
Abstract
Background
Extracellular vesicles (EVs) are attractive candidate drug delivery systems due to their ability to functionally transport biological cargo to recipient cells. However, the apparent lack of target cell specificity of exogenously administered EVs limits their therapeutic applicability. In this study, we propose a novel method to equip EVs with targeting properties, in order to improve their interaction with tumour cells.
Methods
EV producing cells were transfected with vectors encoding for anti-epidermal growth factor receptor (EGFR) nanobodies, which served as targeting ligands for tumour cells, fused to glycosylphosphatidylinositol (GPI) anchor signal peptides derived from decay-accelerating factor (DAF). EVs were isolated using ultrafiltration/size-exclusion liquid chromatography and characterized using western blotting, Nanoparticle Tracking Analysis, and electron microscopy. EV–tumour cell interactions were analyzed under static conditions using flow cytometry and under flow conditions using a live-cell fluorescence microscopy-coupled perfusion system.
Results
V analysis showed that GPI-linked nanobodies were successfully displayed on EV surfaces and were highly enriched in EVs compared with parent cells. Display of GPI-linked nanobodies on EVs did not alter general EV characteristics (i.e. morphology, size distribution and protein marker expression), but greatly improved EV binding to tumour cells dependent on EGFR density under static conditions. Moreover, nanobody-displaying EVs showed a significantly improved cell association to EGFR-expressing tumour cells under flow conditions.
Conclusions
We show that nanobodies can be anchored on the surface of EVs via GPI, which alters their cell targeting behaviour. Furthermore, this study highlights GPI-anchoring as a new tool in the EV toolbox, which may be applied for EV display of a variety of proteins, such as antibodies, reporter proteins and signaling molecules.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Extracellular vesicles (EVs), including exosomes and microvesicles, are submicron lipid bilayer-surrounded vesicles containing proteins and nucleic acids, such as miRNAs and mRNAs. They are released from many, if not all, cell types in the body and are believed to play a role in intercellular communication (Citation1, Citation2). The ability of EVs to selectively convey proteins, lipids and nucleic acids to cells has created excitement in the field of drug delivery, where efficient and targeted delivery of biomolecules is desired (Citation3–Citation8). Multiple studies have shown that the use of EVs for therapeutic purposes is feasible, and EVs have even already been applied in phase I clinical trials (reviewed in (Citation9, Citation10)). However, EVs may also possess unfavourable characteristics that could limit their applicability as drug delivery systems. Their natural bioactive payloads may counteract the desired therapeutic effects, and a lack of targeting specificity may result in uptake by non-targeted, healthy cells. Multiple reports have shown that EVs can be engineered to include specific cargo or express targeting ligands to improve their drug delivery potential (Citation11–Citation17). Previously described targeting strategies have been mainly based on the fusion of targeting ligands with EV membrane proteins, such as Lamp2b (Citation11, Citation15) (Citation16). Albeit effective in these cases, such strategies may have drawbacks. For example, the function of EV membrane proteins (e.g. fusion with cellular membranes or immune regulation (Citation4, Citation18)) may be compromised upon fusion with targeting ligands, and some Lamp2b-fused targeting ligands have been described to undergo premature degradation instead of functional display on EVs (Citation19). To avoid such issues, multiple groups have explored strategies to functionalize EV surfaces after EV secretion, circumventing the need to modify EV producer cells (Citation17, Citation20) (Citation21). For example, Smyth and co-workers grafted alkyne moieties onto isolated EVs to equip these vesicles with fluorescent probes using click chemistry (Citation20). Unfortunately, such modifications may also compromise the functionality of crucial EV components for EV cell interactions and cargo delivery. In this work, we therefore propose an alternative strategy to efficiently decorate EVs with targeting proteins.
EVs have been described to be enriched in lipid raft-associated lipids, including sphingolipids and cholesterol, and proteins, including glycosylphosphatidylinositol (GPI)-anchored proteins (Citation2, Citation22) (Citation23). In fact, the GPI-anchored protein decay-accelerating factor (DAF, also known as CD55) has been described to be selectively secreted in EVs during reticulocyte maturation (Citation24). We hypothesized that this phenomenon could be exploited for the expression of targeting moieties onto EVs. We therefore fused a human DAF-derived GPI-anchor signal peptide to nanobodies, which served as model-targeting ligands. This GPI-anchor peptide has previously been employed to tether a variety of proteins to cell membranes (Citation25–Citation27). Nanobodies are small (15 kD) single variable domains derived from heavy-chain antibodies from Camelidae species. They can be used as versatile targeting tools with binding capacity similar to antibodies. Nanobodies offer several advantages compared with their full-length counterparts, such as straightforward selection and recombinant production, and high chemical and thermal stability (Citation28). In this work, nanobodies were used to target the epidermal growth factor receptor (EGFR), a well-studied oncogene against which a range of clinically approved inhibitors is directed for the treatment of solid tumours (Citation29, Citation30). Here, we investigated whether linkage of nanobodies to GPI-anchors is effective for the display of these proteins on EVs, and how his display influences EV characteristics and in vitro tumour targeting behaviour. Furthermore, we studied the interactions of these EVs with tumour cells under flow conditions using a live-cell imaging perfusion setup.
Materials and methods
Materials
MicroBCA Protein Assay Kit and CellTracker Deep Red dye were obtained from Thermo Fisher Scientific (Waltham, USA). Sepharose CL-4B was ordered from Sigma-Aldrich (Steinheim, Germany). pET28a-EGa1 and pAX51-R2 vectors encoding EGa1 (PDB ID: 4KRN) and R2 (PDB ID: 1QD0) Myc-tagged nanobodies, respectively, were kindly provided by Dr. S. Oliveira (Department of Biology, Utrecht University, Utrecht, The Netherlands).
Molecular cloning
EGa1 and R2 Myc-tagged nanobody sequences were PCR amplified from pET28a-EGa1 and pAX51-R2 vectors with primers designed to flank the nanobody sequences with Sfi and SalI restriction sites. Obtained inserts were Sfi/SalI digested and inserted into a pLNCX vector containing an N-terminal HA-tag, Sfi and SalI cloning sites, and a C-terminal GGGGS2 linker sequence followed by 37 amino acids of human DAF under the control of a CMV promoter (Citation25). The resulting vectors (named pLNCX-DAF-R2 and pLNCX-DAF-EGa1) were sequenced using a BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) according to the manufacturer's instructions to confirm in-frame insertion of the nanobody sequences.
