
Abstract
Exosomes are cell-secreted nanovesicles (40–200 nm) that represent a rich source of novel biomarkers in the diagnosis and prognosis of certain diseases. Despite the increasingly recognized relevance of these vesicles as biomarkers, their detection has been limited due in part to current technical challenges in the rapid isolation and analysis of exosomes. The complexity of the development of analytical platforms relies on the heterogeneous composition of the exosome membrane. One of the most attractive tests is the inmunochromatographic strips, which allow rapid detection by unskilled operators. We have successfully developed a novel lateral flow immunoassay (LFIA) for the detection of exosomes based on the use of tetraspanins as targets. We have applied this platform for the detection of exosomes purified from different sources: cell culture supernatants, human plasma and urine. As proof of concept, we explored the analytical potential of this LFIA platform to accurately quantify exosomes purified from a human metastatic melanoma cell line. The one-step assay can be completed in 15 min, with a limit of detection of 8.54×105 exosomes/µL when a blend of anti-CD9 and anti-CD81 were selected as capture antibodies and anti-CD63 labelled with gold nanoparticles as detection antibody. Based on our results, this platform could be well suited to be used as a rapid exosome quantification tool, with promising diagnostic applications, bearing in mind that the detection of exosomes from different sources may require adaptation of the analytical settings to their specific composition.
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Extracellular vesicles (EVs) consist of small, bubble-like membranous structures that are released from cells in 3 primary types: exosomes, microvesicles and apoptotic bodies (Citation1), and play a pivotal role in intercellular communication. These vesicles have been studied over the years using a variety of isolation strategies and have been classified according to their distinct structural and biochemical properties. Exosomes are defined as 40–200 nm diameter (Citation2) membrane vesicles of endocytic origin that are released by most cell types upon fusion of multivesicular bodies (MVBs) with the plasma membrane (Citation3, Citation4). The protein content of exosomes has been extensively analysed from different cell types and body fluids by mass spectrometry, western blotting, flow cytometry and immunoelectron microscopy and includes both conserved and cell-type-specific proteins. All EVs contain proteins involved in membrane transport and fusion (Rab GTPases and annexins), heat shock proteins (HSP60, HSP70, HSPA5 and HSP90), integrins and variable proportions of tetraspanins (CD63, CD81 and CD9) (Citation4–Citation6). Moreover, EVs also contain a variety of nucleic acids including DNA, mRNA and miRNA. A recent comparative proteomic study between different subtypes of EVs (Citation5) proposed that co-expression of CD63 with at least one other tetraspanin, CD9 and/or CD81, characterized exosomes of endosomal origin, also enriched in proteins involved in MVB biogenesis (e.g. TSG101), whereas other co-isolated small EVs bearing a single or no tetraspanins originated from other intracellular locations.
Exosomes and other EVs are present in many biological fluids, such as serum/plasma, urine, amniotic fluid, cerebrospinal fluid and saliva (Citation7). Since they carry cell-specific signatures, EVs that are secreted into biological fluids have tremendous potential as biomarkers for prognosis and monitoring the response to treatment in a range of diseases including vascular disorders, autoimmune and hematologic diseases, and cancer (Citation1, Citation8). Moreover, analysis of EVs from body fluids may serve as a non-invasive alternative to the current diagnostic tests. In fact, early reports suggest that EVs may be of use as a “liquid biopsy” (Citation9). In general, higher EVs content has been found in patients with advanced cancer compared to healthy donors, although more studies are needed in this context (Citation10, Citation11).
Although this branch of science is growing very fast, there are still limitations in relation to isolation and purification technologies as well as to the ability to measure EVs sizes, concentration and molecular content, and only a few EV-based diagnostic assays are currently available (Citation12–Citation14). The “gold standard” and most commonly used protocol for EVs isolation and purification is differential centrifugation, which involves sequential centrifugation and ultracentrifugation steps (Citation15). Recently, several alternative methods have been introduced for isolation and purification of EVs, including antibody-coated magnetic beads, microfluidic devices (Citation16, Citation17), precipitation methods and filtration technologies (Citation8, Citation18). After EVs isolation, many downstream applications require EVs quantitation. For this, researchers use a standard ELISA approach (Citation8, Citation10) (Citation19). As an alternative to ELISA, lab-on-a-chip devices are useful techniques in clinical care for medical diagnosis, since they allow the use of small volumes of sample and shorter processing times, improve sensitivity and reduce clinical care costs. These miniaturized devices integrate microfluidic approaches that enable on-chip immune isolation and in situ protein analysis of EVs directly from patient plasma (Citation20) and allow multiplex assays (Citation21). Indeed, Chen et al. (Citation22) have developed a point-of-care (POC) system for microvesicle analysis using a paper-based ELISA device, which enables the use of very low sample and reagent volumes and that is completed in 10 min. The Lee and Weissleder group has developed small devices that allow quantification of EVs based on different physicochemical principles, such as nuclear magnetic resonance (Citation23), surface plasmon resonance (Citation24) and electrochemical measurements combined with immunocapture (Citation25).
