Abstract
In recent times the concept of infectious agents playing a role in cardiovascular disease has attracted much attention. Chronic oral disease such as periodontitis, provides a plausible route for entry of bacteria to the circulation. Upon entry to the circulation, the oral bacteria interact with platelets. It has been proposed that their ability to induce platelet aggregation and support platelet adhesion is a critical step in the pathogenesis of the infection process. Many published studies have demonstrated multiple mechanisms through which oral bacteria are able to bind to and activate platelets. This paper will review the various mechanisms oral bacteria use to interact with platelets.
Introduction
The primary function of platelets is their adhesion to endothelium or to matrix protein components at sites of the injured vessel wall in the initiation of haemostasis Citation1. However, platelets are poorly appreciated for their involvement in inflammatory or immune processes in host defence. In this regard, platelets share many similarities with leukocytes well characterised for their role in immuno-protection following invasion by foreign invaders. Just like leukocytes, platelets undergo chemotaxis Citation2, phagocytose foreign particles Citation3 and secrete a multitude of products including inflammatory mediators Citation4, cytokines Citation5 and antimicrobial peptides Citation6. Indeed the concept of platelets playing a role in immuno-protection is not a new theory. Studies by Clawson and White in the 1970s demonstrated that platelets were capable of recognising a foreign invader Citation7 Citation8 Citation9 Citation10 . In doing so, platelets expressed a number of highly adhesive membrane receptors capable of binding injured or infected tissues. Following this, platelets underwent many cytoskeletal rearrangements in the form of extending pseudopodia that stretched around the foreign invader which promoted engulfment, thus clearing the foreign invader from the bloodstream Citation3. Therefore, platelets may provide a bridge between innate immunity and host defence.
The conventional role of platelets in thrombosis
Platelets are small anucleate cell fragments of the larger haematopoietic precursor cell, the megakaryocyte Citation11. Being devoid of a nucleus, the platelet has no control of gene expression but has limited capabilities in translational protein synthesis Citation5. The primary role of platelets in haemostasis is to police the integrity of the endothelium to prevent blood loss Citation12 Citation13. Platelets circulate close to the endothelial cell surface as individual entities that ordinarily do not interact with any other cell types and without any stable interaction with endothelial cells as they exist in an anti-adhesive state. Upon trauma or injury to the vascular endothelium, platelets rapidly accumulate at the site of injury. Recruitment is a highly controlled event that is initiated by the adhesive interaction between the exposed extracellular matrix proteins in damaged endothelium and specific membrane receptors on the platelet. Adhesion to the extracellular matrix requires a synergistic function of several membrane receptors which ultimately results in platelet activation and aggregation. There are several different types of matrix proteins exposed upon vessel injury including collagen, vonWillebrand factor (vWf), fibronectin, laminin and thrombospondin. Other adhesive proteins such as fibrinogen/fibrin and vitronectin are not synthesised by endothelial cells, however, do bind to exposed matrix proteins increasing adhesiveness at the damaged site Citation14 Citation15.
The platelet expresses a number of membrane proteins with specific binding capabilities for one or more of these adhesive matrix proteins. The initial interaction of platelets with the injured vessel wall occurs between GPIb and immobilised vWf Citation16. This interaction initiates the tethering of circulating platelets to the vessel wall. Platelets typically ‘roll’ over the vWf in the direction of flow driven by shear forces experienced by the vasculature Citation1. A loss of interaction between GPIb and vWf on one side of the platelet leads to the formation of another GPIb–vWf interaction on the other side of the platelet which gives rise to a rolling phenomenon. Eventually the platelet will come to a complete stop due to firm adhesion to the injured part of the vessel. This firm adhesion is typically mediated by several membrane receptors, some of which will have become activated as a result of platelet rolling. The adhesion to the extracellular matrix proteins is a result of several platelet receptor interactions; the collagen receptors α2β1 Citation17 Citation18 Citation19 and GPVI Citation20 Citation21, the fibronectin receptor α5β1 Citation22 and the fibrinogen receptor GPIIb/IIIa Citation23. Once firmly adhered, the platelets rearrange cytoskeletal components which results in flattening or spreading of the platelet, this is essential in order to withstand the shear forces experienced in the vasculature Citation1.
At this stage, platelets undergo the release reaction where they release the contents of its stored intracellular granules. These granules contain proteins such as P-selectin which mediates adhesion of platelets to monocytes, neutrophils and lymphocytes, resulting in the formation of platelet leukocyte complexes Citation24 Citation25 Citation26 . The granules also contain many chemotactic agents which lead to the recruitment of various inflammatory cells; platelet derived growth factor (PDGF) and 12-hydroxyeicosatetraenoic acid (12-HETE) which recruit neutrophils Citation27 Citation28; platelet factor 4 and platelet derived histamine releasing factor (PDHRF) which recruit eosinophils in airway disease Citation29 Citation30; PDGF and transforming growth factor β (TGF-β) which recruit monocytes and macrophages and TGF-β which recruits fibroblasts Citation31 Citation32 Citation33 . Platelet granules also contain several mediators of tissue damage such as oxygen-free radicals and hydrolytic enzymes. Platelets also release cationic proteins that initiate vascular permeability and mediators that enhance aggregate formation such as adenosine diphosphate (ADP), serotonin (5-HT), Thromboxane A2 (TxA2) and platelet activating factor (PAF; Citation34,Citation35). More recently it has been shown that platelet granules contain many antimicrobial peptides such as beta-lysin, platelet microbial protein (PMP), neutrophil activating peptide (NAP-2), released upon activation normal T-cell expressed and secreted (RANTES) and fibrinopeptides A and B Citation36 Citation37 Citation38 Citation39 Citation40 .
The flattened part of the platelet forms a new surface for additional platelets to adhere, predominantly through GPIIb/IIIa crosslinking adjacent platelets through a fibrinogen bridge, resulting in aggregate formation. The final step sees an effective plug at the site of injury that is reinforced by the conversion of fibrinogen to fibrin through the coagulation cascade Citation14.
Role of platelets in infection
As well as their established role in haemostasis, there is growing evidence for a role for platelets in immunology. In fact it would appear that platelets have an important role to play in the response to infection. Clinical studies would suggest that serious infection is associated with thrombotic events Citation41 Citation42. Recent use of antibiotics was shown to be associated with myocardial infarction Citation43, although treatment of patients with myocardial infarction with antibiotics provided no benefit Citation44.