Cell culture and generation of stable cell lines
All cells used in this study were maintained at 37°C and 5% CO2 and were tested negative for mycoplasma. Neuro2A cells were cultured in Roswell Park Memorial Institute (RPMI, Gibco) 1640 medium supplemented with 10% foetal bovine serum (FBS) and 100 U/mL penicillin and 100 U/mL streptomycin. A431 and HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM, Gibco) supplemented with 10% FBS and 100 U/mL penicillin and 100 U/mL streptomycin. To generate stable nanobody-DAF expressing cell lines, Neuro2A cells were transfected with pLNCX-DAF-R2 or pLNCX-DAF-EGa1 using TransIT 2020 transfection reagent (Mirius Bio, USA) according to the manufacturer's instructions and selected for at least 2 weeks in medium containing 500 µg/mL G418 (Geneticin, Thermo Fisher Scientific) until cells regained normal growth and morphology. Cells were subsequently maintained in medium containing 250 µg/mL G418.
EV isolation
For EV production, Neuro2A cells were seeded in T175 flasks and cultured for 24 h in normal culture medium, after which medium was replaced by Opti-MEM Reduced Serum medium supplemented with GlutaMAX (Gibco, Thermo Fisher Scientific) and 100 U/mL penicillin, 100 U/mL streptomycin and 250 µg/mL G418 (for transfected cells). After 48 h, when cells reached 90–95% confluency, EVs were isolated using a recently described ultrafiltration/size-exclusion liquid chromatography (UF-LC) method (Citation31). In brief, conditioned medium was centrifuged for 10 min at 300×g and 2,000×g at 4°C to remove cells and debris, respectively. Medium was vacuum filtered through Steritop 0.22 µm filters (Merck Millipore) to remove large vesicles and debris, and concentrated to ≤4 mL using Amicon Ultra-15 Centrifugal Filter Units with a 100 kD MWCO (Merck Millipore) at 4,000×g and 4°C. Concentrated medium samples were loaded onto a HiPrep 16/60 Sephacryl S-400 HR gel filtration column (GE Healthcare Life Sciences), which was equilibrated with phosphate-buffered saline (PBS) and connected to an ÄKTA pure or ÄKTA start chromatography system (GE Healthcare, both maintained at 4°C). EVs were separated from non-vesicular material using PBS as eluent. EV-containing fractions (determined by UV absorbance at 280 nm) were pooled and concentrated to <500 µL samples using 100 kD MWCO Amicon Ultra-15 Centrifugal Filter Units. EV protein yields were determined using a MicroBCA Protein Assay according to manufacturer's instructions.
Western blot analysis
For analysis of protein content of cells, cells in culture flasks were washed once with PBS, trypsinized and lysed in RIPA buffer (Alfa Aesar) supplemented with Protease Inhibitor Cocktail (Sigma-Aldrich). Cell lysates were centrifuged at 20,000×g for 10 min at 4°C to remove insoluble material, and protein concentration was determined using a MicroBCA Protein Assay. EVs in PBS and cell lysates were mixed with sample buffer containing dithiothreitol, heated to 95°C for 10 min and subjected to electrophoresis on 4–12% Bis-Tris polyacrylamide gels (Thermo Scientific). Proteins were blotted on Immobilon-FL polyvinylidene difluoride membranes (Millipore), after which membranes were blocked with 50% v/v Odyssey Blocking Buffer (LI-COR Biosciences) in Tris-buffered saline (TBS). Subsequently, membranes were probed overnight at 4°C with goat anti-HA (1:5,000, A00168-100, Genscript), mouse anti-ALIX (1:1,000, clone 3A9, Abcam), rabbit anti-TSG101 (1:1,000, ab30871, Abcam), mouse anti-β-actin (1:1,000, clone 8H10D10, Cell Signaling Technology), rabbit anti-CD9 (1:2,500, clone EPR2949, Abcam) or rabbit anti-EGFR (1:1,000, clone D38B1, Cell Signaling Technology) antibodies in 50% v/v Odyssey Blocking Buffer in TBS with 0.1% Tween20 (TBS-T). Secondary antibodies included IRDye 800CW donkey anti-goat, IRDye 800CW donkey anti-mouse (LI-COR Biosciences), Alexa Fluor 680 goat anti-mouse or Alexa Fluor 680 goat anti-rabbit (Thermo Fisher Scientific) and were applied at a 1:7,500 dilution. Protein bands were visualized on an Odyssey Infrared Imager (LI-COR Biosciences, Leusden, The Netherlands) at 700 and 800 nm.
Nanoparticle tracking analysis
EV size distribution and concentration was measured using a NanoSight NS500 system equipped with an LM14 405 nm violet laser unit (Malvern Instruments, Worcestershire, UK). Concentrated EV samples were diluted with PBS (confirmed to be particle-free when analysed with the same settings) to appropriate dilutions for analysis (generally 1:1,000–1:5,000) and visualized at camera level 13 under control of a script, which included acquisition of 5 movies of 30 s at a fixed temperature of 22°C. Analysis was performed with NTA 3.1 software. Detection threshold was set at 9 and other settings were kept at default.
Transmission electron microscopy
EVs in PBS were adsorbed to carbon-coated formvar grids for 15 min at room temperature. Unbound EVs were removed by washing with PBS and grids were blocked with 1% BSA in PBS (PBSA) for 10 min. HA epitopes on the grids were immunolabelled with goat anti-HA antibody (1:200, A00168-100, Genscript) in PBSA for 30 min, followed by 10 min incubations with rabbit anti-goat IgG (1:250, RAG/IgG(Fc)/7S, Nordic-MUbio) and 10 nm Protein A gold (CMC, Utrecht, The Netherlands) in PBSA. Grids were thoroughly washed with PBS between incubations. Finally, grids were fixated in 1% glutaraldehyde in PBS for 10 min, counterstained with uranyl-oxalate and embedded in methyl cellulose uranyl-acetate (Citation32). Imaging was performed using a Tecnai T12 electron microscope (FEI, Eindhoven, The Netherlands).
