generated in this studya
EV isolation and characterization
EV-depleted medium was generated by pelleting FBS-derived EVs from DMEM containing 20% FBS by ultracentrifugation at 26,500 rpm in an Optima XP ultracentrifuge (Beckman Coulter) with the SW41Ti rotor for 135 min at 4°C. The cleared supernatant was mixed with serum-free supplemented DMEM to achieve a final concentration of 10% FBS in “EV-depleted medium.” For EV production, cells were incubated for 24 h before conditioned medium harvest, since EV concentration in conditioned medium was not observed to differ significantly at 24, 36 or 48 h of incubation, indicating that a steady state of EV production and uptake was reached by 24 h (Michelle Hung and Joshua Leonard, unpublished observations). For gesicle production, cells were incubated for 48 h before conditioned medium harvest, as previously described (Citation25). EVs were isolated from conditioned medium by differential centrifugation at 4°C. Conditioned medium was centrifuged at 300 g for 10 min, 2,000 g for 10 min and then 10,000 g for 30 min; the supernatant was retrieved at each step to remove cells, cell debris and apoptotic bodies, respectively (Citation26). From this clarified supernatant, EVs were pelleted by ultracentrifugation as described above. Gesicles were isolated from conditioned medium as previously described (Citation25). Briefly, conditioned medium was centrifuged at 300 g for 10 min to remove cells, filtered through a 0.4-µm pore filter and gesicles were pelleted by ultracentrifugation at 30,000 rpm for 1 h at 4°C. EV and gesicle morphology was evaluated by transmission electron microscopy using a 4% uranyl acetate negative stain (Supplementary Fig. 1a and b, respectively). EVs and gesicles displayed round, membrane-bound morphologies and appeared to be 50–200 nm in diameter, as expected. EV and gesicle size distributions and concentrations were profiled by NTA (Supplementary Fig. 1c and d, respectively) using a NanoSight LM10-HS (Malvern) with a laser wavelength of 405 nm and NanoSight NTA software v2.3. Videos were acquired at camera level 14. Ambient temperature (22°C) was used and 100 nm calibration beads (Malvern) were used to check that sizing was accurate. Samples were introduced manually. Three 30 s videos were analysed per sample. EVs were diluted 1:50 and gesicles were diluted 1:100 in PBS to keep concentration between 2 and 9×106 vesicles/mL. Vesicle concentration was defined as the mean of the concentrations determined from each of the 3 videos. Videos were analysed at a detection threshold of 7. The blur, minimum track length and minimum expected particle size were set automatically by the software. EV and gesicle size distributions were approximately normal, with means between 100 and 200 nm, as expected.
Immunoblotting and densitometry
For western blot analysis, EVs and gesicles were heated in Laemmli buffer at 70°C. Protein concentration was measured by BCA assay (Pierce), and several concentrations of each protein were loaded in each lane of a 4–15% gradient polyacrylamide gel (Bio-Rad). After transfer to a PVDF membrane (Bio-Rad) at 100 V for 1 h, membranes were blocked for 1 h in 5% milk at room temperature and blotted with rabbit anti-HA antibody (Cell Signaling Technology C29F4) diluted 1:1,000 and incubated overnight at 4°C. Primary antibodies were detected with horseradish peroxidase-conjugated goat–antirabbit immunoglobulin G secondary antibody (Thermofisher Scientific). Densitometry analysis was performed using ImageJ software (NIH) according to the user guide.
Analysis of RNA in EVs
EVs and gesicles were lysed with Trizol (ThermoFisher Scientific), and RNA was isolated according to the manufacturer's protocol. cDNA synthesis was performed with the M-MuLV reverse transcription kit (NEB) using the included oligo dT primer. The following qPCR primers were used to amplify full-length dTomato: F: TGTCCCGGGGAATGGTGAGCA, R: TCGCCCTCGCCCTCGATCTC; full-length Cas9: F: AGTAGCGGTGGGCCAAAGAAG, R: ACAGAGTTGGTGCCGATGTCC; dTomato and Cas9 3′ fragments: F: TCCCTTCGGCCCTCAATCCA, R: TGAACCCGGGAAGGTACCGA; and GAPDH: F: TGAGCTGAACGGGAAGCTCACTGGC, R: GCAATGCCAGCCCCAGCGTCAAAG. GAPDH-normalized mRNA ratios were statistically tested using a 2-sided Student's t-test and considered significant at p<0.05.