The oral bacterial burden has been shown to be associated with the extent of atherosclerosis Citation45 Citation46 and cardiovascular disease Citation47. Periodontal disease is also associated with ischaemic stroke Citation48 Citation49 and oral pathogens are also strongly associated with infective endocarditis Citation50.
Mechanisms of platelet–bacterial interactions
While it is clear that the increased oral bacteria burden is key in the increased risk of thrombotic events, it is not clear if this is due to increased inflammation leading to endothelial dysfunction Citation51 or is it due directly to the bacteria triggering thrombosis; however, as bacteria can trigger platelet activation, it is likely that this may play a role in triggering the thrombotic events Citation52.
Direct interaction
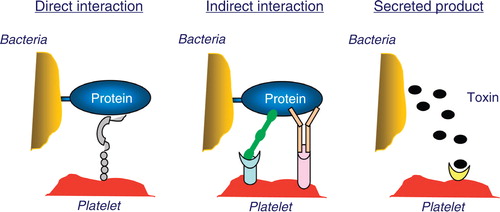
Bacteria can have proteins that can directly interact with a surface receptor on the platelet (). In this case they have ligand-mimetic domains that act as agonists on the platelet receptor. One such direct interaction is that with Streptococcus sanguinis which can directly interact with glycoprotein Ibα (GPIbα), the vWf receptor on the platelet Citation66. Other potential mediators of platelet activation are lipopolysaccharide (LPS) and lipoteichoic acid (LTA). LPS has been shown to activate platelets in a TLR4-dependent process Citation53 Citation54 Citation55 , however, some studies have failed to see this platelet activation Citation56.
Figure 1. Direct interaction: bacteria express proteins that interact directly with a platelet surface receptor. In this case, they have ligand-mimetic domain that act as agonist on the platelet receptor. Indirect interaction: bacteria bind a plasma protein that acts as a bridge and is a natural ligand for a platelet receptor. Secreted products: bacteria also have the potential to secrete products that can in turn directly activate platelets.
Indirect interaction (bridging protein)
One of the more common mechanisms for interacting with the platelet is the use of a bridging protein (). In this case bacteria bind a plasma protein that is a natural ligand for a platelet receptor e.g. Staphylococcus aureus, which has multiple mechanisms for interacting with platelets using a bridging ligand. Clumping factor A and B Citation57 and fibronectin binding protein Citation58 on S. aureus can both bind fibronectin and/or fibrinogen both of which are ligands for GPIIb/IIIa on the platelet. Helicobacter pylori can bind vWf which interacts with GPIbα on the platelet Citation59. The most common bridging molecule for bacteria to use is IgG. IgG bound to the bacterial surface can interact with the platelet FcγRIIa receptor and while it appears that this alone cannot stimulate platelet activation it acts in conjunction with other bridging molecules Citation52 Citation59 Citation60. In the absence of a second bridging, molecule bound antibody can trigger complement formation which can mediate platelet activation via complement receptors in conjunction with the FcγRIIa receptor Citation60 Citation61 Citation62 .
Indirect interaction (secretion)
Bacteria also have the potential to secrete products that can in turn activate platelets (). Porphyromonas gingivalis secretes gingipain, an enzyme that activates the thrombin receptor on platelets which leads to platelet activation Citation63 and Escherichia coli shiga toxin is associated with platelet activation Citation64 via a novel platelet glycosphingolipid Citation65.
Bacterial-induced platelet aggregation is different in some respects to that seen with other platelet agonists. Bacterial-induced aggregation is an all-or-nothing response, in that no matter what concentration of bacteria are added to a platelet preparation, the extent of aggregation will always be maximal (often less than that seen with other agonists) or else there is no aggregation. Unlike other agonists there is a lag time to aggregation. Adjusting the concentration of bacteria shortens the lag time to a minimum, but never eliminates it. There appears to be two categories of bacteria: those that have a short lag time of around 2–5 mins, e.g. S. aureus and those with a long lag time of 15–20 mins, e.g. S. sanguinis or Streptococcus gordonii. The short lag time usually indicates the presence of a direct interaction and is dependent on the levels of expression of the interacting protein on the bacterial surface Citation60. The long lag time usually indicates a complement-dependent aggregation process.
The interaction of platelets with bacteria is not simply one of activation as bacteria are able to either support platelet adhesion or induce platelet aggregation and these are distinct phenomena involving different bacterial proteins. The adhesion process can occur under static conditions or shear conditions and different proteins are often involved in each process Citation66.
Interaction of oral bacteria with platelets
Oral bacteria differ greatly in their ability to interact with platelets. Kerrigan and colleagues have proposed several different phenotypes for platelet interaction Citation67 Citation68. They identified strains of oral bacteria that support platelet adhesion and induce platelet aggregation with a short lag time (S. sanguinis strain 133-79) [Citation67, strains of bacteria that support platelet adhesion and induce platelet aggregation with a long lag time (S. gordonii strain DL1), strains that support platelet adhesion but do not induce platelet aggregation (S. gordonii strain Blackburn, S. sanguinis strain B5.7), strains that cannot support platelet adhesion but can induce platelet aggregation with a short lag time (S. gordonii strain Sk12), strains that cannot support platelet adhesion but can induce platelet aggregation with a long lag time (S. gordonii strain M99 and S. sanguinis strain 7863) and finally strains that cannot support platelet adhesion or induce platelet aggregation (S. gordonii strain M5 and S. sanguinis strain SK96) Citation68.
Several studies have now demonstrated that the platelet bacterial interaction may be a lot more complex than this model suggests. It appears that several bacteria have evolved in such a way that many interactions occur at the same time to mediate the same response. For example, it has been shown that S. gordonii uses three distinct interactions with the platelet to induce platelet aggregation, deletion of one protein interaction does not affect the aggregation, however, deletion of all three interactive proteins ablates the response. Furthermore, platelets exist in an environment where they are exposed to a range of shear stress. Several studies have characterised the interaction between platelet GPIbα and vWf under high shear or between platelet GPIIb/IIIa and fibrinogen under low shear.
Other studies suggest that the virulence of several strains of S. gordonii in the rat model of infective endocarditis does not correlate with their ability to just interact with platelets. Several strains of S. gordonii that were shown to interact with platelets were resistant to polymorphonuclear (PMN) leukocyte-dependent killing Citation69. These results suggest that the ability of S. gordonii to survive in PMN's following phagocytosis may be another important virulence determinant to consider. Therefore, studying the interaction between bacteria, platelets and PMN's is essential to get a more physiological representation of the real picture.