Within the POC systems, lateral flow immunoassay tests (LFIA) represent a well-established technology, which fulfils the characteristics of a rapid, simple and cost-effective test capable of being performed by unskilled operators (Citation26). LFIA is based on the recognition of one or more analytes (multiplex) of interest by using antibodies. These antibodies are immobilized on a nitrocellulose membrane and interact with the analyte when the sample is applied and flows by capillary action. In a sandwich format, protein–target complexes and free capture antibodies, both labelled, accumulate in 2 defined regions forming the test and control line, respectively. The main advantage compared with other immunoassays is that the entire test can be done usually in one step and in a few minutes (Citation27). To the authors’ knowledge, this type of test had not yet been developed for the detection of exosomes. This might reflect the difficulty of dealing with vesicles of this size that have a heterogeneous composition on the membrane, in highly complex biological matrixes. However, LFIAs have been successfully developed for analytes reaching a size of 1 µm, such as spores and bacteria (Citation28, Citation29).
In this study, LFIA platforms for the detection of exosomes have been developed using tetraspanins as targets for capture and detection antibodies. Tetraspanins CD9, CD63 and CD81 are specially enriched in the membrane of exosomes (Citation5, Citation30), and they are often used as exosome biomarkers due to their high abundance in virtually any cell type. However, their single expression on an EV does not demonstrate its endosomal origin (Citation5): the approach developed here allows to analyse specifically EVs bearing at least 2 of these 3 tetraspanins, and thus more specifically bona fide exosomes, rather than other EVs.
Methods
Cell culture
The human melanoma cell line Ma-Mel-86c was established from a stage IV metastatic lymph node lesion (Citation31). Small tissue pieces were distributed in cell culture dishes, and outgrowing cells were split for the first time at 90% cell confluence. Melanoma cell lines were cultured in RPMI-1640 medium supplemented with l-glutamine, 10% foetal bovine serum (FBS) and penicillin/streptomycin. Cells were cultured at 37°C in a 5% CO2 atmosphere. Tumour tissue was collected after approval by the institutional review board and patient informed consent.
Antibodies
Anti-tetraspanin antibodies used for ELISA and LFIA in this study have been previously characterized (Citation32): anti-CD9 VJ1/20 (Citation33) and anti-CD63 Tea3/18 (Citation34). Anti-CD81 5A6 was provided by Dr. S. Levy (Department of Oncology, Stanford University School of Medicine, Stanford, CA). Antibodies were purified by affinity chromatography with Prot-G Sepharose (GE Healthcare Life Sciences) and biotinylated with Sulfo-NHS Biotin (Pierce). Free biotin was eliminated by overnight dialysis against PBS buffer.
Tetraspanin antibodies used for western blot (MEM-63, MEM-81 and MEM-9) were obtained from Vaclav Horejsi (Czech Republic).
Exosomes purification and evaluation of protein concentration
For exosome isolation, cells were cultured in medium supplemented with 0.5% exosome-free FBS for 3–5 days. Cells were centrifuged for 10 min at 200×g and exosomes purified by sequential centrifugation as previously described (Citation35). Exosome-free FBS was prepared by ultracentrifugation of regular FBS, during 16–18 h at 100,000×g followed by filter sterilization. Aliquots were prepared and kept frozen at −20°C.
Briefly, after centrifugation of cells, supernatants were centrifuged twice at 500×g for 10 min. Supernatants were pooled and centrifuged at 10,000×g for 30 min prior to exosomes isolation by ultracentrifugation at 100,000×g for 2 h at 4°C (Beckman Instruments). Exosomes were resuspended in HEPES-buffered saline buffer (HBS: 10 mM HEPES pH 7.2, 150 mM NaCl).
Purified exosomes were supplemented with sucrose to a final concentration of 8% and frozen immediately at −80°C and lyophilized using a Flexi-Dry Lyophilizer (FTS Systems).
Lyophilized commercial exosomes purified from plasma (HBM-PEP) and urine (HBM-PEU) of healthy donors were purchased from HansaBioMed (Tallinn, Estonia) used in LFIAs. The expression of the tetraspanins CD9, CD63 and CD81 was confirmed by western blot and flow cytometry using, respectively, antibodies anti-CD9, anti-CD63 and anti-CD81 (HansaBioMed, Tallinn, Estonia) (Supplementary Figs. 1 and 2). Exosome pellets were resuspended in deionized water (Milli-Q), following the manufacturer's instructions.
For analysis of protein concentration, Bradford assays were performed, following the manufacturer's recommendations.
The concentration of Ma-Mel-86c exosomes was also determined by nanoparticle tracking analysis (NTA) in a NanoSight NS500 (Malvern Instruments Ltd, Malvern, UK). A 405-nm laser beam was used to highlight the particles, which act as point scatters. Analysis was performed using the NTA 3.1 software (Malvern). The experiment was carried out at the laboratory of Dr. H. Peinado, Spanish National Centre for Oncological Research (CNIO).
Western blot
Cell lysates were prepared by incubation in Tris pH 7.6 buffer containing 150 mM NaCl, 5 mM EDTA, 1% NP-40 and the protease inhibitors leupeptin and pepstatin for 30 min at 4°C. Nuclei were eliminated by centrifugation at 13,000×g. Lysates and purified exosomes were resuspended in Laemmli buffer and run on 12% SDS-PAGE gels. For detection of tetraspanins, samples were run under non-reducing conditions. Proteins were transferred to Immobilon-P (Millipore) membrane. The membrane was blocked using PBS containing 0.1% Tween-20 (PBS-T) and 5% non-fat dry milk. Tetraspanins were detected by incubation with the antibodies indicated above, followed by HRP-conjugated secondary antibody. Proteins were visualized using the ECL system (GE Healthcare).