EV and gesicle delivery experiments
Recipient PC-3 cells were plated 24–36 h before EV or gesicle delivery at ~1.5×105 cells/mL in 48-well plates (0.4 mL medium). EV or gesicles were counted by NanoSight, and equal numbers of vesicles were added to each well. Between 6×109 and 2×1010 vesicles were added per well for dTomato delivery. Fluorescence of PC-3 cells after vesicle delivery was analysed on a BDLSRII flow cytometer. FCS files were analysed using FlowJo (TreeStar). Mean fluorescence intensity (MFI) of vesicle-receiving cells was normalized by the MFI of cells receiving only a medium change (i.e. autofluorescence) to facilitate comparisons across different points in a time course. Differences in normalized MFI were statistically evaluated using a 2-sided Student's t-test and considered significant at p<0.05. Where indicated, PC-3 cells were treated with cycloheximide (CHX; Sigma) dissolved in Dimethyl sulfoxide (DMSO) and used at a final concentration of 20 µg/mL. For visualization of dTomato delivery, PC-3 cells cultured in 48-well plates were imaged using an inverted fluorescent Leica DM-IL LED microscope with a Leica N plan 10×/0.25 PH1 objective lens, and images were captured with a high-resolution cooled Q-imaging CCD using an exposure time of 150 ms. Micrographs and overlays presented in Supplementary Fig. 4b were created using ImageJ software (NIH). The brightness and contrast of the brightfield and dTomato channel images were adjusted to facilitate visualization of delivered dTomato in the overlays. Identical adjustments were applied across each corresponding set of images.
Results
Active loading enhances cargo RNA incorporation into EVs
To help elucidate the general rules governing EV-mediated loading and delivery of RNA, we first sought to develop a technology for actively loading any RNA of interest into EVs. To this end, we designed a TAMEL platform, comprising an engineered EV-loading protein and a cargo RNA. The EV-loading protein is a fusion protein between an EV-enriched protein (EEP) and an RNA-binding domain (RBD). The cargo RNA displays the binding motif recognized by the RBD (a). To investigate the TAMEL strategy, we first selected the exosomal protein, Lamp2b (Citation27–Citation29), as a candidate EEP, and the MS2 bacteriophage coat protein dimer (a well-characterized RNA-binding protein (Citation21)) as the RBD. The MS2 dimer was fused to the EV-luminal C-terminus of Lamp2b, such that RNA bound to the MS2 dimer would be incorporated into the EV interior. An HA affinity tag was also added to the C-terminus of the MS2 dimer, resulting in the candidate EV-loading protein Lamp2b–MS2–HA.
Fig. 1. Evaluation of RNA loading into EVs via the TAMEL platform. (a) This cartoon summarizes the concept of facilitating active loading of cargo RNA into EVs via our TAMEL platform. A TAMEL EV-loading protein comprises an EV-enriched protein (EEP, blue) fused to an RNA-binding domain (RBD, green), which localizes to EVs. Actively loaded RNA (green) contains a motif that binds to the RBD, resulting in enhanced loading into EVs relative to passively loaded RNA (orange). (b) RNA cargo design impacts active loading. The “fold enrichment of cargo mRNA +/−MS2” is defined as the ratio of cargo RNA/GAPDH mRNA in EVs derived from cells expressing Lamp2b–MS2–HA divided by the same RNA ratio in EVs derived from cells expressing Lamp2b–HA. All experiments were performed in biological triplicates. (c) Cartoon illustrating the 3′ RNA fragment analysis technique. Cargo RNA is first reverse transcribed using an oligo dT primer, and amplicons corresponding to the RNA 5′ or 3′ ends (the latter is located ~500 bases upstream of the polyA site) are then quantified by qPCR using the primer pairs indicated. Note that the amplicon near the RNA 3′ end will be present in cDNA derived from both full-length RNA and 3′ RNA fragments. (d) Analysis of 3′ RNA fragment loading into EVs. Cargo RNA levels were quantified as depicted in panel c and normalized to GAPDH. Passive loading: cells transfected with Lamp2b–HA; active loading: cells transfected with Lamp2b–MS2–HA. (e) Full-length RNA and 3′ fragment RNA levels in EVs were quantified following incubation at 37°C; experiments were performed in technical duplicate with a biological replicate shown in Supplementary Fig. 2a. Error bars indicate 1 standard deviation, throughout. MVB, multivesicular body.
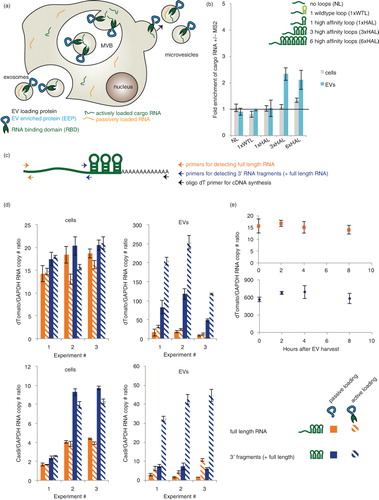
We next investigated whether the candidate-loading protein could facilitate active, specific loading of a cargo RNA into EVs. To this end, a series of HEK293FT cell lines was lentivirally transduced to stably express variants of a model cargo RNA encoding nuclear-targeted dTomato (NLS dTomato) (Table ). Each cargo RNA was engineered to include 0, 1, 3 or 6 MS2 loops in the 3′UTR, and both the wild-type MS2 loop (WTL, Kd~1 nM) and a mutated high-affinity loop (HAL, Kd~0.2 nM) (Citation21) were tested. Each line was transfected with plasmids encoding Lamp2b–MS2–HA, or Lamp2b–HA as a control for the effects of Lamp2b overexpression on EV production and loading. There was no difference in the number of EVs produced by HEK293FT cells transfected with Lamp2b–HA or Lamp2b–MS2–HA (Supplementary Fig. 2a). Cargo RNA levels were quantified in both cell lysates and EVs isolated from these cells. Transfection of Lamp2b–HA into the EV-producing cells did not significantly impact cargo RNA/GAPDH ratios in EVs or cells relative to non-transfected cells (tested in cell line MH97, expressing cargo RNA with 3 high-affinity MS2 loops) (Supplementary Fig. 2b).
