
Abstract
Rapamycin inhibits mechanistic (or mammalian) target of rapamycin (mTOR) that promotes protein production in cells by facilitating ribosome biogenesis (RiBi) and eIF4E-mediated 5'cap mRNA translation. Chronic treatment with encapsulated rapamycin (eRapa) extended health and life span for wild-type and cancer-prone mice. Yet, the long-term consequences of chronic eRapa treatment are not known at the organ level. Here, we report our observations of chronic eRapa treatment on mTORC1 signaling and RiBi in mouse colon and visceral adipose. As expected, chronic eRapa treatment decreased detection of phosphorylated mTORC1/S6K substrate, ribosomal protein (rpS6) in colon and fat. However, in colon, contrary to expectations, there was an upregulation of 18S rRNA and some ribosomal protein genes (RPGs) suggesting increased RiBi. Among RPGs, eRapa increases rpl22l1 mRNA but not its paralog rpl22. Furthermore, there was an increase in the cap-binding protein, eIF4E relative to its repressor 4E-BP1 suggesting increased translation. By comparison, in fat, there was a decrease in the level of 18S rRNA (opposite to colon), while overall mRNAs encoding ribosomal protein genes appeared to increase, including rpl22, but not rpl22l1 (opposite to colon). In fat, there was a decrease in eIF4E relative to actin (opposite to colon) but also an increase in the eIF4E/4E-BP1 ratio likely due to reductions in 4E-BP1 at our lower eRapa dose (similar to colon). Thus, in contrast to predictions of decreased protein production seen in cell-based studies, we provide evidence that colon from chronically treated mice exhibited an adaptive ‘pseudo-anabolic’ state, which is only partially present in fat, which might relate to differing tissue levels of rapamycin, cell-type-specific responses, and/or strain differences.
To access the supplementary material for this article, please see Supplementary files under ‘Article Tools’
Encapsulated rapamycin (eRapa) releases drug directly into the intestine, and chronic treatment extended health (Citation1, Citation2) and life span in mice (Citation2–Citation6). Impressively, chronic eRapa exposure suppressed intestinal adenomas to restore a normal life span for Apc Min/ + mice (Citation7), a model for human familial adenomatous polyposis (FAP). This was a fivefold extension and illustrates the potential clinical value of chronic eRapa treatment since the alternative for FAP patients is prophylactic colectomy at an early age. eRapa and other rapamycin formulations and derivatives (rapalogs) have been shown to ameliorate other forms of cancer as well (Citation8–Citation19). Therefore, a better understanding of the changes produced by chronic eRapa exposure is imperative for its potential long-term use in people for cancer prevention and possibly other age-associated diseases.
Rapamycin impairs protein synthesis (Citation20) by interacting with the rapamycin-FK506 binding protein 12 (FKBP12) and allosterically inhibiting the activity of the mechanistic (or mammalian) target of rapamycin (mTOR) complex 1 (mTORC1) by either destabilizing mTORC1 (Citation21) or limiting access to its kinase active site (Citation22). Rapamycin has two modes of action to inhibit protein synthesis. First, rapamycin reduces eIF4E-sensitive protein synthesis by increasing the binding of its repressor, 4E-BP1. eIF4E, a component of the eIF4F initiation complex, enables protein synthesis by binding to capped messenger RNAs (Citation23). The 4E-BP1 repressor inhibits the assembly of the eIF4F complex [reviewed in (Citation24)]. By reducing the phosphorylation of 4E-BP1 by mTORC1, the rapamycin-FK506BP complex inhibits eIF4F assembly and eIF4E-sensitive protein synthesis. Second, rapamycin reduces ribosome biogenesis (RiBi, reviewed in (Citation25)). Ribosome subunits are produced in the nucleolus and are composed of ribosomal proteins (rp) and ribosomal RNAs (rRNAs). The mTORC1/S6K pathway enables RiBi via phosphorylation of a ribosomal protein, rpS6, at Ser240/244 (Citation26), which controls the RiBi transcription program (e.g. ribosome protein mRNAs). It also promotes non-nucleolar transcription of 5S rRNA (by RNA Pol III) by phosphorylation of Maf1, and transcription of nucleolar 47S rRNA (by RNA Pol I and a precursor to 18S, 5.8S, and 28S rRNAs) (Citation27, Citation28). Finally, the mTORC1/eIF4E pathway regulates translation of mRNAs containing 5′TOP sequences including those encoding all the ribosome subunit proteins (Citation29). Thus, rapamycin could reduce all of these processes in RiBi as part of its anti-cancer and anti-aging mechanism.