Transmission electron microscopy
Lyophilized exosomes were reconstituted and diluted 1:20 with PBS. Electron microscope examination of exosomes was carried out by floating a carbon-coated 400-mesh Formvar EM grid on top of 1 drop of freshly prepared exosomes (60 µg/mL in PBS) for about 1 min. The grid was then briefly washed with deionized water and floated on a drop of 2% uranyl acetate. Samples were examined using a Jeol JEM 1011 electron microscope operating at 100 kV with a CCD camera Gatan Erlangshen ES1000W. The experiment was carried out at the Electron Microscopy Facility, Spanish National Centre for Biotechnology (CNB).
Size distribution and ζ-potential
Size distribution and ζ-potential assays were carried out with both fresh and lyophilized samples using a Zetasizer Nano ZS ZEN3600 (Malvern Instruments, Malvern, UK) equipped with a solid-state He-Ne laser (λ=633) for monitoring the conjugation process. Diluted (50- to 100-fold) fractions were loaded in the cell. A total of 3 readings were performed at 25°C. Each reading was composed of 15 measurements of the backscattering (173°) intensity. Zetasizer software version 7.03 was used for data processing and analysis.
ELISA for exosome detection
In order to test the recognition of different molecules in the same exosome sample, and before testing the LFIA system, different combinations of tetraspanin-specific antibodies were tested by ELISA. Ninety-six well plates (Falcon) were coated with the indicated antibodies (100 µL at 5.0 µg/mL) in either 100 mM sodium borate buffer, pH 9, or 100 mM sodium carbonate buffer, pH 9.6, overnight at 4°C. Wells were washed with HBS and blocked with 200 µL/well (2% BSA in HBS) for 1 h at 37°C. Following 3 washes, exosomes purified from Ma-Mel-86c cell culture supernatants were added (100 µL/well) and incubated at 37°C for 2 h. HBS was used as a negative control. After 3 washes with HBS, the indicated amount of secondary biotinylated antibodies (anti-CD9, anti-CD81 or anti-CD63) was added and incubated for 1 h at 37°C. After 3 washes with HBS, the plate was incubated with 100 µL of HRP-conjugated streptavidin (Biolegend) diluted 1:2,000 at 37°C for 1 h. After 3 final washes, the reaction was developed with ABTS for 1 h (Roche) and optical densities were recorded at 405 nm with a reference of 490 nm, using a Sunrise absorbance reader (Tecan).
Lateral flow immunoassay
Gold nanoparticles of size 40 nm (AuNP) were purchased from BB International (UK). Nitrocellulose membranes (HF07504XSS) and glass fibre sample pads (GFCP001000) were purchased from Millipore (Germany). Other materials used were backing cards (KN-V1080, Kenoshatapes, the Netherlands) and absorbent pads (Whatman, USA). The running buffer consisted of 10 mM HEPES, pH 7.4, with 150 mM NaCl, 0.05% Tween-20 and 1% BSA.
An IsoFlow reagent dispensing system (Imagene Technology, USA) was used to dispense the detection lines (dispense rate 0.100 µL/mm) and the strips were cut with a guillotine Fellowes Gamma (Spain).
Labelling antibody with colloidal gold
Anti-CD9 monoclonal antibody (Clone VJ1/20) and anti-CD63 monoclonal antibody (Clone Tea3/18) were conjugated to AuNP. In order to find the optimal concentration of the antibody to stabilize the gold nanoparticles, a gold colloid titration procedure was followed (Citation36). In the case of anti-CD9, aggregation of gold nanoparticles occurred even though different concentrations of antibody and different buffers were tested. For this reason, 2 different conjugation protocols were carried out: (a) a non-covalent process for anti-CD63 and (b) a covalent protocol for anti-CD9.
Non-covalent process: 100 µL of 150 µg/mL anti-CD63 (Clone Tea3/18) was added to 1.5 mL of AuNP suspension. After shaking for 1 h, 100 µL of blocking solution (1 mg/mL BSA in PBS 10 mM, pH 7.4) was added to block the residual surfaces of antibody-colloidal gold conjugate. After 20 min of reaction, the mixture was centrifuged at 6,800×g for 20 min. The supernatant was discarded and the pellet was resuspended in 2 mM borate buffer, pH 7.4, with 10% sucrose and 1% BSA. The product (AuNPs-anti-CD63 conjugate) was then stored at 4°C until used.
Covalent protocol: Conjugation of AuNP with anti-CD9 monoclonal antibody (Clone VJ1/20) was carried out using 3,3′-dithiobis[sulfosuccinimidylpropionate] (DTSSP) as a bifunctional cross linker, adapted from a previously reported procedure (Citation37). 10 µL of 1 mM DTSSP was added to 1.0 mL of AuNP and mixed for 30 min. The suspension was then centrifuged at 3,300×g for 5 min. The supernatant was discarded and the pellet was resuspended in 2 mM borate buffer (pH 8.9). Then, 15 µg of anti-CD9 was added to the AuNP, shaken for 1.5 h and the solution centrifuged at 6,800×g for 20 min. The supernatant was discarded and the pellet was resuspended in 2 mM borate buffer containing 1% BSA. The product (AuNPs–anti-CD9 conjugate) was then stored at 4°C until use.
Preparation of immunostrips
The LFIA was carried out in a dipstick format. The nitrocellulose membrane (25 mm wide) was incorporated into the plastic backing to give robustness to the membrane. The test zone of the strip was prepared dispensing a desired volume of 1 mg/mL anti-tetraspanin and anti-IgG to form the test and control lines, respectively, with the dispenser IsoFlow onto NC membrane at a dispensing rate of 0.100 µL/mm and was dried for 20 min at 37°C. The sample pad and the absorbent pad were then settled onto the backing card with an overlap between them of around 2 mm. The complete strip was cut into individual 4-mm strips.