To quantify the effect of active loading, we defined the “fold enrichment of cargo RNA +/−MS2” as the ratio of cargo RNA/GAPDH mRNA in EVs derived from cells expressing Lamp2b–MS2–HA divided by the same RNA ratio in EVs derived from cells expressing Lamp2b–HA. Cargo RNA bearing a single copy of either MS2 loop did not exhibit active loading (b). However, cargo RNA bearing 3 or 6 HAL motifs exhibited similar significant degrees of active loading. This indicated that active loading required a high-affinity and high avidity interaction between the EV-loading protein and the cargo RNA, with 3 loops being sufficient to mediate active loading. In most cases, expression of Lamp2b–MS2–HA did not result in any change in the level of the cargo RNA in cells, as expected. Thus, the observed enrichment of cargo RNA in EVs was due to active RNA loading rather than increased concentration-driven passive loading. Surprisingly, expression of Lamp2b–MS2–HA significantly (but modestly) also enhanced levels of 6× HAL cargo RNA in cells, potentially via Lamp2b–MS2–HA-binding induced stabilization of the cargo RNA. Based on these observations, 3× HAL cargo RNAs were used for all subsequent active loading experiments.
Small RNAs are actively loaded more efficiently than are long RNAs
Given the observation that EV contents are enriched in small RNA fragments (Citation1, Citation12), we next investigated whether RNA size also impacts the efficiency of active RNA loading, using the TAMEL system. We first hypothesized that if some cargo RNA were partially degraded in EV-producing cells, then some smaller cargo RNA fragments may still bear HAL motifs and exhibit active loading. To test this question, we used qPCR to compare the levels of full-length cargo RNA (~1.8 kb, as quantified in b) to the levels of 3′ cargo RNA domains (i.e. the terminal 500 bases including the 3× HAL motif, which is present in both 3′ RNA fragments of various lengths and full-length cargo RNA, as illustrated in c) in both cells and EVs (d, upper row). While most cargo RNAs in cells appeared to be full length, EVs were vastly enriched in 3′ RNA fragments compared to full-length cargo RNA. Such 3′ RNA fragments also exhibited significant active loading when EV-producing cells expressed Lamp2b–MS2–HA. Note that the 3′ RNA fragment pool actually represents a distribution of RNA sizes spanning 500 bases up to full cargo RNA length. To further investigate the impact of cargo RNA size, we conducted similar experiments using a longer cargo RNA encoding the Cas9 protein (~5.4 kb) (d, lower row). In this case of longer cargo RNA, some 3′ fragments were observed in EV-producing cells. However, EVs were enriched in 3′ cargo RNA fragments, compared to full-length cargo RNA, to an extent greater than can be explained by this moderate difference in concentration driving force. Active loading also resulted in a greater ratio of 3′ Cas9 fragments to full-length RNA than 3′ dTomato fragments to full-length RNA. To investigate whether cargo RNA fragments were generated in cells, prior to EV production, or via degradation of full-length cargo RNA in secreted EVs, we incubated EVs at 37°C and monitored levels of both full-length cargo RNA and 3′ fragments (e and Supplementary Fig. 2c). Levels of both RNA species held constant over 8 h of incubation, suggesting that fragmentation occurred prior to loading cargo RNA into EVs. Altogether, these observations indicate that (Citation1) smaller RNAs were loaded into EVs at much greater efficiency than were longer RNA, via either passive or active mechanisms, and (Citation2) active loading had a greater effect on the loading efficiency of longer cargo RNA compared to that of smaller cargo RNA.
Choice of EEP strongly impacts RNA-loading efficiency
We next investigated whether our choice of EEP (Lamp2b in the above experiments) impacts the degree to which the TAMEL mechanism confers active loading of cargo RNA. We hypothesized that EEP choice could impact loading efficiency if different EEPs are expressed in cells and loaded into EVs to different extents. To test this hypothesis, we investigated the use of alternative EEPs CD63, an exosome-enriched tetraspanin (Citation30, Citation31), and Hspa8, an EV-enriched cytosolic protein (Citation31). We also investigated a modified version of Lamp2b bearing an N-terminal glycosylation motif (GNSTM), which we previously showed to accumulate in EVs at higher levels than does unmodified Lamp2b (Citation28). MS2 fusions of GNSTM–Lamp2b and CD63 conferred greater active loading of full-length (~1.8 kb) cargo RNA than did Lamp2b, resulting in an average of ~4-fold and ~6-fold cargo RNA enrichment in EVs for GNSTM–Lamp2b and CD63, respectively (a). In contrast, Hspa8 fusions conferred no cargo RNA enrichment in EVs. None of these EEPs modulated cargo RNA levels in EV-producing cells. Transfection of EV-producing cells with the different EEPs did not significantly impact the number of EVs generated (Supplementary Fig. 2d).