EV labelling and purification
For functional assays, EVs were labelled with CellTracker Deep Red immediately after isolation. CellTracker Deep Red dye was dissolved at 2 mM concentration in dimethyl sulfoxide (DMSO) and mixed with fixed EV amounts (based on protein concentration) at a final dye concentration of 16 µM. EV/dye mixtures were incubated for 1 h at 37°C and stored at 4°C until purification (maximally 16 h). For removal of unincorporated label, Sepharose CL-4B (Sigma-Aldrich) was packed in a XK-16/20 column (GE Healthcare) according to the manufacturer's instructions. Column was connected to a refrigerated ÄKTA pure chromatography system, equilibrated with PBS, and EV/dye mixtures were injected. Pooled EV fractions were concentrated to 100–200 µL using Vivaspin ultrafiltration tubes with 100 kD cut-off (Sartorius, UK). To determine whether labelling efficiency differed among EV samples, protein concentrations were measured using a MicroBCA Protein Assay, and fluorescence of 60 µL samples in a black 96-well plate was determined using a SpectraMax M2e microplate reader (Molecular Devices) at 630 nm excitation and 650 nm emission. Relative labelling efficiency was defined as relative fluorescence intensity/µg EV protein, and deviations of <10% between samples were considered acceptable before proceeding to cell association studies.
Cell binding assays
Neuro2A, HeLa or A431 cells were trypsinized and resuspended in ice-cold culture medium. Cell suspensions were transferred to round-bottom 96-well plates at a concentration of 40,000 cells/well on ice. CellTracker Deep Red-labelled EVs were added at a concentration of 5 µg/mL in triplicates, and cells were incubated for 1 h at 4°C to allow EV binding. Plates were centrifuged at 500×g for 5 min, medium was removed and cells were resuspended in ice-cold wash buffer (0.3% BSA in PBS). This process was repeated twice for a total of 3 washes, and finally, cells were resuspended in 0.2% formaldehyde in PBS. Mean fluorescence intensity (MFI) values were measured using a FACSCanto II flow cytometer (BD Biosciences, USA) and normalized to untreated cells.
Cell uptake assays
Neuro2A or A431 cells were cultured in normal culture medium in flat-bottom 96-well plates until a confluency of 80–90% was reached. CellTracker Deep Red-labelled EVs were added at a concentration of 5 µg/mL in triplicates and incubated for 1, 3 or 6 h at 37°C. Medium was removed, and cells were washed once with PBS, trypsinized and transferred to round-bottom 96-well plates in normal culture medium. Cells were pelleted for 5 min at 500×g and resuspended in wash buffer. Subsequently, cells were washed once with acid wash buffer (0.5 M NaCl, 0.2 M acetic acid, pH 3) to remove cell-bound EVs and once more with wash buffer . Finally, cells were resuspended in 0.2% formaldehyde in PBS, and MFI values were determined by flow cytometry.
Perfusion experiments
Perfusion experiments were performed using perspex perfusion chambers containing a sample inlet, sample outlet connected to a syringe pump and a connector for a vacuum pump (see Supplementary Fig. 1). A silicone sheet with a thickness of 0.125 mm was placed on top of the perspex frame, thereby forming a flow channel of 2×30 mm between sample in- and outlets. A431 cells cultured on glass coverslips (24×50 mm) were placed over the silicone sheet, and vacuum was applied to the chamber to seal the flow channel. Cell-covered perfusion surfaces were prepared by placing sterilized glass coverslips in 4-well slide tray plates. Coverslips were covered with 0.9 mL of 1% gelatin for 20 min at 37°C. Subsequently, 1.8 mL of 0.5% glutaraldehyde was added and plates were incubated for 20 min at room temperature. Liquid was replaced with 1.8 mL of 1 M glycine and plates were incubated for 20 min at room temperature. Coverslips were washed with PBS, and A431 cells were seeded in normal culture medium. When cells reached a confluency of 80–90%, medium was removed and cells were stained with 4',6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich) in PBS for 5 min at room temperature. Coverslips were washed once with plain DMEM and assembled on the perfusion chamber which was pre-equilibrated with DMEM, preventing air from entering the flow channel. Perfusion chamber was mounted on an Axio Observer fluorescence microscope (Carl Zeiss, Oberkochen, Germany), and cells were visualized using differential interference contrast (DIC) and DAPI channels with a 40× objective. A syringe was connected to the sample outlet of the perfusion chamber, and liquid was slowly withdrawn using a Pump 22 Multiple Syringe Pump (Harvard Apparatus, USA) at 25 µL/min, corresponding with a shear rate of 82.5 s−1. When flow rate stabilized, sample inlet was connected to a 2 µg/mL suspension of CellTracker Deep Red-labelled EVs in DMEM. EV perfusion was performed for 40 min, while pictures in DAPI, DIC and Cy5 fluorescence channels were taken with 10 s intervals. Cells were washed by perfusion with plain DMEM and fixated with 2% paraformaldehyde in PBS (PFA). Coverslips were removed and stored overnight in 1% PFA at 4°C. For semi-quantitative analysis, fixated coverslips were washed with PBS and mounted on microscopy slides using Vectashield HardSet Antifade mounting medium with DAPI (Vector Laboratories Inc.). Cell-associated EVs were imaged with a fluorescence microscope. Thirty to sixty images were randomly acquired across the entire area of the flow channel while focusing on the DAPI channel to avoid selection bias. The number of fluorescent EVs in each picture was analysed by Zen 2 pro analysis software (Carl Zeiss) and normalized to the number of cells (DAPI-stained nuclei) in each picture.
Statistical analysis
When applicable, statistical analysis was performed using IBM SPSS Statistics, version 21. Multiple-group testing was performed using one-way ANOVA with Tukey post-hoc tests and comparisons between 2 groups were made using independent samples t tests. Differences with p values <0.05 were considered statistically significant.