The interaction between streptococci and platelets
At sites of trauma in the oral cavity, streptococci can gain access to the bloodstream and interact with circulating platelets Citation70. Septic thrombi (oral streptococci encased in activated platelets) can be found at several sites in the circulatory system, for example, on heart valves, endocardium or indeed in atherosclerotic plaques.
Several approaches have been used to identify the factor responsible for inducing thrombus or aggregate formation (). Early studies demonstrated that bacteria can either secrete products capable of activating platelets or bind directly to platelets to induce an intracellular signal, either way the outcome is the same – aggregate formation.
Table 1. Identified molecular interactions between oral pathogens and platelets.
S. sanguinis was the first oral bacteria shown to have the capability to induce platelet aggregation and to support platelet adhesion Citation67 Citation71 Citation72. S. sanguinis-induced platelet aggregation appeared to be dependent on calcium and fibrinogen and in some cases non-specific antibody, whereas platelet adhesion to S. sanguinis occurred independently of calcium, fibrinogen or non-specific antibody. This led to the initial classification of the response, a class 1 component-mediated adhesion to the platelet, a class 2 component mediates a calcium-dependent activation and a class 3 component amplifies the response.
Early studies identified components of S. sanguinis thought to play a role in binding to and activating platelets. Platelet-associated activating protein (PAAP) was first identified in 1990 Citation73. PAAP is a 115 kDa glycoprotein, which contains a collagen-like epitope and is constitutively expressed on the surface of S. sanguinis Citation74 Citation75. PAAP is possibly environmentally regulated during infection in response to high temperature (fever) or exposed collagen (exposed on damaged heart valves) Citation70. Partial sequence alignment shows homology to the heat shock family of proteins of Myobacterium tuberculosis and E. coli Citation76. PAAP interacts with an unknown platelet receptor which is capable of transducing an intracellular signal leading to platelet activation. Several attempts that have been made to identify the platelet receptor and potential candidates include the integrin α2β1 Citation77 or an integrin-associated protein of 175 kDa Citation78. As not all donors respond to S. sanguinis, it was reported that the ability of PAAP to induce platelet aggregation may be donor specific Citation79. Platelets release several mediators from stored granules when they become activated of which includes ATP. S. sanguinis has been shown to have ectoATPase activity and is capable of hydrolysing ATP to ADP Citation80 Citation81. There are several ADP receptors expressed on the platelets, therefore the conversion of ATP to ADP may serve as an amplification step essential for platelet aggregate formation.
Further classification of the streptococcal-induced aggregation response has been described by Kerrigan et al. Type 1 streptococci induce platelet aggregation with a short lag time (<5 mins), type 2 streptococcus induce platelet aggregation with a long lag time (10–15 mins) and finally type 3 streptococci fail to induce platelet aggregation (>20 mins). S. sanguinis strain 133-79 is an example of type 1 streptococcus and interacts with platelets via platelet receptor glycoprotein Ibα (the vWf receptor) Citation67. S. sanguinis binds to the N-terminal portion of GPIbα between amino acids 1 and 282. Platelets from patients with Bernard Soulier Syndrome (patients who fail to express GPIbα on the surface of their platelets) fail to interact with S. sanguinis Citation67. S. sanguinis lysates were passed through a GPIbα affinity column in order to identify the S. sanguinis protein binding to GPIbα Citation77. This led to the identification of a serine-rich glycoprotein called serine-rich protein A (SrpA). Deletion of SrpA failed to have any effect on the percent platelet aggregation, however, significantly prolonged the lag time. Furthermore, deletion of SrpA from S. sanguinis significantly reduced its ability to support platelet adhesion Citation82. These results highlight the complexity of the interactions between S. sanguinis and platelets as multiple proteins must be involved in supporting platelet adhesion and inducing platelet aggregation.
S. sanguinis strain NCTC 7863 is an example of a type 2 streptococcus. Ford et al. demonstrated that S. sanguinis strain NCTC 7863 induced aggregation of normal platelets suspended in plasma, removal of plasma protein abolished aggregation Citation62. The long lag time of S. sanguinis strain 7863 was progressively shortened by incubating the bacteria in plasma for increasing lengths of time prior to addition to platelets. Since the lag time was due to addition of plasma rather than donor platelets, it was suggested that the rate limiting step for platelet aggregation was assembly or binding of plasma factors on the bacterial cell surface. Subsequent experiments demonstrated that complement assembly on the surface of the bacteria was necessary for aggregation of platelets. Further experiments demonstrated that complement assembly was not enough to trigger platelet aggregation which led to the discovery that IgG and fibrinogen was necessary to complete the aggregation process Citation61. More recently, McNicol and colleagues demonstrated that depletion of S. sanguinis-specific antibodies from plasma prevented platelet aggregation Citation83. Addition of antibodies led to rapid phosphorylation of the platelet antibody receptor, FcγRIIa, occurred following S. sanguinis binding Citation84.
S. gordonii is a distinct but close relative of S. sanguinis and has been extensively examined for its interactions with platelets. Platelet binding to S. gordonii is predominantly mediated by the cell surface glycoprotein, glycosylated streptococcal protein B (GspB). This is a large protein of over 3,000 amino acids and has a predicted molecular weight of 286 kDa. It is heavily glycosylated, primarily with glucose and glucosamine in the cytoplasm and is transported to the cell surface via an accessory system comprising of the SecA2 and SecY2 proteins Citation85 Citation86. Sequence alignment demonstrates that GspB is a member of a highly glycosylated serine-rich family of proteins which include S. gordonii haemaglutinin streptococcal antigen (Hsa) Citation87 Citation88, S. sanguinis SrpA Citation82 and Streptococcus parasanguinis fimbria associated protein 1 (FAP1) Citation89. Hsa is a GspB homologue of S. gordonii that was originally characterised as a sialic acid binding haemaglutinin Citation90. Like GspB, Hsa has also been shown to bind to GPIbα on human platelets Citation91. Interestingly, even though both GspB and Hsa bind sialic acid residues on the platelet GPIbα, there are subtle differences in the binding specificities. For example, GspB specifically binds O-linked sialic residues and the membrane proximal mucin-rich core of GPIbα Citation92, whereas Hsa specifically binds N-linked sialic residues on GPIbα Citation92 and GPIIb/IIIa Citation91. Similar to the effect seen using the S. sanguinis SrpA mutant, deletion of either GspB or Hsa from S. gordonii had no effect on platelet aggregation, however, significantly reduced platelet adhesion Citation68. Thus, other factors are most likely involved in supporting platelet adhesion and inducing platelet aggregation.