Fig. 2. Impact of EEP choice on TAMEL-mediated active RNA loading into vesicles. (a) Effects of EEP choice on cargo RNA loading into EVs. Experiments were performed in biological triplicate. (b) Effects of EEP choice on cargo loading into gesicles. Experiments were performed in biological triplicate. (c) Protein abundance was quantified by densitometry analysis of anti-HA western blots (Supplementary Fig. 3), and each blot was internally normalized by the intensity for VSVG–MS2–HA in gesicles (maximal intensity case). This experiment was performed in biological duplicate. The y-axis is in log scale to enable visualization of all values. Error bars indicate 1 standard deviation, throughout.
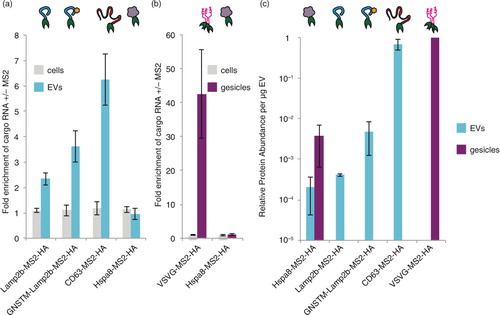
To further elucidate the impact of EEP choice, we next investigated whether the TAMEL approach could confer active loading of cargo RNA into gesicles, which are vesicles produced from cells expressing the VSVG (Citation25). Since Hspa8, but not Lamp2b or CD63, is enriched in gesicles (Citation25), both VSVG and Hspa8 were investigated as MS2 fusion EEPs in gesicles. Notably, VSVG–MS2–HA was a highly effective EEP, mediating an average of 42-fold enrichment of full-length cargo RNA (~1.8 kb) in gesicles (b). As was observed in EVs, however, Hspa8–MS2–HA did not mediate active loading of cargo RNA in gesicles. Also as observed in EVs, EEP choice did not impact the number of gesicles produced (Supplementary Fig. 2d). Why EEP choice so dramatically impacted loading efficiency in both EVs and gesicles was not yet clear.
We hypothesized that if the efficiency with which EEP–MS2 fusion proteins are incorporated into EVs limits the degree of active cargo RNA loading, then EEP protein abundance in EVs would correlate with the observed extents of active loading. Since the TAMEL approach specifically enables such direct comparisons, we quantified the extent to which each engineered EEP protein was incorporated into EVs or gesicles (c; primary western blot images and corresponding densitometry analyses are presented in Supplementary Fig. 3a–c). As predicted, the rate of engineered EEP incorporation into EVs correlated well with the extent to which these proteins conferred active cargo RNA loading, with CD63–MS2–HA and VSVG–MS2–HA being both most efficiently incorporated and most effective for loading RNA into EVs and gesicles, respectively. Altogether, these results indicate that active cargo RNA loading via the TAMEL approach is a robust strategy that is adaptable to multiple EEPs.
Fate of EV cargo RNA upon delivery to recipient cells
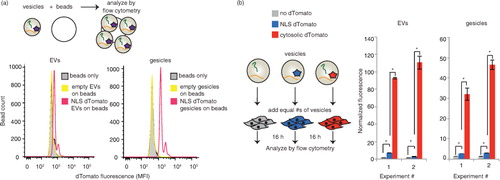
EVs are capable of delivering mRNA and protein cargo to recipient cells, yet as discussed above, the extent to which this cargo is released into the cytoplasm in recipient cells is generally less clear and requires further study. To this end, we next leveraged the TAMEL system as a novel experimental method for investigating this question by selectively enhancing the loading of cargo mRNA. Since such delivery phenomena may well be system-specific, we chose to investigate this question in the context of a clinically motivated model system – delivery of mRNA to prostate cancer cells (PC-3), which we previously observed to robustly internalize EVs from HEK293FT cells (Supplementary Fig. 4a). To help distinguish between delivery of cargo protein and delivery of cargo mRNA, we built a cargo RNA encoding a nuclear-targeted protein (NLS–dTomato) as well as a 3′ 3×HAL motif to facilitate active loading. We hypothesized that minimal NLS–dTomato protein from EV-producing cells would be loaded into EVs, such that this strategy should facilitate the observation of any cargo mRNA translated after delivery to recipient cells. To test whether vesicles from cells expressing NLS–dTomato–3× HAL contain dTomato protein, 5×109 EVs or gesicles were bound to latex beads and analysed by flow cytometry (a). Both EVs and gesicles exhibited some protein loading, with gesicles appearing to be more enriched in dTomato protein than were EVs. We next evaluated whether these vesicles could confer fluorescence upon recipient PC-3 cells (b and Supplementary Fig. 4b and c). As a control, we also evaluated vesicles from cells expressing dTomato (no NLS), since this modification was expected to enhance passive loading of protein (but not mRNA) into EVs. As expected, delivery of EVs containing cytoplasmically targeted dTomato conferred much greater fluorescence than did delivery of EVs containing NLS–dTomato. Thus, we concluded that while EV-mediated delivery of some NLS–dTomato protein could be possible, protein loading into EVs was indeed substantially reduced by engineered subcellular localization.