Results
Characterization of EVs derived from DAF-nanobody expressing cells
The EGa1 nanobody is a high-affinity ligand for EGFR, which competitively inhibits binding of the natural ligand epidermal growth factor and sterically prevents receptor activation (Citation33, Citation34). In contrast, the R2 nanobody, which was raised against the azo-dye Reactive Red (RR6) (Citation35), was used as a control nanobody (Citation36, Citation37). It was hypothesized that nanobodies could be displayed on EVs via fusion to a C-terminal GPI signal peptide derived from human DAF. When expressed in cells, the DAF peptide is cleaved off by GPI transamidase enzymes, thereby driving nanobody attachment to GPI anchors (Citation27). This way, nanobodies would localize to GPI-rich lipid rafts in cell membranes (Citation25, Citation38) (Citation39) and possibly in EV membranes. To test this hypothesis, R2 or EGa1 sequences were cloned into pLNCX vectors designed to drive the expression of HA-tagged proteins fused to DAF peptides (schematically shown in ). The resulting constructs (pLNCX-DAF-R2 and pLNCX-DAF-EGa1) were stably transfected into Neuro2A cells to create DAF-R2 and DAF-EGa1 cells, respectively. EVs were isolated from these cells using a previously described “ultrafiltration followed by liquid chromatography” method (Citation31). EV yields (as determined by protein quantification) typically ranged between 0.4 and 0.9 µg per mL of conditioned medium from Neuro2A and DAF-R2 cells, but were slightly lower for DAF-EGa1 cells (0.3–0.5 µg/mL of conditioned medium). Expression of GPI-anchored HA-tagged nanobodies in cells and EVs was analysed by western blotting (a and Supplementary Fig. 2). DAF-R2 and DAF-EGa1 proteins were hardly detectable in cell lysates from their respective cell lines. Remarkably, both proteins were found to be highly enriched in EVs (termed EV-DAF-R2 and EV-DAF-EGa1) compared with their parent cells. Both showed bands of approximately 27 kD, which was slightly bigger than their calculated molecular weight (25.3 kD for DAF-R2 and 25.7 kD for DAF-EGa1), which may be explained by the successful attachment of GPI to the proteins. Commonly used EV markers ALIX, TSG101 and CD9 were also clearly detectable in all EV samples, and ALIX and CD9 were highly enriched compared with parent cells, in accordance with previous reports (Citation6, Citation40) (Citation41). To investigate whether the expression of GPI-anchored nanobodies altered EV size, EVs were analysed by Nanoparticle Tracking Analysis (NTA, b). For both unmodified and DAF-nanobody EVs, a typical Neuro2A-derived EV size distribution was observed (Citation31, Citation42), with a mode size of approximately 100 nm and a small population of EVs sizing around 150–250 nm (possibly suggesting that the latter mainly originate from the plasma membrane (Citation1)). Of note, particle concentrations correlated well with protein concentrations among different EVs (typically 1 µg of EVs corresponded with 5–10×108 particles), indicating a similar protein load per particle. To assess the orientation of the nanobodies in the EV membranes and to assess whether expression of GPI-anchored nanobodies affected EV morphology, EVs were analysed by whole-mount transmission electron microscopy (TEM) after immunogold labelling of HA tags (c). Control EVs showed a “cup-shaped” morphology and were mostly unstained by anti-HA antibodies. EV-DAF-R2 and EV-DAF-EGa1 appeared similar to controls when comparing morphology and electron density, but a substantial amount of EV membranes stained positive for HA. Interestingly, some EVs showed patches of clustered gold particles on their membrane, possibly indicating the concentration of multiple GPI-anchored nanobodies in lipid rafts. Based on immunogold-TEM analysis, it was estimated that at least 15–25% of EVs displayed at least 1 nanobody, regardless of nanobody sequence. Taken together, these data show that GPI-anchors can be used to display nanobodies on EV surfaces, without affecting other EV characteristics (i.e. protein composition, size and morphology).
Fig. 1. Schematic presentation of nanobody-DAF fusion proteins. Nanobody-DAF protein expression was driven by a CMV promoter in pLNCX vectors. Recombinant proteins comprise an Igκ leader sequence (LS), N-terminal HA-tag (HA), nanobody sequence, Myc tag (Myc), GGGGS2 linker and a C-terminal GPI-anchor signal peptide (DAF) (a). Nanobodies fused to GPI-anchors were hypothesized to localize to lipid rafts in cellular membranes upon expression (b).
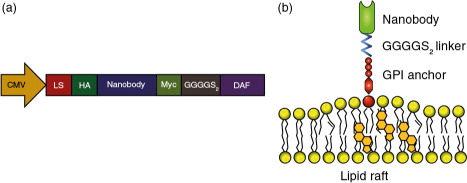
Fig. 2. GPI-anchored nanobodies are enriched in EVs compared with parent cells and are displayed on EV surfaces without affecting EV characteristics. (a) Western blot analysis of Neuro2A cells after stable transfection with pLNCX-DAF-EGa1 (DAF-EGa1) and pLNCX-DAF-R2 (DAF-R2) vectors compared with untransfected controls and EVs secreted by these cell lines. HA-tag was used to detect nanobody expression, and ALIX, TSG101 and CD9 were used as EV markers. Equal amounts of protein were analysed. Actin was included as a loading control. (b) Size distribution of normal EVs and DAF-nanobody EVs as determined by Nanoparticle Tracking Analysis. Data are displayed as mean±SD of 5 measurements. (c) Transmission electron microscopy images of EVs and DAF-nanobody EVs after immunogold labelling with anti-HA antibodies (arrowheads indicate membrane-associated gold). Scale bars represent 100 nm.