LFIA procedure
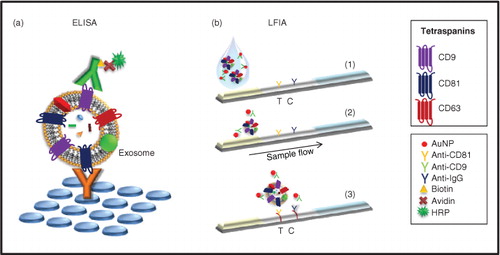
For dipstick analysis, purified exosome samples prepared in running buffer were transferred into the microtube containing AuNPs–antibody conjugate and homogenized (final volume 100 µL). The dipstick was added and the sample allowed to run for 15 min. The performance of the immunostrip relied on non-competitive assay formats. Exosomes in the sample were sandwiched between an anti-tetraspanin antibody immobilized on the strip (Test line, T) and the AuNP-conjugated antibody. The unbound AuNP-conjugates migrated further to be captured with the anti-mouse immunoglobulin antibodies (control, C) for system functional verification ().
Fig. 1. Schematic view of the experimental procedure for exosomes detection. (a) Schematic representation of the ELISA set up for exosomes detection. Here, each reagent is added in sequential steps. (b) Schematic representation of the lateral flow immunoassay dipstick. (Citation1) Specific antibodies against tetraspanins (test, T) and anti-mouse immunoglobulin antibodies (control, C) are immobilized on the membrane. (Citation2) Exosomes, if present in the sample, are detected by the detection probes (AuNP-conjugated antibodies). (Citation3) As the complexes flow, they are captured onto the membrane by the immobilized antibodies.
Results and discussion
Exosome characterization
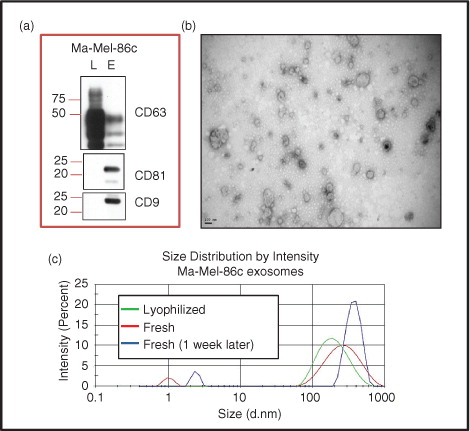
Exosomes purified from cell culture supernatants of the human melanoma cell line Ma-Mel-86c by sequential centrifugation were characterized for expression of different tetraspanins. Ma-Mel-86c exosomes were highly enriched in CD81 and CD9, but contained lower amounts of CD63 than whole cell lysates (a). Visualization of the purified vesicles by transmission electron microscopy (TEM) showed a round shape with a diameter ranging between 50 and 150 nm (b). The average size recorded by dynamic light scattering (DLS) was 206 nm (PDI=0.290) for fresh samples and 177 nm (PDI=0.226) for lyophilized samples. The results showed a ζ-potential of −30 mV and −34 mV for fresh and lyophilized samples, respectively, indicating that lyophilization did not cause exosome aggregation. Exosome size and stability during storage in fresh and lyophilized samples were analysed, at 4°C and −20°C. The size of exosomes did not change during the first week of storage at 4°C in lyophilized samples but some aggregates appeared in fresh samples during that period of time (680 nm, PDI=0.587; ζ-potential: −12 mV) (c). When samples were stored at −20°C, the exosomes remained stable for at least 9 months (data not shown). Both plasma and urine commercial exosomes were also analysed by DLS (Supplementary Fig. 3).
Fig. 2. Metastatic melanoma exosome characterization. (a) Western blot: Ma-Mel-86c lysates (L) and exosomes (E) were prepared, run in SDS-PAGE and analysed by western blot using the indicated antibodies. (b) TEM image: Exosomes from Ma-Mel-86c cells were negatively stained with 2% phosphotungstic acid and analysed by electron microscopy. Bar: 100 nm. (c) Hydrodynamic size distribution profiles: Lyophilized and fresh Ma-Mel-86c-derived exosomes (as indicated) were analysed in a Zetasizer for their size distribution. The graph shows 1 reading representative of 3.
Exosomes detection by in-house ELISA
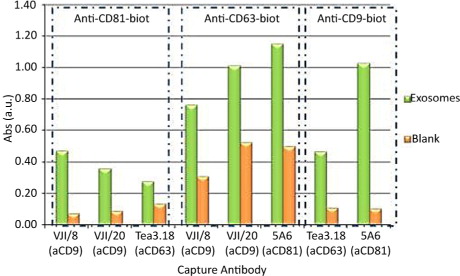
In order to test the availability of different tetraspanin epitopes simultaneously in the same sample of exosomes, several combinations of antibodies recognizing CD9, CD81 and CD63 were assayed in ELISA experiments with a constant concentration of exosomes derived from Ma-Mel-86c. Different concentrations of capture and detection antibodies were assessed, and different wash and incubation buffers were also tested. Optimal results were obtained when the capture antibody was used at 5 µg/mL in 100 mM sodium borate buffer, pH 9, while the exosomes were more stable in HEPES-buffered saline, both for washes and incubations. Different biotinylated antibody concentrations were also tested for detection. Biotinylated anti-CD9 (VJ1/20), anti-CD81 (5A6) or anti-CD63 (Tea3/18) mAbs were compared as shown in ; the pair of antibodies that produced the best signal-to-noise ratio (sample/blank) was anti-CD81 as capture mAb and anti-CD9 (1:1000) for detection. Mixtures of several antibodies against tetraspanins were tested as capture agents, but they did not improve the results obtained when anti-CD81 alone was used as capture antibody (data not shown).