Fig. 3. NLS–dTomato protein delivery by actively or passively loaded vesicles. (a) Fluorescence of vesicles adsorbed to latex beads. 5×109 vesicles were adsorbed to latex beads for 2 h and analysed by flow cytometry. EVs, left; gesicles, right. (b) Comparison of nuclear-targeted (blue) and cytosolic (red) dTomato delivery by EVs (left) and gesicles (right). EVs from cells expressing no dTomato (grey) was a negative control. “Normalized fluorescence” is defined as the mean fluorescence of cells receiving vesicles divided by the mean fluorescence of cells receiving a medium change only. Duplicate wells of vesicle-receiving cells were analysed. Within each experiment, the number of vesicles added to each well was normalized (via vesicle counts determined NanoSight) such that each well received the same number of vesicles. *Significant difference was evaluated with a Student's t-test using a cut-off of p<0.05.
We next investigated the fate of NLS–dTomato protein and cargo RNA following EV-mediated delivery to recipient cells. First, we evaluated dTomato delivery by EVs in which cargo RNA was loaded either actively or passively by expression of CD63–MS2–HA or CD63–HA, respectively. To initially investigate whether cargo molecules were delivered to the cytoplasm of recipient cells, we quantified EV-mediated dTomato delivery after 4 h, changed medium in some groups and evaluated fluorescence again 12 h later (a and Supplementary Fig. 3a). We hypothesized that if mRNA and/or protein were delivered to the cytoplasm, then fluorescence would persist or increase due to evasion of lysosomal degradation and, potentially, new translation of delivered mRNA. Moreover, if mRNA escape were possible, then actively loaded EVs may confer a greater persistent signal than would passively loaded EVs. If recipient cells were continuously incubated with EVs across this interval, conferred fluorescence was relatively constant between 4 and 16 h post-delivery (a and Supplementary Fig. 5a), and in subsequent experiments this pattern extended to at least 24 h post-delivery (Supplementary Fig. 5b). Several observations suggested that cargo delivery to the cytoplasm was minimal. First, for cells in which medium was changed at 4 h post-delivery, conferred fluorescence diminished substantially in the following 12 h (a and Supplementary Fig. 5a). This loss is not readily attributable to cell division [PC-3 cell doubling time is ~38 h (Citation32)] or protein degradation in the cytoplasm, where dTomato half-life is expected to be at least 24 h (Citation33). Moreover, active cargo mRNA loading conferred no enhancement in persistent dTomato fluorescence in recipient cells. Thus, it is likely that sustained incubation of recipient cells with EVs represents a steady state between continuous uptake and degradation of EV contents, with this particular model system exhibiting no evidence of endosomal escape.
Fig. 4. Comparative analysis of dTomato delivery by actively or passively loaded vesicles. For panels a–c, the cartoons at left summarize the experimental designs, and in the panels at right, each data point represents the average of duplicate wells of cells treated with the same type of vesicle. Error bars indicate 1 standard deviation. “Normalized fluorescence” is defined as the mean fluorescence of cells receiving vesicles divided by the mean fluorescence of cells receiving a medium change only. (a) Time course of EV delivery to cells. Grey squares: CD63–HA EVs; orange circles: CD63–MS2–HA EVs. The solid arrow represents cells that received a medium change after 4 h of EV treatment. The dashed arrow indicates that cells did not receive a medium change. Statistically significant differences (p<0.05, not shown for clarity): CD63–MS2–HA +/− medium change. An independent repeat of this experiment is shown in Supplementary Fig. 3a. (b) Time course of gesicle delivery to cells. Purple squares: VSVG–HA gesicles; green circles: VSVG–MS2–HA gesicles. Solid and dashed arrows carry the same meaning as in panel (a). Statistically significant differences (p < 0.05, not shown for clarity): VSVG–HA versus VSVG–MS2–HA at 4 and 16 h (comparisons were made for each time point), VSVG–HA +/− medium change, and VSVG–MS2–HA +/− medium change. An independent repeat of this experiment is shown in Supplementary Fig. 3c. (c) Comparison of delivery by gesicles from cells transfected with VSVG–HA (purple), VSVG–MS2–HA (green) or a 50:50 mix of VSVG–HA and VSVG–MS2–HA (hybrid gesicles, magenta). (d) dTomato RNA levels (normalized to GAPDH) in VSVG–HA gesicles (purple), VSVG–MS2–HA gesicles (green) or hybrid gesicles (magenta). Error bars indicate 1 standard deviation of technical duplicate samples. *Significant difference was evaluated with a Student's t-test using a cut-off of p < 0.05.