Possibly the best characterised streptococcal adhesins are those of the antigen I/II family of proteins. Initially identified in Streptococcus mutants, antigens I/II have now been detected on most oral streptococcal species Citation93. In S. gordonii, the antigen I/II polypeptides have been designated SspA (172 kDa) and SspB (164 kDa). These polypeptides are oligospecific adhesins recognising multiple ligands such as collagen type 1 Citation94, beta 1 integrins Citation93, salivary agglutinin glycoprotein (gp-340; Citation95), as well as several other bacteria including P. gingivalis, Candida albicans and Actinomyces naeslundii Citation96 Citation97 Citation98 . Using a proteomic approach to identify differential cell wall protein expression between an aggregating strain of S. gordonii (DL1) and a non-aggregating strain of S. gordonii (Blackburn), Kerrigan and colleagues identified SspA and SspB as proteins differentially expressed Citation68. Deletion of SspA and SspB from S. gordonii DL1 induced platelet aggregation and supported platelet adhesion as normal. However, deletion of SspA and SspB along with Hsa abolished platelet aggregation and reduced platelet adhesion by 50%, similar to the Hsa mutant alone. Furthermore, overexpression of SspA and SspB in the non-platelet reactive surrogate host Lactococcus lactis, induced platelet aggregation but failed to support platelet adhesion Citation68. These results suggest that S. gordonii-induced platelet aggregation is a multifactorial event mediated by several surface proteins.
A major limitation in our current understanding in platelet–bacterial interactions stems from the fact that all of the previous studies have been carried out under static conditions. It has been argued in the literature that data obtained in vitro using static binding assays may not be relevant to the fluid dynamic environment encountered in the vasculature Citation99. There are a growing number of papers in the literature in the last number of years suggesting that the local fluid environment of the circulation critically affects the molecular pathways of cell–cell interactions Citation100 Citation101. Platelets perfused over immobilised S. sanguinis or S. gordonii interacted with a typical rolling behaviour followed by firm adhesion. Deletion of SrpA or Hsa abolished this rolling behaviour, suggesting that these proteins are essential for the initial contact of platelets with the bacteria Citation68 Citation82. Platelet rolling with subsequent firm adhesion is characteristic of the platelet interaction with vWf and collagen at the site of injury. Typically, platelets roll along immobilised vWf under high shear conditions. The high shear is necessary to induce a conformational change in vWf thereby making it recognisable by its corresponding receptor on platelets, GPIbα. As platelets roll along immobilised S. gordonii or S. sanguinis under low shear conditions, it suggests that the bacterial proteins involved, Hsa or SrpA, are in a conformation already recognisable by platelet GPIbα and possibly mediate the initial interaction leading to thrombus formation. Furthermore, deletion of Hsa from S. gordonii reduced thrombus formation in a catheterised rat model of infective endocarditis Citation102. The S. sanguinis or S. gordonii protein that mediates firm adhesion has yet to be identified.
Streptococcus mitis is less well characterised in its interaction with platelets. In 2001, Bensing et al. described two surface expressed proteins that were involved in binding platelets namely, PblA (107 kDa) and PblB (121 kDa; Citation103,104). The genes encoding these proteins reside in the temperate bacteriophage SM1, a member of the siphoviridae family. PblA and PblB are unusual because neither of the protein expresses homology to any other bacterial adhesins described, however, do resemble structural components of bacteriophages Citation104 Citation105. Typically, PblA and PblB are released from S. mitis are then capable of binding to the bacterial cell wall via the choline residues. Once presented on the cell wall they are capable of mediating platelet binding and also enhance activation of platelet aggregation. Deletion of PblA or PblB reduced thrombus formation in an animal model of infective endocarditis, therefore suggesting that these proteins may be important for platelet deposition onto the infected valve Citation105.
S. mutans has been shown to induce platelet aggregation and support adhesion of human platelets via a soluble serotype-specific rhamnose–glucose polymer Citation106. This is a direct interaction with the platelets as S. mutans induced aggregation in a plasma-free environment in a rapid and saturable manner. Rhamnose polymers are also found in other glycopeptides such as ristocetin. Ristocetin is used to induce agglutination of platelets. The mechanism for ristocetin-induced agglutination involves the binding and bridging of the vWF to platelet GPIbα. Cleavage of the rhamnose tetrasaccharide of ristocetin abolished its ability to induce platelet aggregation in plasma. This suggests that the polymer rhamnose plays an important role in platelet aggregation. Furthermore, depletion of the rhamnose–glucose polymers from the cell wall of S. mutans, aggregation was reduced down to 50% of the level of the wild-type strain. These results suggest that the rhamnose backbone play an essential role in inducing platelet aggregation Citation106.
Streptococcus oralis and S. parasanguinis do support platelet adhesion, however, not as efficiently as S. gordonii or S. sanguinis Citation107 Citation108. S. oralis and S. parasanguinis express a protein homologous to the highly glycosylated serine-rich family of proteins, similar to S. gordonii Hsa and S. sanguis SrpA Citation109. The lack of efficient adherence to platelets may be due to the absence of the sialic acid-binding moieties necessary to interact with platelet GPIbα. Alternatively the protein may not be readily accessible on the cell surface, for example, if it is blocked by other surface structures such as a capsule Citation109. Some strains of S. oralis and S. parasanguinis did induce platelet aggregation, however, the mechanisms involved have not been elucidated.
The interaction between other oral bacteria and platelets
P. gingivalis contains small vesicles on the outside of their surface that have been shown to interact with many host cells. These vesicles contain factors that have cytotoxic, haemagglutinin, proteolytic and platelet activating activities. Pham et al. identified that gingipain-R was the factor responsible for inducing platelet aggregation Citation110. Lourbakos et al. reported that two forms of purified gingipain-R (50 kDa RgpB and a 95 kDa RgpA) caused aggregation of human platelets with efficiency similar to thrombin through the activation of protease-activated receptors 1 and 4 (PAR1 and PAR4; 63). Naito and colleagues demonstrated that P. gingivalis-induced platelet aggregation depends on HagA-encoding genes that intragenically code for adhesins such as Hgp44 Citation111. Hgp44 adhesin on the bacterial cell surface is processed from haemagglutinin A by the Rgp and Kgp proteinases. Specific antibodies against P. gingivalis binds to the platelet antibody receptor FcγRIIa is also thought to be an essential step leading to platelet aggregation. Cross-reacting antibodies recognising platelet GPIbα prevented platelet aggregation, suggesting that P. gingivalis may be binding to GPIbα. More recent studies investigated the ultrastructural properties of P. gingivalis-induced platelet aggregation Citation112. This study demonstrated that platelets have the ability to engulf or phagocytose P. gingivalis during or after platelet activation thereby removing the bacteria from the circulation.