Fig. 3. Comparison of exosome detection using different antibodies by ELISA. A constant concentration of Ma-Mel-86c purified exosomes was used in ELISA experiments in which different combinations of capture and detection antibodies (indicated at the bottom and top of the figure, respectively) were used. The concentrations used for the different capture (5 µg/mL) and detection (anti-CD9 1:1000, anti-CD81 1:1000 and anti-CD63 1:500) antibodies had been optimized in a different experiment. Blank wells had incubation with both capture and detection antibodies but not with exosome samples.
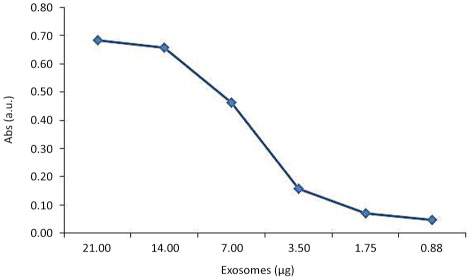
Different concentrations of exosomes purified from cell culture supernatants were tested using anti-CD81 capture antibody and biotinylated anti-CD9 detection antibody, showing a linear dose response in the range of 14 to 2 µg/mL of exosomes ().
Fig. 4. Titration of exosome detection by ELISA. Varying amounts of Ma-Mel-86c-derived exosomes were analysed by ELISA using anti-CD81 (5A6) as capture antibody and biotinylated anti-CD9 (J1/20) as detection antibody. The data points are the average of 2 replicates.
Characterization of nanoparticle–mAb conjugates
Since the biotinylated anti-CD9 antibody (VJ1/20) was the most efficient detection antibody for exosomes in the ELISA experiments, this antibody was selected for detection for the LFIA. The protocol followed to conjugate the anti-CD9 antibody with AuNP is described in the Methods section.
DLS measurements were carried out to confirm the conjugation reaction between the gold nanoparticles and the antibody. This technique allows monitoring the size variation of the nanoparticles after the conjugation reaction. The results show that the hydrodynamic size of the AuNP–anti-CD9 conjugate was 56 nm (PDI 0.172). To determine the stability of the conjugated antibody, ζ-potential measures were carried out. The results showed a ζ-potential of −17 mV for AuNP–anti-CD9, indicating that the conjugates had enough repulsion to keep the particles apart and, thus, were stable.
LFIA development
Initially, an LFIA system was developed following the optimal pattern obtained by ELISA. Thus, anti-CD81 monoclonal antibody (Clone 5A6) was dispensed onto the membrane as capture antibody and anti-CD9 monoclonal antibody (Clone VJ1/20) was conjugated with AuNP as detection probe. The system was tested with plasma exosomes and Ma-Mel-86c exosomes. Surprisingly, despite the ELISA results, this system was unable to detect exosomes isolated from melanoma cell culture supernatant. In contrast, exosomes isolated from human plasma could be detected with the naked eye at levels as low as 5 µg, by using LFIA system as shown in .
Fig. 5. Photograph of the test strips of LFIA for melanoma cell culture supernatant and plasma exosomes using anti-CD81 as capture antibody and AuNP–anti-CD9 as detection probe. (a) Blank: running buffer. (b) Photograph of the strip after adding 20 µg of Ma-Mel-86c exosomes. (c) Photograph of the strip corresponding to 5 µg of plasma exosomes (HBM).

Different competition events and steric effects may occur in ELISA and LFIA, which could explain this discrepancy. In ELISA, exosomes are captured by antibodies immobilized on a surface, so that epitopes on the opposite side of the exosome would be available for binding with the detection antibody. In contrast, in LFIA, exosomes are first bound to the detection antibody, which is normally present in excess. This step occurs in the buffer solution, and therefore, the whole surface of the exosome is accessible to be covered by antibody. In the case of Ma-Mel-86c cells, exosomes are expected to be completely surrounded by antibody, since CD9 is especially abundant in these EVs (a) and the CD81 capture antibody may not be able to access epitopes due to steric hindrance. In fact, when Ma-Mel-86c exosomes were present in the assay, the intensity of the control line was in general lower than in the blank under the same conditions (a and b), suggesting that AuNP–CD9 had been depleted by an extensive binding to the exosome and, in consequence, there was less excess of antibody available to bind at the control line.
Therefore, the differences in protein composition, localization and density of tetraspanins at the exosome surface between these types of exosomes can markedly influence their detection in LFIA.
In view of these results, an alternative tetraspanin LFIA system was developed. ELISA data suggested that the antibody combination of anti-CD9 as capture and anti-CD63 as detection could also be functional, despite a higher background signal in ELISA (). The conjugation of anti-CD63 antibody with AuNP was carried out following the protocol described in the Methods section, and the corresponding increase in hydrodynamic diameter over the naked AuNP and the absence of aggregates were checked by DLS. The results show that the hydrodynamic size of the AuNP–anti-CD63 was 64 nm (PDI=0.117), with a ζ-potential of −23 mV, suggesting that, as in the case of AuNP–anti-CD9, the complexes were stable.