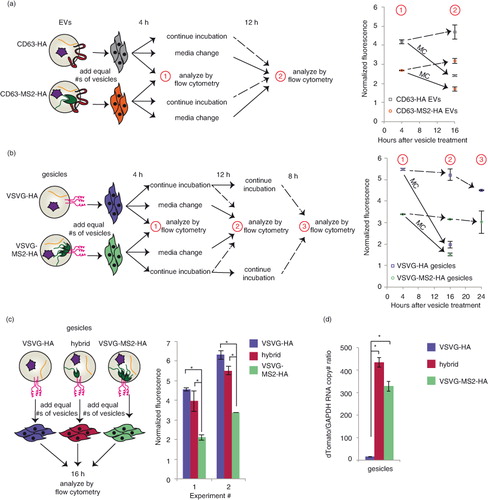
We next performed a parallel analysis to investigate the fate of cargo molecules when delivered by gesicles, which express the fusogenic protein VSVG. As with EVs, cargo mRNAs were loaded into gesicles by active (VSVG–MS2–HA) or passive (VSVG–HA) means, and a medium change strategy was applied to investigate whether dTomato-associated fluorescence would persist (b and Supplementary Fig. 5c). As was observed with EVs, gesicle-mediated dTomato fluorescence was relatively constant when cells were incubated continuously with vesicles over 24 h, and this fluorescence decreased substantially upon medium change. Surprisingly, PC-3 cells receiving passively loaded gesicles were consistently more fluorescent than actively loaded gesicles. This effect was independent of the interaction between the MS2 protein and the cargo RNA, since a similar difference was observed between VSVG–MS2–HA and VSVG–HA gesicles from cells expressing cargo RNA including no loops (Supplementary Fig. 5d). Thus, VSVG appeared to play a role in gesicle uptake, but not necessarily in endosomal escape. One likely explanation is that VSVG generally mediates uptake via binding the low-density lipoprotein (LDL) receptor, which is expressed in PC-3 cells (Citation34); this mechanism was recently identified to confer VSVG-mediated lentiviral delivery to many cells (Citation35). Since fusing VSVG to a second protein (e.g. VSVG–GFP) impairs the formation of VSVG–GFP trimers (but not VSVG–GFP/VSVG heterotrimers) (Citation36), we hypothesized that VSVG–MS2–HA may inefficiently form trimers and thus mediate reduced binding to the LDL receptor. Moreover, we hypothesized that VSVG–HA and VSVG–MS2–HA might be able to form heterotrimers, recovering the delivery effect mediated by VSVG–HA, and this was tested by generating “hybrid” gesicles from cells expressing both VSVG–HA and VSVG–MS2–HA (c). As anticipated, hybrid gesicles conferred dTomato delivery comparable to that conferred by VSVG–HA gesicles. Moreover, active loading of cargo RNA into hybrid gesicles was just as robust as was active loading into VSVG–MS2–HA gesicles (d). Thus, while the hybrid gesicle approach achieved both active mRNA loading (~40× enhanced cargo RNA loading) and enhanced VSVG-mediated uptake, no additional dTomato fluorescence was observed by actively loaded versus passively loaded gesicles. Altogether then, in this model system, endosomal escape appears to limit gesicle-mediated delivery as was observed for EV-mediated delivery.
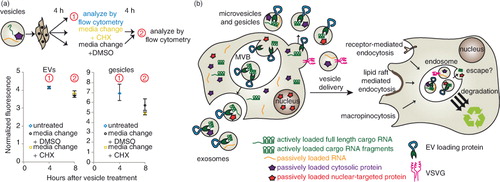
Given the substantial evidence suggesting that cargo mRNA was not translated in recipient cells, we performed one final investigation to rule out the possibility that cargo mRNA translation in recipient cells contributed to the steady-state fluorescence observed in recipient cells cultured continuously with EVs. To address this possibility, cells were treated with 20 µg/mL of the translation inhibitor CHX at 4 h after vesicle delivery was initiated, and fluorescence was reassessed after another 4 h had elapsed (a and Supplementary Fig. 5e). This time scale is relevant because significant accumulation of dTomato protein occurs within 4 h of inducing transcription (Citation37) and because CHX concentrations greater than 104 nM (~3 µg/mL) completely inhibit protein synthesis in mammalian cells within at least 2 h (Citation38). Overall, there was no significant difference in fluorescence between CHX-treated and control cells, indicating that translation of mRNA into dTomato protein in recipient cells was not significant. Altogether, our observations indicate that in this model system (delivery of cargo molecules via HEK293FT-derived EVs and gesicles to PC-3 cells), robust cargo delivery to the endosomal compartment may be achieved, but escape from the endosomal/lysosomal pathway is likely the limiting step in vesicle-mediated delivery (b). Moreover, this comparative analysis illustrates the utility of harnessing the TAMEL platform for active RNA loading to elucidate the process of EV-mediated intercellular transfer of biomolecules.
Fig. 5. Fate of cargo delivered by HEK293FT-derived EVs and gesicles to PC-3 recipient cells. (a) Evaluating the role of translation in EV-mediated conferral of fluorescence to recipient cells. PC-3 cells were treated with vesicles for 4 h, and then fresh medium containing cycloheximide (CHX, gold square) or DMSO (black circle) was added. EVs (left); gesicles (right). An independent repeat of this experiment is shown in Supplementary Fig. 3e. (b) Proposed conceptual summary of the conclusions drawn for this model system: cargo molecule delivery via HEK293FT-derived vesicles to PC-3 recipient cells.