Clinical implications
It is clear that oral bacteria especially Streptococci spp. and P. gingivalis are capable of triggering platelet activation. Thrombus formation plays a critical role in infective endocarditis, atherosclerosis, myocardial infarction and stroke, all of which have been associated with periodontal disease. Thus, it is likely that inflammation in the oral cavity allows oral bacteria to access the circulation. Once in the blood, they interact with platelets forming platelet–bacterial aggregates, which can bind to heart valves or interact with sites of atherosclerosis. In the case of a high level of infection, it can lead to septicaemia with systemic activation of platelets, which are cleared from the circulation leading to thrombocytopenia. Reducing bacterial load in the mouth is an important step to preventing thrombus formation, as lesser bacteria will be available to enter the circulation. Several strategies have been employed to address this in the form of oral healthcare programmes. However, in more severe cases root canal or tooth extraction may be more appropriate.
Conventional therapy focuses on the use of antibiotics to treat overt infection and as prophylaxis for patients at high risk, such as patients with damaged or replacement cardiac valves undergoing dental procedures. However, the increase in the incidence of antibiotic resistant strains of bacteria has made it more difficult to treat these infections. The latest guidelines from American Heart Association no longer recommend the prophylactic use of antibiotics except in those patients with very high risk of complications Citation113.
Targeting the platelet–bacterial interaction may also play an important role in the treatment and prevention of the serious complications of infection. Thus, if the interaction between streptococci and platelets could be prevented, the thrombotic complications of infection could be reduced. While septicaemia may still occur, it would prevent the formation of infected thrombi on the cardiac valve and the formation of septic emboli. It would also prevent the thrombocytopenia associated with sepsis.
There are two approaches to targeting the bacteria–platelet interaction: blocking the bacterial protein or the platelet receptor. The difficulty with blocking the bacterial proteins is that there are so many different proteins involved as each bacterial species uses a different protein and some use three or four different proteins. Targeting the platelet may be a more effective approach as all of the bacteria that activate platelets do so by interacting with three platelet receptors: GPIb, GPIIb/IIIa and FcγRIIa. The FcγRIIa receptor is a very promising target as recent reports in the literature suggest that FcγRIIa acts as a co-signalling receptor for platelet membrane receptors. These reports demonstrate that inhibition of FcγRIIa blocks downstream signal events upon engagement of either GPIIb/IIIa or GPIbα Citation114 thus preventing platelet activation. An added advantage is that unlike inhibition of GPIbα or GPIIb/IIIa, inhibition of FcγRIIa does not affect normal platelet function. As a result there is a lot of interest in developing small molecule inhibitors of FcγRIIa. However, as the Fcγ receptor is a crucial player in leukocyte activation in the immune system, small molecule inhibitors may adversely affect the immune function of these cells. In vivo experiments will address this caveat.
When oral bacteria gain entry to the circulation as well as coming into contact with platelets, they also come into contact with polymorphonucleocytes such as neutrophils and monocytes. Recent studies have demonstrated that platelets and monocytes from patients with periodontitis are more sensitive to activation with oral bacteria over control patients Citation115. In addition, platelet complexes with neutrophils and monocytes bound more oral bacteria than uncomplexed neutrophils or monocytes Citation51. The net result of these platelet leukocyte complexes is cellular priming with increased release of inflammatory mediators and unwanted accelerated thrombosis at sites of vascular injury.
Conflict of interest and funding
There is no conflict of interest in the present study for any of the authors.
References
- Ruggeri ZM. Platelet adhesion under flow. Microcirculation. 2009; 16: 58–83.
- Clemetson KJ, Clemetson JM, Proudfoot AE, Power CA, Baggiolini M, Wells TN. Functional expression of CCR1, CCR3, CCR4, and CXCR4 chemokine receptors on human platelets. Blood. 2000; 96: 4046–54.
- Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. 2002; 99: 4021–9.
- Klinger MH. Platelets and inflammation. Anat Embryol (Berlin). 1997; 196: 1–11.
- Lindemann S, Tolley ND, Eyre JR, Kraiss LW, Mahoney TM, Weyrich AS. Integrins regulate the intracellular distribution of eukaryotic initiation factor 4E in platelets. A checkpoint for translational control. J Biol Chem. 2001; 276: 33947–51.
- Yeaman MR, Puentes SM, Norman DC, Bayer AS. Partial characterization and staphylocidal activity of thrombin-induced platelet microbicidal protein. Infect Immun. 1992; 60: 1202–9.
- Clawson CC, White JG. Platelet interaction with bacteria. I. Reaction phases and effects of inhibitors. Am J Pathol. 1971; 65: 367–80.
- Clawson CC, White JG. Platelet interaction with bacteria. II. Fate of the bacteria. Am J Pathol. 1971; 65: 381–97.
- Clawson CC. Platelet interaction with bacteria. 3. Ultrastructure. Am J Pathol. 1973; 70: 449–71.
- Clawson CC, Rao GH, White JG. Platelet interaction with bacteria. IV. Stimulation of the release reaction. Am J Pathol. 1975; 81: 411–20.
- Patel SR, Hartwig JH, Italiano Jr, JE. The biogenesis of platelets from megakaryocyte proplatelets. J Clin Invest. 2005; 115: 3348–54.
- Weiss HJ. Platelet physiology and abnormalities of platelet function (first of two parts). N Engl J Med. 1975; 293: 531–41.
- Weiss HJ. Platelet physiology and abnormalities of platelet function (second of two parts). N Engl J Med. 1975; 293: 580–8.
- Ruggeri ZM, Orje JN, Habermann R, Federici AB, Reininger AJ. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood. 2006; 108: 1903–10.
- Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007; 100: 1673–85.
- Chesterman CN, Berndt MC. Platelet and vessel wall interaction and the genesis of atherosclerosis. Clin Haematol. 1986; 15: 323–53.