Cell association of targeted EVs
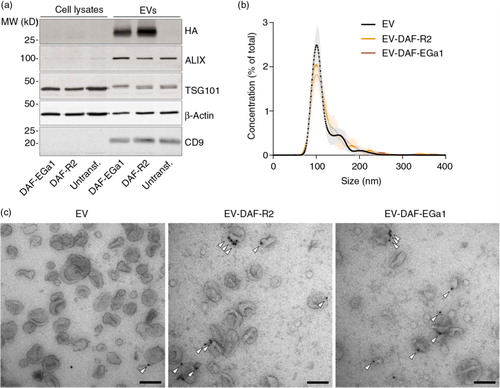
To investigate whether GPI-anchored nanobodies could influence the targeting behaviour of EVs, a cell association assay was performed. For this assay, EVs were stained with the far-red fluorescent dye CellTracker Deep Red. Importantly, the labelling efficiency (defined as fluorescence intensity per µg of EVs) was determined for each batch of EVs prior to experiments. Higher EV protein concentration during labelling resulted in higher labelling efficiency, while increased EV storage time prior to labelling tended to decrease labelling efficiency (Supplementary Fig. 3). To study cell association of nanobody-decorated EVs, labelled EVs were incubated with Neuro2A, HeLa or A431 cells for 1 h at 4°C to allow binding, but inhibit EV uptake by these cells (a) (Citation14, Citation18) (Citation43). These cell types vary in EGFR expression level (Citation36, Citation44), with undetectable expression in Neuro2A and clear overexpression in A431 (western blot in a). As shown in a, EVs and EV-DAF-R2 associated with all 3 cell types with similar efficiency. In contrast, EV-DAF-EGa1 showed more than 10-fold higher association with A431 cells compared with controls. In addition, a small increase in binding to HeLa cells was observed, while binding to Neuro2A cells was similar compared with control EVs and EV-DAF-R2. Together, these data demonstrate that the GPI-anchored nanobodies are functional and facilitate EV binding to tumour cells in an EGFR-dependent manner. To investigate whether this altered binding behaviour of EV-DAF-EGa1 also translated in altered uptake kinetics, uptake assays were performed in Neuro2A and A431 cells. EVs were incubated with cells for 1, 3 or 6 h at 37°C and cells were acid washed prior to flow cytometry analysis to remove surface-bound EVs. Surprisingly, uptake kinetics were similar between EV-DAF-EGa1, control EVs and EV-DAF-R2 and did not differ between EGFR-negative (Neuro2A) and EGFR-positive (A431) cells (b). Moreover, these data suggest that, under these conditions, binding of EVs to cells does not result in the same degree of cellular uptake.
Fig. 3. Display of GPI-anchored anti-EGFR nanobodies on EVs increases EV binding to EGFR-expressing tumour cells, but does not increase uptake by these cells. (a) Binding of CellTracker Deep Red-labelled control EVs and EVs with GPI-anchored nanobodies to Neuro2A, HeLa and A431 cells for 1 h at 4°C, as quantified by flow cytometry (left panel). EGFR expression in these cells was analysed by western blot (right panel). (b) Uptake of CellTracker Deep Red-labelled control EVs and EVs with GPI-anchored nanobodies by Neuro2A and A431 cells at 37°C for 1, 3 or 6 h, as determined by flow cytometry. All data are displayed as mean±SD and are representative of 3 independent experiments. ns=not significant and *** indicates p<0.001 as determined by one-way ANOVA with Tukey post-hoc test.
Cell association under flow conditions
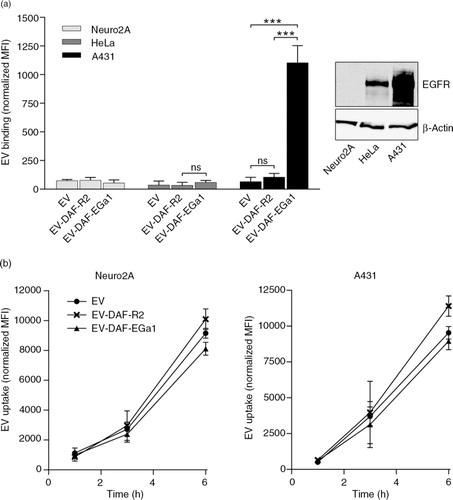
We next reasoned that cell-specific binding of EVs under flow conditions could be of greater physiological relevance than binding under static conditions, given that EV-carrying liquids (e.g. blood and interstitial fluid) are constantly in motion. This is also the case in solid tumour tissue, given that high interstitial fluid pressure drives macromolecules and nanoparticles to the tumour periphery (Citation45–Citation47). Efficient capture of EVs by tumour cells or tissues could greatly improve EV retention in such tissues and thereby facilitate targeted delivery of encapsulated cargo. To investigate whether the display of nanobodies on EVs could improve cell interaction under flow conditions, live-cell perfusion experiments were performed. A431 cells were grown on glass coverslips, which were subsequently mounted onto perspex perfusion chambers (see also Supplementary Fig. 1). This setup allowed for controlled perfusion of EV-containing medium over the cells in a 2×30 mm flow channel and real-time monitoring of EV cell association using a fluorescence microscope. For perfusion experiments, EVs were labelled with CellTracker Deep Red fluorescent dye and perfused over cells at a rate of 25 µL/min for 40 min at room temperature. It was observed that control EVs and EV-DAF-R2 were not efficiently captured by the cells over the course of the experiment, while EV-DAF-EGa1 rapidly associated with cell surfaces (a). To verify that this process occurred throughout the entire perfused area and not just in the recorded fields of view, cells were washed after perfusion, fixated and mounted on microscopy slides. EV cell association was analysed by acquiring fluorescence microscopy pictures at low magnification over the entire area of the perfusion channel (b). Again, it was evident that cell association was higher for EV-DAF-EGa1 than for control EVs and EV-DAF-R2, regardless of position in the channel. Quantification of the number of retained EVs in flow channels revealed that under these flow conditions, cell association of EV-DAF-EGa1 was approximately 2-fold higher than the cell association of EVs and EV-DAF-R2 (c).
Fig. 4. Display of GPI-anchored nanobodies on EVs increases association with tumour cells under flow conditions. (a) Fluorescence microscopy pictures of DAPI (blue)-stained A431 cells perfused for 40 min with CellTracker Deep Red-labelled EVs (red) with and without GPI-anchored nanobodies. DIC overlays are shown in grey as a reference for cell boundaries. EVs were perfused over cells at a rate of 25 µL/min. After perfusion, cells in flow channels were washed, fixated and mounted on microscopy slides. Fluorescence microscopy pictures were randomly taken at low magnification across the entire area of the flow channel (35–60 pictures per channel). Representative CellTracker Deep Red/DAPI overlay pictures are shown in (b). The number of EVs (n EV) and the cell-covered area (DAPI area) in each picture were quantified with Zen 2 software and displayed in (c). Results are shown as mean±SD and *** indicates p<0.001 as determined by one-way ANOVA with Tukey post-hoc test.