Discussion
A central motivation for this investigation is the general need for approaches enabling modulation of RNA loading to elucidate the rules governing both loading and delivery of cargo RNA and proteins across different types of cells. To meet this need, we developed the TAMEL platform. Importantly, the TAMEL platform is both modular and generalizable. Many types of RNA may be engineered to harness this active loading approach, and an EV-loading protein may be engineered from a variety of EEPs. Choice of EEP strongly impacted the degree of active loading mediated by the TAMEL platform, and this variation is likely explained by variations in the abundance (in EVs) of each engineered EEP. This concentration-driven phenomenon may also apply to natural active loading mechanisms and may regulate the degree to which RNAs are actively loading into EVs.
Our results provide the first direct evidence that RNA loading into EVs is at least partially regulated by size alone. 3′ Cargo RNA fragments (≥0.5 kb) were heavily enriched in EVs relative to full-length cargo RNA (≥1.8 kb). An alternative hypothesis – that mRNA fragments are actively loaded into EVs by a native mechanism – could explain the elevated levels of fragments in EVs compared to EV-producing cells. However, such a hypothesis would not explain why the TAMEL platform-mediated active loading of 3′ cargo RNA fragments to such a greater extent than full-length cargo RNA, nor would it explain why the shorter cargo RNA (1.8 kb dTomato) was loaded into EVs more efficiently than was the longer cargo RNA (5.4 kb Cas9). Therefore, we propose that 3′ cargo RNA fragments bound to EV-loading proteins are more efficiently loaded into EVs than are large full-length RNAs bound to EV-loading proteins, such that loading efficiency decreases with cargo RNA size.
The combination of EV source and recipient cell type may well play an important role in determining the fate of EV-delivered biomolecules. Through this lens, our observation of rapid EV cargo degradation in our model system may help to interpret prior reports of RNA and protein delivery by HEK293 cell line-derived vesicles delivered to a variety of recipient cell types. In general, our findings are consistent with those of Kanada et al., which held that protein delivered by HEK293FT-derived EVs to HEK293FT cells localized to endosomes, and that mRNA delivered by HEK293FT-derived EVs to HEK293FT cells was not translated (Citation17). Likewise, Mizrak et al., demonstrated delivery, but not endosomal escape, of CD-UPRT protein and mRNA by HEK293T-derived EVs to HEI-193 schwannoma cells (Citation16). Collectively, these studies suggest that endosomal escape of cargo delivered via HEK293-derived EVs is limited, and that an important step in harnessing these and potentially other EVs as delivery vehicles will be to increase vesicle content escape from the endosome. By comparison, studies that have concluded that EVs mediate functional mRNA delivery utilized other EV sources: MDA–MB-231-derived EVs delivered RNA to MDA–MB-231, MCF-7 or T47D cells (Citation18), and TU2449-derived EVs and LLC2-derived EVs delivered RNA to murine MDSCs in vivo (Citation19). For these systems in which there is some evidence for functional RNA delivery, whether RNA loading into EVs is limiting remains unknown. However the TAMEL platform could be applied to answer this question and, if needed, to enhance active loading of cargo RNA into EVs.
We anticipate that the TAMEL platform will serve as a useful tool for the EV community. This modular approach should be applicable to EVs from any cell type, and for loading many RNA types of interest. As EVs move increasingly towards clinical applications, we would posit that characterizing the limiting steps in any new EV source/recipient cell system will be of increasing importance. This study also suggests that for applications in which cargo RNA delivery is desired, active RNA loading could substantially increase the number of cargo RNA molecules per EV. Thus, the TAMEL platform could both enable a better understanding of EV-mediated intercellular communication and facilitate the development of EV-based therapeutics.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Supplementary figure 1
Download PDF (12 MB)Supplementary figure 2
Download PDF (854.6 KB)Supplementary figure 3
Download PDF (1.6 MB)Supplementary figure 4
Download PDF (1,019.2 KB)Supplementary figure 5
Download PDF (19.1 MB)Acknowledgements
The authors acknowledge Charlene Wilke for her assistance with transmission electron microscopy. TEM was performed at the Northwestern University Biological Imaging Facility on a JEOL 3200 FETEM supported by the NU Office for Research and NCRR 1S10RR025092. The authors thank Dr. Arabela Grigorescu for assistance with NanoSight analysis. NanoSight analysis was performed at the Northwestern University Keck Biophysics Facility. Traditional sequencing services were performed at the Northwestern University Genomics Core Facility. BCA assays were performed at the Northwestern University High Throughput Analysis Lab, which is supported by the Chicago Biomedical Consortium and The Searle Funds at The Chicago Community Trust. Flow cytometry was performed at the Northwestern University Flow Cytometry Facility, which is supported by a Cancer Center Support Grant (NCI CA060553). The authors acknowledge support from a 3M Non-tenured Faculty Award, the Lynn Sage Breast Cancer Foundation and the Northwestern University Prostate Cancer Specialized Program of Research Excellence (SPORE) through NIH award P50 CA090386 (to JNL). This work was also supported by the National Science Foundation's Graduate Research Fellowship Program (NSF GRFP) award DGE-0824162 (to MEH).