- Santoro SA. Identification of a 160,000 dalton platelet membrane protein that mediates the initial divalent cation-dependent adhesion of platelets to collagen. Cell. 1986; 46: 913–20.
- Kunicki TJ, Nugent DJ, Staats SJ, Orchekowski RP, Wayner EA, Carter WG. The human fibroblast class II extracellular matrix receptor mediates platelet adhesion to collagen and is identical to the platelet glycoprotein Ia-IIa complex. J Biol Chem. 1988; 263: 4516–9.
- Nieuwenhuis HK, Akkerman JW, Houdijk WP, Sixma JJ. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature. 1985; 318: 470–2.
- Moroi M, Jung SM, Okuma M, Shinmyozu K. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989; 84: 1440–5.
- Moroi M, Jung SM, Shinmyozu K, Tomiyama Y, Ordinas A, Diaz-Ricart M. Analysis of platelet adhesion to a collagen-coated surface under flow conditions: the involvement of glycoprotein VI in the platelet adhesion. Blood. 1996; 88: 2081–92.
- Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998; 94: 657–66.
- Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996; 84: 289–97.
- Diacovo TG, Puri KD, Warnock RA, Springer TA, von Andrian UH. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 1996; 273: 252–5.
- Diacovo TG, Roth SJ, Buccola JM, Bainton DF, Springer TA. Neutrophil rolling, arrest, and transmigration across activated, surface-adherent platelets via sequential action of P-selectin and the beta 2-integrin CD11b/CD18. Blood. 1996; 88: 146–57.
- Larsen E, Celi A, Gilbert GE, Furie BC, Erban JK, Bonfanti R, et al.. PADGEM protein: a receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989; 59: 305–12.
- Herd CM, Page CP. Pulmonary immune cells in health and disease: platelets. Eur Respir J. 1994; 7: 1145–60.
- Mannaioni PF, Di Bello MG, Masini E. Platelets and inflammation: role of platelet-derived growth factor, adhesion molecules and histamine. Inflamm Res. 1997; 46: 4–18.
- Frigas E, Gleich GJ. The eosinophil and the pathophysiology of asthma. J Allergy Clin Immunol. 1986; 77: 527–37.
- Brindley LL, Sweet JM, Goetzl EJ. Stimulation of histamine release from human basophils by human platelet factor 4. J Clin Invest. 1983; 72: 1218–23.
- Deuel TF, Senior RM, Huang JS, Griffin GL. Chemotaxis of monocytes and neutrophils to platelet-derived growth factor. J Clin Invest. 1982; 69: 1046–9.
- Tzeng DY, Deuel TF, Huang JS, Baehner RL. Platelet-derived growth factor promotes human peripheral monocyte activation. Blood. 1985; 66: 179–83.
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, et al.. Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci USA. 1987; 84: 5788–92.
- McIntyre TM, Prescott SM, Weyrich AS, Zimmerman GA. Cell–cell interactions: leukocyte–endothelial interactions. Curr Opin Hematol. 2003; 10: 150–8.
- Patrono C, Patrignani P, Garcia Rodriguez LA. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J Clin Invest. 2001; 108: 7–13.
- Kameyoshi Y, Dorschner A, Mallet AI, Christophers E, Schroder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med. 1992; 176: 587–92.
- Krijgsveld J, Zaat SA, Meeldijk J, van Veelen PA, Fang G, Poolman B, et al.. Thrombocidins, microbicidal proteins from human blood platelets, are C-terminal deletion products of CXC chemokines. J Biol Chem. 2000; 275: 20374–81.
- Yeaman MR, Tang YQ, Shen AJ, Bayer AS, Selsted ME. Purification and in vitro activities of rabbit platelet microbicidal proteins. Infect Immun. 1997; 65: 1023–31.
- Johnson FB, Donaldson DM. Purification of staphylocidal beta-lysin from rabbit serum. J Bacteriol. 1968; 96: 589–95.
- Donaldson DM, Tew JG. Beta-lysin of platelet origin. Bacteriol Rev. 1977; 41: 501–13.
- Levine RL, LeClerc JR, Bailey JE, Monberg MJ, Sarwat S. Venous and arterial thromboembolism in severe sepsis. Thromb Haemost. 2008; 99: 892–8.
- Lindsberg PJ, Grau AJ. Inflammation and infections as risk factors for ischemic stroke. Stroke. 2003; 34: 2518–32.
- Meier CR, Derby LE, Jick SS, Vasilakis C, Jick H. Antibiotics and risk of subsequent first-time acute myocardial infarction. JAMA. 1999; 281: 427–31.
- Zahn R, Schneider S, Frilling B, Seidl K, Tebbe U, Weber M, et al.. Antibiotic therapy after acute myocardial infarction: a prospective randomized study. Circulation. 2003; 107: 1253–9.
- Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DRJr, Sacco RL, et al.. Periodontal microbiota and carotid intima-media thickness: the oral infections and vascular disease epidemiology study (INVEST). Circulation. 2005; 111: 576–82.
- Tonetti MS. Periodontitis and risk for atherosclerosis: an update on intervention trials. J Clin Periodontol. 2009; 36: 15–9.
- Beck JD, Offenbacher S. Systemic effects of periodontitis: epidemiology of periodontal disease and cardiovascular disease. J Periodontol. 2005; 76: 2089–100.
- Dorfer CE, Becher H, Ziegler CM, Kaiser C, Lutz R, Jorss D, et al.. The association of gingivitis and periodontitis with ischemic stroke. J Clin Periodontol. 2004; 31: 396–401.
- Grau AJ, Becher H, Ziegler CM, Lichy C, Buggle F, Kaiser C, et al.. Periodontal disease as a risk factor for ischemic stroke. Stroke. 2004; 35: 496–501.
- Moreillon P, Que YA. Infective endocarditis. Lancet. 2004; 363: 139–49.
- Nicu EA, Van der Velden U, Nieuwland R, Everts V, Loos BG. Elevated platelet and leukocyte response to oral bacteria in periodontitis. J Thromb Haemost. 2009; 7: 162–70.
- Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol. 2006; 4: 445–57.
- Zhao L, Ohtaki Y, Yamaguchi K, Matsushita M, Fujita T, Yokochi T, et al.. LPS-induced platelet response and rapid shock in mice: contribution of O-antigen region of LPS and involvement of the lectin pathway of the complement system. Blood. 2002; 100: 3233–9.