This pair of antibodies (anti-CD9 as capture antibody and anti-CD63 as detection antibody) had a high background in the absence of exosomes, in ELISA, but not in the LFIA system. With this combination, we were able to detect exosomes from melanoma cell supernatants and human plasma (b and c). Moreover, this combination of anti-tetraspanin antibodies also allowed the detection of urine exosomes (d), which have been reported to contain very small amounts of the tetraspanin CD81 (Citation38). Exosome-depleted plasma was used as negative control (e).
Fig. 6. Photograph of the test strips of LFIA using anti-CD9 as capture antibody and AuNP–anti-CD63 as detection probe, corresponding to: (a) Blank: running buffer. (b) 20 µg Ma-Mel-86c exosomes. (c) 20µg of plasma exosomes (HBM). (d) 20 µg of urine exosomes (HBM). (e) Exosome-depleted plasma.

Given the recent discovery of EVs as potential biomarkers, convenient diagnosis devices need to be developed. The results shown in establish a proof of concept for the development of rapid tests for the detection of exosomes with clinical applications.
With the aim of giving an estimate of the analytical potential of these platforms, we have carried out a quantitation of how the increase in intensity at the test line varies with the concentration of exosomes. This correlation allows us to estimate the linear useful range and sensitivity of the measurements. Since most of the time the final end-user of the in vitro test will not have information about the abundance of tetraspanin domains on the surface of the exosomes tested, the immunoassay was developed immobilizing a mixture of anti-CD9 and anti-CD81 (1:1) at the test line, in order to ensure the capture at all the possible exosome sources, and keeping AuNP–anti-CD63 as detection probe. This guarantees that our LFIA strip will be able to detect exosomes purified from a wide range of fluids, although for Ma-Mel-86c exosomes, this combination of mAbs in the test line did not show any advantage when compared with anti-CD9 alone (Supplementary Fig. 4).
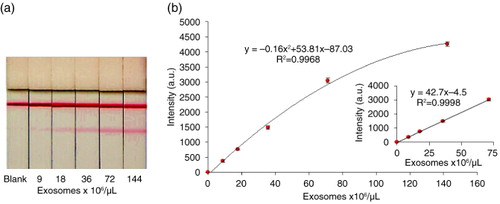
Once the test with exosomes purified from Ma-Mel-86c supernatants was run, the line intensities were recorded by scanning the images using a Lexmark 4800 scan and the optical density was measured using ImageJ 1.48 v software. All the assays were performed in triplicates and scanned in grey scale with a scan resolution of 2,400 ppp (). The colour of test lines displayed clear gradient according to the concentration of exosomes, although the test line started to saturate at quantities of 1.44×108 exosomes/µL. Non-specific binding was not observed in the absence of exosomes. The limit of detection (LOD), calculated from the calibration curve (the concentration corresponding to 3 times the standard deviation of the intercept) (Citation39), was 8.54×105 exosomes/µL.
Fig. 7. Effect of Ma-Mel-86c purified exosomes concentration on the optical density by LFIA. A blend of anti-CD9 and anti-CD81 was used as capture antibodies and AuNP-anti-CD63 as detection probe. (a) Representative example of results obtained in the strips. (b) Calibration curve obtained at the LFIA device. The data points were obtained in triplicate and error bars represent the standard deviations of triplicate runs. Inset shows the expanded view of low concentration data and linear regression lines.
Our approach shows a positive correlation between the concentration of exosomes purified from melanoma cell culture supernatant and the intensity of signal. Similar behaviour is expected with exosomes from the commercial source or purified tissue fluid. Hence, to our knowledge, this is the first time that such a novel method for the detection of exosomes had been developed (Citation13, Citation14). Although the analytical features described here lay the ground for the development of clinical applications at screening, diagnostics or prognostics, the aim of this manuscript was to introduce for the first time the use of LFIA to detect vesicles and more work should follow to improve the applications in a POC setting. In any case, in order to have a POC, clinically suited device, specific markers for specific diseases, when appropriately described, should also be included in the LFIA device. Lastly, in our system, the possibility that EVs could be associated with aggregated proteins from the culture media or plasma materials like LDL (Citation40) could not be excluded. However, the data presented here prove that, as the sample solution flows through the strip, EVs can be recognized by a labelled tetraspanin and that this immunocomplex can be captured afterwards by a second tetraspanin immobilized on nitrocellulose.
Conclusions
In this study, we have described an in-house ELISA to detect exosomes purified from cell culture supernatants and developed several LFIA to detect exosomes from cell culture supernatants as well as from commercial exosomes previously enriched from human plasma and urine. Here, we have presented a novel lateral flow with a wide range of applications. This assay has been applied for the detection of exosomes purified from a metastatic melanoma cell line, with an LOD of 8.54×105 exosomes/µL. The assay is easy to conduct, including by unskilled operators, and the results can be checked in 15 min. This work was carried out taking into account the heterogeneous composition of exosomes, in order to achieve a universal wide-broad application LFIA system. With this aim, we have chosen to use a blend of anti-CD9 and anti-CD81 as capture antibodies to detect exosomes from different sources. However, we have shown too that a specialized LFIA for a specific exosome subpopulation could be developed. In this sense, strips with anti-CD9 alone as capture antibody and AuNP–anti-CD63 as detection probe lead to better results for Ma-Mel-86c metastatic melanoma cells.
Conflict of interest and funding
The authors declare no conflict of interest.