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654–9.
- Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, etal. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006; 20: 847–56.
- Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, Harding CV, Melief CJ, etal. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996; 183: 1161–72.
- Kosaka N, Yoshioka Y, Hagiwara K, Tominaga N, Katsuda T, Ochiya T. Trash or treasure: extracellular microRNAs and cell-to-cell communication. Front Genet. 2013; 4: 173.
- Saha B, Momen-Heravi F, Kodys K, Szabo G. MicroRNA cargo of extracellular vesicles from alcohol-exposed monocytes signals naive monocytes to differentiate into M2 macrophages. J Biol Chem. 2016; 291: 149–59.
- van Balkom BW, de Jong OG, Smits M, Brummelman J, den Ouden K, de Bree PM, etal. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood. 2013; 121: 3997–4006. [PubMed Abstract] S1–15.
- van der Vos KE, Abels ER, Zhang X, Lai C, Carrizosa E, Oakley D, etal. Directly visualized glioblastoma-derived extracellular vesicles transfer RNA to microglia/macrophages in the brain. Neuro Oncol. 2016; 18: 58–69.
- Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, etal. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013; 4: 2980.
- Zhang J, Li S, Li L, Li M, Guo C, Yao J, etal. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics. 2015; 13: 17–24.
- Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, etal. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015; 527: 100–4.
- Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, etal. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10: 1470–6.
- Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J Biol Chem. 2010; 285: 17442–52.
- van Balkom BW, Eisele AS, Pegtel DM, Bervoets S, Verhaar MC. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J Extracell Vesicles. 2015; 4: 26760.
- Hagiwara K, Katsuda T, Gailhouste L, Kosaka N, Ochiya T. Commitment of annexin A2 in recruitment of microRNAs into extracellular vesicles. FEBS Lett. 2015; 589: 4071–8.
- Bolukbasi MF, Mizrak A, Ozdener GB, Madlener S, Strobel T, Erkan EP, etal. miR-1289 and “Zipcode”-like sequence enrich mRNAs in microvesicles. Mol Ther Nucleic Acids. 2012; 1: e10.
- Mizrak A, Bolukbasi MF, Ozdener GB, Brenner GJ, Madlener S, Erkan EP, etal. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol Ther. 2013; 21: 101–8.
- Kanada M, Bachmann MH, Hardy JW, Frimannson DO, Bronsart L, Wang A, etal. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc Natl Acad Sci USA. 2015; 112: E1433–42. [PubMed Abstract] [PubMed CentralFull Text].
- Zomer A, Maynard C, Verweij FJ, Kamermans A, Schafer R, Beerling E, etal. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015; 161: 1046–57.
- Ridder K, Sevko A, Heide J, Dams M, Rupp AK, Macas J, etal. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology. 2015; 4: e1008371.
- Peabody DS, Ely KR. Control of translational repression by protein-protein interactions. Nucleic Acids Res. 1992; 20: 1649–55.
- Keryer-Bibens C, Barreau C, Osborne HB. Tethering of proteins to RNAs by bacteriophage proteins. Biol Cell. 2008; 100: 125–38.
- Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, etal. Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol. 2003; 13: 161–7.
- Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008; 28: 7025–30.
- Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984; 39: 499–509.
- Mangeot PE, Dollet S, Girard M, Ciancia C, Joly S, Peschanski M, etal. Protein transfer into human cells by VSV-G-induced nanovesicles. Mol Ther. 2011; 19: 1656–66.
- Thery C , Amigorena S , Raposo G , Clayton A . Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006. Chapter 3:Unit 3:22.
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011; 29: 341–5.
- Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. 2015; 290: 8166–72.
- Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, etal. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014; 35: 2383–90.
- Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998; 273: 20121–7.
- Mathivanan S, Simpson RJ. ExoCarta: a compendium of exosomal proteins and RNA. Proteomics. 2009; 9: 4997–5000.
- Sheng X, Li Z, Wang DL, Li WB, Luo Z, Chen KH, etal. Isolation and enrichment of PC-3 prostate cancer stem-like cells using MACS and serum-free medium. Oncol Lett. 2013; 5: 787–92. [PubMed Abstract] [PubMed CentralFull Text].
- Snapp EL. Fluorescent proteins: a cell biologist's user guide. Trends Cell Biol. 2009; 19: 649–55.
- Sekine Y, Koike H, Nakano T, Nakajima K, Takahashi S, Suzuki K. Remnant lipoproteins induced proliferation of human prostate cancer cell, PC-3 but not LNCaP, via low density lipoprotein receptor. Canc Epidemiol. 2009; 33: 16–23.
- Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA. 2013; 110: 7306–11.
- Dalton KP, Rose JK. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology. 2001; 279: 414–21.
- Clontech_Laboratories. [PubMed CentralFull Text].
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, etal. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 2010; 6: 209–17.