Discussion
EVs harbour favourable characteristics for the transfer of biological cargo to recipient cells, which increasingly highlights them as possible candidates for drug delivery. Unfortunately, engineering of EVs for targeted drug delivery is less straightforward than for synthetic systems (e.g. liposomes). While liposomes can be easily modified with targeting ligands and loaded with therapeutic cargoes, similar strategies are often not applicable to EVs due to their cellular origin (Citation5). Previously described EV targeting strategies are based on fusion of targeting ligands to EV membrane proteins, such as transmembrane domains of platelet-derived growth factor (Citation14, Citation48) and Lamp2b (Citation11, Citation16). Albeit various reports have demonstrated the feasibility of this approach, it remains unclear whether these strategies result in high-level expression (i.e. enrichment) of targeting ligands in EVs compared with parent cells. In this work, we present a novel method to display targeting ligands (i.e. nanobodies) on EV surfaces by linkage to GPI anchors. It was observed that nanobody expression was highly enriched in EVs compared with their parent cells, suggesting a selective secretion of GPI-anchored proteins in EVs. This finding is supported by previous reports, which showed efficient incorporation of GPI-anchored prion proteins in EVs (Citation49) and selective release of GPI-anchored proteins in EVs during reticulocyte maturation (Citation24). Moreover, lipid raft microdomains, in which GPI-anchored proteins are highly enriched, are believed to be involved in EV biogenesis and have often been identified as abundant constituents of EV membranes (Citation22, Citation23),Citation50–(Citation55). Hence, GPI-anchoring of targeting ligands may be an appealing alternative to previously described targeting methods. Besides recombinant protein enrichment in EVs, this strategy may offer several advantages. The DAF GPI-anchoring signal peptide is relatively small (37 amino acids), is highly soluble and is not expected to interfere with proper recombinant protein folding, especially given that the peptide is cleaved off and replaced by a GPI anchor during post-translational modification of the protein (Citation25–Citation27). This could be beneficial when expressing proteins with complex tertiary structures (e.g. antibodies or enzymes), which are expected to lose functionality when fused to membrane proteins. Furthermore, EV membrane proteins, such as tetraspanins, may be crucial for proper EV functioning and influence binding, membrane fusion or signalling in recipient cells (Citation4, Citation18). Upon fusion with (targeting) proteins, the functionality of these natural EV constituents may be compromised. Such issues may be avoided when introducing new proteins onto EV membranes via GPI-anchors.
Furthermore, we showed in the present study that EV binding to EGFR-overexpressing cells dramatically increased upon display of GPI-anchored EGa1, both under static and flow conditions.
We hypothesize that EV cell binding under flow conditions better represents in vivo physiology than cell binding under static conditions. In vitro experiments are often performed under static conditions, which potentially leads to improbable and non-specific interactions between EVs and cells. Such experiments may poorly translate to in vivo behaviour of EVs, given that flow of EV-containing body fluids may significantly impact the exposure of EVs to their target tissue and affect their functionality. In this study, EVs were perfused over cells in a perfusion chamber with a shear rate of 82.5 s−1, resembling a typical venous shear rate (20–200 s−1 (Citation56)). EV association with A431 cells (visualized as immobilized EVs on cell surfaces) could be traced in real time with a fluorescence microscope and quantified. It was observed that EV-DAF-EGa1 displayed enhanced association with EGFR-expressing cells under these conditions compared with control EVs and EV-DAF-R2, which could have major implications for their in vivo retention in tumour tissues.
Neuro2A EVs appeared to exert limited cell type specificity (illustrated by similar uptake kinetics for A431 and Neuro2A cells under static conditions), a phenomenon also reported for other EV types (Citation57, Citation58). Remarkably, under the conditions in our study, uptake of EGa1-displaying EVs by EGFR-overexpressing cells was unaltered compared with control EVs. These results indicate that EV binding to cells does not necessarily result in EV uptake. A possible explanation for this phenomenon could be that monovalent EGa1 nanobodies bind EGFR with high affinity, but do not trigger receptor clustering and internalization (Citation59). However, previous studies showed that when EGa1 nanobodies were grafted on the surface of liposomes, these liposomes promoted EGFR sequestration and were internalized by EGFR-expressing cells (Citation37, Citation59). Hence, it appears that uptake of EGa1-displaying nanoparticles is only triggered when the nanobody surface density is sufficient to promote receptor clustering. It could be that in our study, the achieved nanobody surface density was too low, resulting in cell binding, but not uptake. To further investigate this phenomenon, we decorated the surface of isolated EVs with nanobodies using a chemical linker. Using this procedure, nanobodies are expected to be non-reducibly conjugated to all EV proteins with accessible primary amine groups, resulting in a high nanobody surface density and a smear pattern as analysed by western blotting (Supplementary Fig. 4a). When cell association experiments were performed with nanobody-conjugated EVs, both cell binding and cell uptake of EGa1-conjugated EVs were greatly increased when compared with untreated EVs and EVs conjugated with R2 nanobodies (Supplementary Fig. 4b and c). Interestingly, EV conjugation with R2 nanobodies decreased cell binding and uptake, illustrating that EV proteins containing exposed primary amines – whose functionality is likely to be compromised upon chemical conjugation with nanobodies – play a major role in these processes. Furthermore, these results suggest that nanobody surface density may affect internalization of nanobody-decorated EVs after binding. This phenomenon has also been described for other targeted nanoparticulate systems (Citation60, Citation61) and may be worth taking into account when designing a strategy to introduce targeting ligands in EVs. Additionally, it is important to note that EVs may be directed away from their natural uptake route (e.g. clathrin-, caveolin-, or lipid raft-mediated endocytosis, macropinocytosis or phagocytosis (Citation18)) through the use of targeting ligands, which may alter functional delivery of cargo such as mRNA or miRNA. However, whether this is the case remains unclear, given that mechanisms through which natural EVs functionally transfer their cargo are yet to be elucidated.