Supplementary Material
Download PDF (160.2 KB)Acknowledgments
The authors thank Dr. V. Horejsi, Dr. S. Levy and Dr. Sánchez-Madrid for the gift of antibodies; Dr. H. Peinado for the use of NanoSight; and the electron microscopy service at the CNB. Funding from projects CTQ2013-47396-R, SAF2012-32293, SAF2014-58752-R, BFU2014-55478-R (Spanish Ministry of Economy and Competitivity), FC15-GRUPIN14-022 (Regional Government of Asturias) and S2010/BMD-2326-INMUNOTHERCAN (Regional Government of Madrid) is acknowledged. M. Oliveira-Rodríguez thanks FICYT for her pre-doctoral grant. SLC is a recipient of an FPU fellowship from the Spanish Ministry of Education.
Notes
To access the supplementary material to this article, please see Supplementary files under ‘Article Tools’.
Related Research Data
References
- Gyorgy B, Szabo TG, Paszto M, Pal Z, Misjak P, Aradi B, etal. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011; 68: 2667–88. doi: http://dx.doi.org/10.1007/s00018-011-0689-3.
- Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014; 30: 255–89. doi: http://dx.doi.org/10.1146/annurev-cellbio-101512-122326.
- Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200: 373–83. doi: http://dx.doi.org/10.1083/jcb.201211138.
- Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010; 73: 1907–20. doi: http://dx.doi.org/10.1016/j.jprot.2010.06.006.
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, etal. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 2016; 113: E968–77. doi: http://dx.doi.org/10.1073/pnas.1521230113.
- Azmi AS, Bao B, Sarkar FH. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013; 32: 623–42. doi: http://dx.doi.org/10.1007/s10555-013-9441-9.
- Properzi F, Logozzi M, Fais S. Exosomes: the future of biomarkers in medicine. Biomark Med. 2013; 7: 769–78. doi: http://dx.doi.org/10.2217/bmm.13.63.
- Peterson MF, Otoc N, Sethi JK, Gupta A, Antes TJ. Integrated systems for exosome investigation. Methods. 2015; 87: 31–45,. doi: http://dx.doi.org/10.1016/j.ymeth.2015.04.015.
- Santiago-Dieppa DR, Steinberg J, Gonda D, Cheung VJ, Carter BS, Chen CC. Extracellular vesicles as a platform for “liquid biopsy” in glioblastoma patients. Expert Rev Mol Diagn. 2014;; 14: 819–25,. doi: http://dx.doi.org/10.1586/14737159.2014.943193.
- Logozzi M, De Milito A, Lugini L, Borghi M, Calabro L, Spada M, etal. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One. 2009; 4: e5219. doi: http://dx.doi.org/10.1371/journal.pone.0005219.
- Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, etal. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015; 523: 177–82,. doi: http://dx.doi.org/10.1038/nature14581.
- Momen-Heravi F, Balaj L, Alian S, Tigges J, Toxavidis V, Ericsson M, etal. Alternative methods for characterization of extracellular vesicles. Front Physiol. 2012; 3: 354. doi: http://dx.doi.org/10.3389/fphys.2012.00354.
- Sunkara V, Woo HK, Cho YK. Emerging techniques in the isolation and characterization of extracellular vesicles and their roles in cancer diagnostics and prognostics. Analyst. 2016; 141: 371–81,. doi: http://dx.doi.org/10.1039/c5an01775k.
- Ko J, Carpenter E, Issadore D. Detection and isolation of circulating exosomes and microvesicles for cancer monitoring and diagnostics using micro-/nano-based devices. Analyst. 2016; 141: 450–60,. doi: http://dx.doi.org/10.1039/c5an01610j.
- Momen-Heravi F, Balaj L, Alian S, Mantel PY, Halleck AE, Trachtenberg AJ, etal. Current methods for the isolation of extracellular vesicles. Biol Chem. 2013; 394: 1253–62,. doi: http://dx.doi.org/10.1515/hsz-2013-0141.
- Chen C, Skog J, Hsu CH, Lessard RT, Balaj L, Wurdinger T, etal. Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab Chip. 2010; 10: 505–11,. doi: http://dx.doi.org/10.1039/b916199f.
- Davies RT, Kim J, Jang SC, Choi EJ, Gho YS, Park J. Microfluidic filtration system to isolate extracellular vesicles from blood. Lab Chip. 2012; 12: 5202–10,. doi: http://dx.doi.org/10.1039/c2lc41006k.
- Zarovni N, Corrado A, Guazzi P, Zocco D, Lari E, Radano G, etal. Integrated isolation and quantitative analysis of exosome shuttled proteins and nucleic acids using immunocapture approaches. Methods. 2015; 87: 46–58,. doi: http://dx.doi.org/10.1016/j.ymeth.2015.05.028.
- Ueda K, Ishikawa N, Tatsuguchi A, Saichi N, Fujii R, Nakagawa H. Antibody-coupled monolithic silica microtips for high throughput molecular profiling of circulating exosomes. Sci Rep. 2014; 4: 6232,. doi: http://dx.doi.org/10.1038/srep06232.
- He M, Crow J, Roth M, Zeng Y, Godwin AK. Integrated immunoisolation and protein analysis of circulating exosomes using microfluidic technology. Lab Chip. 2014; 14: 3773–80.