- Wachowicz B, Saluk J, Kaca W. Response of blood platelets to Proteus mirabilis lipopolysaccharide. Microbiol Immunol. 1998; 42: 47–9.
- Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, et al.. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood. 2006; 108: 167–76.
- Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, et al.. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost. 2005; 94: 831–8.
- O'Brien L, Kerrigan SW, Kaw G, Hogan M, Penades J, Litt D, et al.. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol Microbiol. 2002; 44: 1033–44.
- Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, et al.. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol Microbiol. 2006; 59: 212–30.
- Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, et al.. Helicobacter pylori binds von Willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology. 2003; 124: 1846–54.
- Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, et al.. Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol. 2005; 57: 804–18.
- Ford I, Douglas CW, Cox D, Rees DG, Heath J, Preston FE. The role of immunoglobulin G and fibrinogen in platelet aggregation by Streptococcus sanguis. Br J Haematol. 1997; 97: 737–46.
- Ford I, Douglas CW, Heath J, Rees C, Preston FE. Evidence for the involvement of complement proteins in platelet aggregation by Streptococcus sanguis NCTC 7863. Br J Haematol. 1996; 94: 729–39.
- Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R, et al.. Activation of protease-activated receptors by gingipains from Porphyromonas gingivalis leads to platelet aggregation: a new trait in microbial pathogenicity. Blood. 2001; 97: 3790–7.
- Proulx F, Seidman EG, Karpman D. Pathogenesis of Shiga toxin-associated hemolytic uremic syndrome. Pediatr Res. 2001; 50: 163–71.
- Cooling LL, Walker KE, Gille T, Koerner TA. Shiga toxin binds human platelets via globotriaosylceramide (Pk antigen) and a novel platelet glycosphingolipid. Infect Immun. 1998; 66: 4355–66.
- Kerrigan SW, Clarke N, Loughman A, Meade G, Foster TJ, Cox D. Molecular basis for Staphylococcus aureus-mediated platelet aggregate formation under arterial shear in vitro. Arterioscler Thromb Vasc Biol. 2008; 28: 335–40.
- Kerrigan SW, Douglas I, Wray A, Heath J, Byrne MF, Fitzgerald D, et al.. A role for glycoprotein Ib in Streptococcus sanguis-induced platelet aggregation. Blood. 2002; 100: 509–16.
- Kerrigan SW, Jakubovics NS, Keane C, Maguire P, Wynne K, Jenkinson HF, et al.. Role of Streptococcus gordonii surface proteins SspA/SspB and Hsa in platelet function. Infect Immun. 2007; 75: 5740–7.
- Young Lee S, Cisar JO, Bryant JL, Eckhaus MA, Sandberg AL. Resistance of Streptococcus gordonii to polymorphonuclear leukocyte killing is a potential virulence determinant of infective endocarditis. Infect Immun. 2006; 74: 3148–55.
- Heimdahl A. Hall G. Hedberg M. Sandberg H. Soder P.O. Tuner K, et al.. Detection and quantitation by lysis-filtration of bacteremia after different oral surgical procedures. J Clin Microbiol. 1990; 28: 2205–9.
- Scheld WM, Valone JA, Sande MA. Bacterial adherence in the pathogenesis of endocarditis. Interaction of bacterial dextran, platelets, and fibrin. J Clin Invest. 1978; 61: 1394–404.
- Herzberg MC, Brintzenhofe KL, Clawson CC. Aggregation of human platelets and adhesion of Streptococcus sanguis. Infect Immun. 1983; 39: 1457–69.
- Erickson PR, Herzberg MC. Purification and partial characterization of a 65-kDa platelet aggregation-associated protein antigen from the surface of Streptococcus sanguis. J Biol Chem. 1990; 265: 14080–7.
- Erickson PR, Herzberg MC. Evidence for the covalent linkage of carbohydrate polymers to a glycoprotein from Streptococcus sanguis. J Biol Chem. 1993; 268: 23780–3.
- Erickson PR, Herzberg MC, Tierney G. Cross-reactive immunodeterminants on Streptococcus sanguis and collagen. Predicting a structural motif of platelet-interactive domains. J Biol Chem. 1992; 267: 10018–23.
- Herzberg MC. Platelet-streptococcal interactions in endocarditis. Crit Rev Oral Biol Med. 1996; 7: 222–36.
- Soberay AH, Herzberg MC, Rudney JD, Nieuwenhuis HK, Sixma JJ, Seligsohn U. Responses of platelets to strains of Streptococcus sanguis: findings in healthy subjects, Bernard-Soulier, Glanzmann's, and collagen-unresponsive patients. Thromb Haemost. 1987; 57: 222–5.
- Gong K, Wen DY, Ouyang T, Rao AT, Herzberg MC. Platelet receptors for the Streptococcus sanguis adhesin and aggregation-associated antigens are distinguished by anti-idiotypical monoclonal antibodies. Infect Immun. 1995; 63: 3628–33.
- Herzberg MC, Nobbs A, Tao L, Kilic A, Beckman E, Khammanivong A, et al.. Khammanivong, et al. Oral streptococci and cardiovascular disease: searching for the platelet aggregation-associated protein gene and mechanisms of Streptococcus sanguis-induced thrombosis. J Periodontol. 2005; 76: 2101–5.
- Herzberg MC, Brintzenhofe KL. ADP-like platelet aggregation activity generated by viridans streptococci incubated with exogenous ATP. Infect Immun. 1983; 40: 120–5.
- MacFarlane GD, Sampson DE, Clawson DJ, Clawson CC, Kelly KL, Herzberg MC. Herzberg. Evidence for an ecto-ATPase on the cell wall of Streptococcus sanguis. Oral Microbiol Immunol. 1994; 9: 180–5.
- Plummer C. Wu H. Kerrigan S.W. Meade G. Cox D. Ian Douglas. C.W. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol. 2005; 129: 101–9.
- McNicol A. Zhu R. Pesun R. Pampolina C. Jackson E.C. Bowden G.H, et al.. A role for immunoglobulin G in donor-specific Streptococcus sanguis-induced platelet aggregation. Thromb Haemost. 2006; 95: 288–93.
- Pampolina C, McNicol A. Streptococcus sanguis-induced platelet activation involves two waves of tyrosine phosphorylation mediated by FcgammaRIIA and alphaIIbbeta3. Thromb Haemost. 2005; 93: 932–9.