It is possible that GPI-anchored proteins are not evenly distributed among secreted EVs, but concentrated in specific subpopulations. The basal uptake of “blank” EVs may in this case have masked any beneficial effects of EGa1 anchorage on EVs in cell uptake experiments. Using TEM analysis, we could only detect nanobodies on the surface of a minority of the secreted EVs. This may be explained by technical limitations of the TEM technique, but it is conceivable that lipid rafts (and associated proteins) are predominantly incorporated in a subset of EVs. Previous studies have shown that detergent-resistance and membrane lipid order differ between subpopulations of EVs, supporting the idea that lipid rafts (which exhibit high resistance to detergents (Citation62)) are not evenly distributed among EV subsets (Citation63, Citation64). Interestingly, it was recently suggested that lipid rafts may be involved in RNA loading of EVs (Citation65), implying that RNA may be specifically enriched in lipid raft containing subpopulations of EVs. Hence, expression of GPI-anchored targeting proteins on EVs may open possibilities for co-purification of both targeted and RNA-enriched EVs (e.g. using an immunoprecipitation procedure).
In conclusion, we developed a novel approach to display targeting ligands on EV surfaces. Through fusion with GPI-anchors, nanobodies were strongly enriched in EVs compared with parent cells, dramatically improving EV cell interactions under static and flow conditions. GPI-anchoring may potentially be used as a versatile tool to incorporate a variety of proteins on EVs, including antibodies, enzymes, reporter proteins and (immune-stimulatory) signalling molecules.
Conflict of interest and funding
The authors have not received any funding or benefits from industry or elsewhere to conduct this study.
Supplementary Material
Download PDF (313.8 KB)Acknowledgements
The authors declare no competing financial interests. The work of SAAK, CGA, PV and RMS on cell-derived membrane vesicles is supported by a European Research Council starting grant (260627) “MINDS” in the FP7 ideas program of the European Union. The work of SRR is supported by a grant from the Academia Sinica Research Program on Nanoscience and Nanotechnology. PV is supported by a VENI Fellowship (# 13667) from the Netherlands Organisation for Scientific Research (NWO).
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
References
- Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014; 30: 255–89.
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, etal. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015; 4: 27066, doi: http://dx.doi.org/10.3402/jev.v4.27066.
- van Dommelen SM, Vader P, Lakhal S, Kooijmans SA, van Solinge WW, Wood MJ, etal. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release. 2012; 161: 635–44.
- Kooijmans SA, Vader P, van Dommelen SM, van Solinge WW, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. Int J Nanomed. 2012; 7: 1525–41.
- van der Meel R, Fens MH, Vader P, van Solinge WW, Eniola-Adefeso O, Schiffelers RM. Extracellular vesicles as drug delivery systems: lessons from the liposome field. J Control Release. 2014; 195: 72–85.
- El-Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013; 12: 347–57.
- Fuhrmann G, Herrmann IK, Stevens MM. Cell-derived vesicles for drug therapy and diagnostics: opportunities and challenges. Nano Today. 2015; 10: 397–409.
- Batrakova EV, Kim MS. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J Control Release. 2015; 219: 396–405.
- Gyorgy B, Hung ME, Breakefield XO, Leonard JN. Therapeutic applications of extracellular vesicles: clinical promise and open questions. Annu Rev Pharmacol Toxicol. 2015; 55: 439–64.
- Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G, etal. Applying extracellular vesicles based therapeutics in clinical trials – an ISEV position paper. J Extracell Vesicles. 2015; 4: 30087, doi: http://dx.doi.org/10.3402/jev.v4.30087.
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011; 29: 341–5.
- Tang K, Zhang Y, Zhang H, Xu P, Liu J, Ma J, etal. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat Commun. 2012; 3: 1282.
- Gujrati V, Kim S, Kim SH, Min JJ, Choy HE, Kim SC, etal. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano. 2014; 8: 1525–37.
- Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, etal. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther. 2013; 21: 185–91.
- Cooper JM, Wiklander PB, Nordin JZ, Al-Shawi R, Wood MJ, Vithlani M, etal. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord. 2014; 29: 1476–85.
- Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, etal. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014; 35: 2383–90.
- Kooijmans SAA, Fliervoet LAL, van der Meel R, Fens MHAM, Heijnen HFG, van Bergen en Henegouwen PMP, etal. PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release. 2016; 224: 77–85.
- Mulcahy LA, Pink RC, Carter DR. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014; 3: 24641, doi: http://dx.doi.org/10.3402/jev.v3.24641.
- Hung ME, Leonard JN. Stabilization of exosome-targeting peptides via engineered glycosylation. J Biol Chem. 2015; 290: 8166–72.
- Smyth T, Petrova K, Payton NM, Persaud I, Redzic JS, Graner MW, etal. Surface functionalization of exosomes using click chemistry. Bioconjug Chem. 2014; 25: 1777–84.
- Wang M, Altinoglu S, Takeda YS, Xu Q. Integrating protein engineering and bioorthogonal click conjugation for extracellular vesicle modulation and intracellular delivery. PLoS One. 2015; 10: e0141860.
- Wubbolts R, Leckie RS, Veenhuizen PT, Schwarzmann G, Mobius W, Hoernschemeyer J, etal. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J Biol Chem. 2003; 278: 10963–72.
- de Gassart A, Geminard C, Fevrier B, Raposo G, Vidal M. Lipid raft-associated protein sorting in exosomes. Blood. 2003; 102: 4336–44.
- Rabesandratana H, Toutant JP, Reggio H, Vidal M. Decay-accelerating factor (CD55) and membrane inhibitor of reactive lysis (CD59) are released within exosomes during in vitro maturation of reticulocytes. Blood. 1998; 91: 2573–80.
- Chen CP, Hsieh YT, Prijovich ZM, Chuang HY, Chen KC, Lu WC, etal. ECSTASY, an adjustable membrane-tethered/soluble protein expression system for the directed evolution of mammalian proteins. Protein Eng Des Sel. 2012; 25: 367–75.
- Chou WC, Liao KW, Lo YC, Jiang SY, Yeh MY, Roffler SR. Expression of chimeric monomer and dimer proteins on the plasma membrane of mammalian cells. Biotechnol Bioeng. 1999; 65: 160–9.