- Vaidyanathan R, Naghibosadat M, Rauf S, Korbie D, Carrascosa LG, Shiddiky MJ, etal. Detecting exosomes specifically: a multiplexed device based on alternating current electrohydrodynamic induced nanoshearing. Anal Chem. 2014; 86: 11125–32,. doi: http://dx.doi.org/10.1021/ac502082b.
- Chen C, Lin BR, Hsu MY, Cheng CM. Paper-based devices for isolation and characterization of extracellular vesicles. J Vis Exp. 2015; 98: 52722. doi: http://dx.doi.org/10.3791/52722.
- Issadore D, Min C, Liong M, Chung J, Weissleder R, Lee H. Miniature magnetic resonance system for point-of-care diagnostics. Lab Chip. 2011; 11: 2282–7,. doi: http://dx.doi.org/10.1039/c1lc20177h.
- Im H, Shao H, Park YI, Peterson VM, Castro CM, Weissleder R, etal. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotechnol. 2014; 32: 490–5,. doi: http://dx.doi.org/10.1038/nbt.2886.
- Jeong S, Park J, Pathania D, Castro CM, Weissleder R, Lee H. Integrated magneto-electrochemical sensor for exosome analysis. ACS Nano. 2016; 10: 1802–9,. doi: http://dx.doi.org/10.1021/acsnano.5b07584.
- Gordon J, Michel G. Analytical sensitivity limits for lateral flow immunoassays. Clin Chem. 2008; 54: 1250–1,. doi: http://dx.doi.org/10.1373/clinchem.2007.102491.
- Posthuma-Trumpie GA, Korf J, van Amerongen A. Lateral flow (immuno)assay: its strengths, weaknesses, opportunities and threats. A literature survey. Anal Bioanal Chem. 2009; 393: 569–82,. doi: http://dx.doi.org/10.1007/s00216-008-2287-2.
- Wang DB, Tian B, Zhang ZP, Deng JY, Cui ZQ, Yang RF, etal. Rapid detection of Bacillus anthracis spores using a super-paramagnetic lateral-flow immunological detection system. Biosens Bioelectron. 2013; 42: 661–7,. doi: http://dx.doi.org/10.1016/j.bios.2012.10.088.
- Zhang L, Huang Y, Wang J, Rong Y, Lai W, Zhang J, etal. Hierarchical flowerlike gold nanoparticles labelled immunochromatography test strip for highly sensitive detection of Escherichia coli O157:H7. Langmuir. 2015; 31: 5537–44,. doi: http://dx.doi.org/10.1021/acs.langmuir.5b00592.
- Andreu Z, Yáñez-Mó M. Tetraspanins in extracellular vesicle formation and function. Front Immunol. 2014; 5: 442,. doi: http://dx.doi.org/10.3389/fimmu.2014.00442.
- Zhao F, Sucker A, Horn S, Heeke C, Bielefeld N, Schrörs B, etal. Melanoma lesions independently acquire T-cell resistance during metastatic latency. Cancer Res. 2016. 76(15):4347–4358, doi: http://dx.doi.org/10.1158/0008-5472.CAN-16-0008.
- Gordón-Alonso M, Yañez-Mó M, Barreiro O, Álvarez S, Muñoz-Fernández MA, Valenzuela-Fernández A, etal. Tetraspanins CD9 and CD81 modulate HIV-1-induced membrane fusion. J Immunol. 2006; 177: 5129–37,. doi: http://dx.doi.org/10.4049/jimmunol.177.8.5129.
- Yanez-Mo M, Tejedor R, Rousselle P, Sanchez-Madrid F. Tetraspanins in intercellular adhesion of polarized epithelial cells: spatial and functional relationship to integrins and cadherins. J Cell Sci. 2001; 114: 577–87.
- Peñas PF, García-Díez A, Sánchez-Madrid F, Yáñez-Mó M. Tetraspanins are localized at motility-related structures and involved in normal human keratinocyte wound healing migration. J Invest Dermatol. 2000; 114: 1126–35.
- Ashiru O, Boutet P, Fernández-Messina L, Agüera-González S, Skepper JN, Valés-Gómez M, etal. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA*008 that is shed by tumor cells in exosomes. Cancer Res. 2010; 70: 481–9,. doi: http://dx.doi.org/10.1158/0008-5472.CAN-09-1688.
- Thobhani S, Attree S, Boyd R, Kumarswami N, Noble J, Szymanski M, etal. Bioconjugation and characterisation of gold colloid-labelled proteins. J Immunol Methods. 2010; 356: 60–9,. doi: http://dx.doi.org/10.1016/j.jim.2010.02.007.
- Driskell JD, Jones CA, Tompkins SM, Tripp RA. One-step assay for detecting influenza virus using dynamic light scattering and gold nanoparticles. Analyst. 2011; 136: 3083–90,. doi: http://dx.doi.org/10.1039/c1an15303j.
- Lozano-Ramos I, Bancu I, Oliveira-Tercero A, Armengo MP, Menezes-Neto A, Del Portillo HA, etal. Size-exclusion chromatography-based enrichment of extracellular vesicles from urine samples. J Extracell Vesicles. 2015; 4: 27369,. doi: http://dx.doi.org/10.3402/jev.v4.27369.
- Miller N, Miller JC. Statistics and chemometrics for analytical chemistry. 2000; Harlow: Pearson Education Limited. 121–3.
- Sódar BW, Kittel Á, Pálóczi K, Vukman KV, Osteikoetxea X, Szabó-Taylor K, etal. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Sci Rep. 2016; 6: 24316,. doi: http://dx.doi.org/10.1038/srep24316.