- Takahashi Y, Yajima A, Cisar JO, Konishi K. Functional analysis of the Streptococcus gordonii DL1 sialic acid-binding adhesin and its essential role in bacterial binding to platelets. Infect Immun. 2004; 72: 3876–82.
- Bensing BA, Sullam PM. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol. 2002; 44: 1081–94.
- Takahashi Y, Sandberg AL, Ruhl S, Muller J, Cisar JO. A specific cell surface antigen of Streptococcus gordonii is associated with bacterial hemagglutination and adhesion to alpha2-3-linked sialic acid-containing receptors. Infect Immun. 1997; 65: 5042–51.
- Takahashi Y, Konishi K, Cisar JO, Yoshikawa M. Identification and characterization of Hsa, the gene encoding the sialic acid-binding adhesin of Streptococcus gordonii DL1. Infect Immun. 2002; 70: 1209–18.
- Wu H, Zeng M, Fives-Taylor P. The glycan moieties and the N-terminal polypeptide backbone of a fimbria-associated adhesin, Fap1, play distinct roles in the biofilm development of Streptococcus parasanguinis. Infect Immun. 2007; 75: 2181–8.
- Takahashi Y, Ruhl S, Yoon JW, Sandberg AL, Cisar JO. Adhesion of viridans group streptococci to sialic acid-, galactose- and N-acetylgalactosamine-containing receptors. Oral Microbiol Immunol. 2002; 17: 257–62.
- Yajima A, Takahashi Y, Konishi K. Identification of platelet receptors for the Streptococcus gordonii DL1 sialic acid-binding adhesin. Microbiol Immunol. 2005; 49: 795–800.
- Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, Lopez JA, et al.. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol. 2005; 58: 380–92.
- Nobbs AH, Shearer BH, Drobni M, Jepson MA, Jenkinson HF. Adherence and internalization of Streptococcus gordonii by epithelial cells involves beta1 integrin recognition by SspA and SspB (antigen I/II family) polypeptides. Cell Microbiol. 2007; 9: 65–83.
- Heddle C, Nobbs AH, Jakubovics NS, Gal M, Mansell JP, Dymock D, et al.. Host collagen signal induces antigen I/II adhesin and invasin gene expression in oral Streptococcus gordonii. Mol Microbiol. 2003; 50: 597–607.
- Prakobphol A, Xu F, Hoang VM, Larsson T, Bergstrom J, Johansson I, et al.. Salivary agglutinin, which binds Streptococcus mutans and Helicobacter pylori, is the lung scavenger receptor cysteine-rich protein gp-340. J Biol Chem. 2000; 275: 39860–6.
- Jakubovics NS, Stromberg N, van Dolleweerd CJ, Kelly CG, Jenkinson HF. Differential binding specificities of oral streptococcal antigen I/II family adhesins for human or bacterial ligands. Mol Microbiol. 2005; 55: 1591–605.
- Egland PG, Du LD, Kolenbrander PE. Identification of independent Streptococcus gordonii SspA and SspB functions in coaggregation with Actinomyces naeslundii. Infect Immun. 2001; 69: 7512–6.
- Demuth DR, Iroine DC, Costerton JW, Cook GS, Lamont RJ. Discrete protein determinant directs the species-specific adherence of Porphyromonas gingivalis to oral Streptococci. Infect Immun. 2001; 69: 5736–41.
- Varki A. Selectin ligands. Proc Natl Acad Sci USA. 1994; 91: 7390–7.
- Lehoux S, Tedgui A. Shear and signal transduction in the endothelial cell. Med Sci (Paris). 2004; 20: 551–6.
- Bahou WF, Scudder L, Rubenstein D, Jesty J. A shear-restricted pathway of platelet procoagulant activity is regulated by IQGAP1. J Biol Chem. 2004; 279: 22571–7.
- Takahashi Y, Takashima E, Shimazu K, Yagishita H, Aoba T, Konishi K. Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by Streptococcus gordonii DL1. Infect Immun. 2006; 74: 740–3.
- Bensing BA, Rubens CE, Sullam PM. Genetic loci of Streptococcus mitis that mediate binding to human platelets. Infect Immun. 2001; 69: 1373–80.
- Bensing BA, Siboo IR, Sullam PM. Proteins PblA and PblB of Streptococcus mitis, which promote binding to human platelets, are encoded within a lysogenic bacteriophage. Infect Immun. 2001; 69: 6186–92.
- Mitchell J, Siboo IR, Takamatsu D, Chambers HF, Sullam PM. Mechanism of cell surface expression of the Streptococcus mitis platelet binding proteins PblA and PblB. Mol Microbiol. 2007; 64: 844–57.
- Chia JS, Lin YL, Lien HT, Chen JY. Platelet aggregation induced by serotype polysaccharides from Streptococcus mutans. Infect Immun. 2004; 72: 2605–17.
- Douglas CW, Brown PR, Preston FE. Platelet aggregation by oral streptococci. FEMS Microbiol Lett. 1990; 60: 63–7.
- Douglas CW, Heath J, Hampton KK, Preston FE. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol. 1993; 39: 179–82.
- Stephenson AE, Wu H, Novak J, Tomana M, Mintz K, Fives-Taylor P. The Fap1 fimbrial adhesin is a glycoprotein: antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol Microbiol. 2002; 43: 147–57.
- Pham K, Feik D, Hammond BF, Rams TE, Whitaker EJ. Aggregation of human platelets by gingipain-R from Porphyromonas gingivalis cells and membrane vesicles. Platelets. 2002; 13: 21–30.
- Naito M, Sakai E, Shi Y, Ideguchi H, Shoji M, Ohara N, et al.. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not Rgp proteinase. Mol Microbiol. 2006; 59: 152–67.
- Li X, Iwai T, Nakamura H, Inoue Y, Chen Y, Umeda M, et al.. An ultrastructural study of Porphyromonas gingivalis-induced platelet aggregation. Thromb Res. 2008; 122: 810–9.
- Wilson W, Taubert KA, Gewitz M, Lockhart PB, Baddour LM, Levison M, et al.. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association rheumatic fever, endocarditis, and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation. 2007; 116: 1736–54.
- Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 2008; 112: 2780–6.
- Papapanagiotou D, Nicu EA, Bizzarro S, Gerdes VE, Meijers JC, Nieuwland R, et al.. Periodontitis is associated with platelet activation. Atherosclerosis. 2009; 202: 605–11.