- Cheng TL, Roffler S. Membrane-tethered proteins for basic research, imaging, and therapy. Med Res Rev. 2008; 28: 885–928.
- Revets H, De Baetselier P, Muyldermans S. Nanobodies as novel agents for cancer therapy. Expert Opin Biol Ther. 2005; 5: 111–24.
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008; 358: 1160–74.
- Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013; 13: 663–73.
- Nordin JZ, Lee Y, Vader P, Mager I, Johansson HJ, Heusermann W, etal. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine. 2015; 11: 879–83.
- Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999; 94: 3791–9.
- Hofman EG, Ruonala MO, Bader AN, van den Heuvel D, Voortman J, Roovers RC, etal. EGF induces coalescence of different lipid rafts. J Cell Sci. 2008; 121(Pt 15): 2519–28.
- Schmitz KR, Bagchi A, Roovers RC, van Bergen en Henegouwen PM, Ferguson KM. Structural evaluation of EGFR inhibition mechanisms for nanobodies/VHH domains. Structure. 2013; 21: 1214–24.
- Frenken LG, van der Linden RH, Hermans PW, Bos JW, Ruuls RC, de Geus B, etal. Isolation of antigen specific llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J Biotechnol. 2000; 78: 11–21.
- Oliveira S, van Dongen GA, Stigter-van Walsum M, Roovers RC, Stam JC, Mali W, etal. Rapid visualization of human tumor xenografts through optical imaging with a near-infrared fluorescent anti-epidermal growth factor receptor nanobody. Mol Imaging. 2012; 11: 33–46.
- van der Meel R, Oliveira S, Altintas I, Haselberg R, van der Veeken J, Roovers RC, etal. Tumor-targeted nanobullets: anti-EGFR nanobody-liposomes loaded with anti-IGF-1R kinase inhibitor for cancer treatment. J Control Release. 2012; 159: 281–9.
- Udenfriend S, Kodukula K. How glycosylphosphatidylinositol-anchored membrane proteins are made. Annu Rev Biochem. 1995; 64: 563–91.
- Levental I, Grzybek M, Simons K. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry. 2010; 49: 6305–16.
- Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Chapter 3:Unit 3.22..
- Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009; 9: 581–93.
- Li J, Lee Y, Johansson HJ, Mager I, Vader P, Nordin JZ, etal. Serum-free culture alters the quantity and protein composition of neuroblastoma-derived extracellular vesicles. J Extracell Vesicles. 2015; 4: 26883, doi: http://dx.doi.org/10.3402/jev.v4.26883.
- Montecalvo A, Larregina AT, Shufesky WJ, Beer Stolz D, Sullivan ML, Karlsson JM, etal. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012; 119: 756–66.
- Zhang F, Wang S, Yin L, Yang Y, Guan Y, Wang W, etal. Quantification of epidermal growth factor receptor expression level and binding kinetics on cell surfaces by surface plasmon resonance imaging. Anal Chem. 2015; 87: 9960–5.
- Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994; 271: 58–65.
- Jain RK, Tong RT, Munn LL. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: insights from a mathematical model. Cancer Res. 2007; 67: 2729–35.
- Ozturk D, Yonucu S, Yilmaz D, Unlu MB. Influence of vascular normalization on interstitial flow and delivery of liposomes in tumors. Phys Med Biol. 2015; 60: 1477–96.
- Lai CP, Mardini O, Ericsson M, Prabhakar S, Maguire CA, Chen JW, etal. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano. 2014; 8: 483–94.
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, etal. Cells release prions in association with exosomes. Proc Natl Acad Sci USA. 2004; 101: 9683–8.
- Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005; 106: 1604–11.
- Staubach S, Razawi H, Hanisch FG. Proteomics of MUC1-containing lipid rafts from plasma membranes and exosomes of human breast carcinoma cells MCF-7. Proteomics. 2009; 9: 2820–35.
- Laulagnier K, Motta C, Hamdi S, Roy S, Fauvelle F, Pageaux JF, etal. Mast cell– and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochem J. 2004; 380(Pt 1): 161–71.
- Tan SS, Yin Y, Lee T, Lai RC, Yeo RW, Zhang B, etal. Therapeutic MSC exosomes are derived from lipid raft microdomains in the plasma membrane. J Extracell Vesicles. 2013; 2: 22614, doi: http://dx.doi.org/10.3402/jev.v2i0.22614.
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, etal. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008; 319: 1244–7.
- Thery C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, etal. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999; 147: 599–610.
- Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996; 88: 1525–41.
- Zech D, Rana S, Buchler MW, Zoller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun Signal. 2012; 10: 37.
- Svensson KJ, Christianson HC, Wittrup A, Bourseau-Guilmain E, Lindqvist E, Svensson LM, etal. Exosome uptake depends on ERK1/2-heat shock protein 27 signaling and lipid Raft-mediated endocytosis negatively regulated by caveolin-1. J Biol Chem. 2013; 288: 17713–24.
- Oliveira S, Schiffelers RM, van der Veeken J, van der Meel R, Vongpromek R, van Bergen En Henegouwen PM, etal. Downregulation of EGFR by a novel multivalent nanobody-liposome platform. J Control Release. 2010; 145: 165–75.
- Zhong Y, Meng F, Deng C, Zhong Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014; 15: 1955–69.
- Reuter KG, Perry JL, Kim D, Luft JC, Liu R, DeSimone JM. Targeted PRINT Hydrogels: the role of nanoparticle size and ligand density on cell association, biodistribution, and tumor accumulation. Nano Lett. 2015; 15: 6371–8.
- Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998; 14: 111–36.
- Osteikoetxea X, Sodar B, Nemeth A, Szabo-Taylor K, Paloczi K, Vukman KV, etal. Differential detergent sensitivity of extracellular vesicle subpopulations. Org Biomol Chem. 2015; 13: 9775–82.
- Osteikoetxea X, Balogh A, Szabo-Taylor K, Nemeth A, Szabo TG, Paloczi K, etal. Improved characterization of EV preparations based on protein to lipid ratio and lipid properties. PLoS One. 2015; 10: e0121184.
- Janas T, Janas MM, Sapon K. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015; 589: 1391–8.