Following up on our observation of eRapa effects in the small bowel (Citation7), here we measured the impact chronic eRapa treatment has on the colon in mice and compared it to visceral adipose. In accordance with our previous observations (Citation7), we found that chronic eRapa exposure reduced the detection of rpS6 Ser240/244 phosphorylation in the colon and fat. However, in colon, contrary to our expectations, chronic eRapa exposure led to an mRNA profile suggesting an increase in messages that encode ribosomal proteins including two paralogs implicated in immunology and cancer, rpl22 and rpL22l1, and an increase in 18S rRNA. There was also an increase in eIF4E relative to its repressor 4E-BP1. By comparison to fat, the response profile to FKBP-rapamycin was similar but with distinct differences that include opposite responses for rpl22 and rpl22l1 and a decrease in 18S rRNA, an increase in ribosome protein mRNAs, and decreases in eIF4E and 4E-BP1. These unexpected findings suggest differential compensatory responses to maintain protein production that could be important for long-term survival and possibly anti-aging effects of rapamycin.
Materials and methods
Mice
We generously obtain C57BL/6 mice from a breeding overseen by the Nathan Shock Animal Aging Animal Models and Longevity Assessment Core. The original breeding pairs were obtained from the Jackson Laboratory.
UM-HET3 mice were bred in the Intervention Testing Center animal facility in the Barshop Institute for Longevity and Aging Studies. They are a 4-way cross, the first-generation offspring of BALB/cByJ×C57BL/6J)F1 mothers (CByB6F1/J; JAX Mice stock #100009) and C3H/HeJ×DBA/2J)F1 fathers (C3D2F1/J; JAX Mice stock #100004). Each HET3 mouse in the test population is genetically unique but shares one half of its (nuclear) genome, on average, with every other mouse in the population. We treated and housed mice according to IACUC standards. We used colon from wild-type B6.129S2 mice (Citation8) in our microarray experiments.
Rapamycin diets
C57BL/6 mice were fed microencapsulated rapamycin-containing diets containing a concentration of either 14 mg/kg food (14 ppm), which provided a dose of ~2.24 mg of rapamycin/kg body weight/day (Citation3, Citation4), or 42 mg/kg food (42 ppm) (Citation1), which provided a dose of ~6.72 mg of rapamycin/kg body weight/day. Control diet was the same but with empty capsules. We housed mice in accordance with the NIH guide for the care and use of lab animals. For the data in Supplementary Fig. 2, we fed mice diets with variable levels of eRapa as indicated. For our microarray data in , we used UM-HET3 mice treated the same as above. We previously reported life span extension in this strain (Citation8).
Measurement of rapamycin in blood, intestine, and fat
We previously described the procedure for measuring rapamycin blood levels (Citation8) and our HPLC/MS system used for rapamycin quantification in tissue assays (Citation7).
Immunoblots
We used snap frozen colon, visceral adipose tissues for immunoblot assay lysate preparation and for the isolation of RNA used in microarray and qRT-PCR assays below. Because of difficulties encountered with immunoblot assays of intestinal (including colon) tissue, we optimized a procedure, which we described earlier (Citation7). Fluorescent signals were quantified and ratios calculated as previously described (Citation7). Actin, pan Ab-5 (clone ACTN05) mouse monoclonal primary antibody was obtained from Thermo Fisher, and all other primary antibodies were obtained from Cell Signaling Technologies.
RNA preparation for microarray
We extracted total RNA from flash frozen colon from C57BL/6 female tissues using a miRNeasy mini kit (Qiagen). We measured RNA concentrations using a Nanodrop Spectrophotometer (Thermo Scientific) before quality was assessed using a 2100 Bioanalyzer (Agilent). We generated biotinylated, amplified cRNAs using Ambion's Illumina TotalPrep RNA Amplification Kit (Life Technologies), which were then hybridized with Mouse WG6 Whole-Genome Expression BeadChips (Illumina). All kits were used according to manufacturer's instructions. The Genomics Core at the University of Texas Health Science Center at San Antonio performed all Gene Expression Profiling. GenomeStudio (Illumina) and GeneSpring (Agilent) software were used to normalize, filter, and analyze gene expression data. Some differentially expressed genes were then annotated according to protein–protein interactions, disease associations, and bio-pathways using IPA software (Ingenuity).
Quantitative real-time PCR
Colon and small intestine
Frozen tissues (10–35 mg) were cryofractured as previously described (Citation7), and total RNA was extracted using a mirVana miRNA Isolation Kit (Life Technologies). Respective cDNAs were then synthesized from 2 µg total RNA using ABI's TaqMan RNA-to-Ct Kit, and qRT-PCR was performed on 25 ng cDNA in a 10 µl reaction (2.5 ng for 18S only) using single tube inventoried TaqMan probes and TaqMan Gene Expression Master Mix (Life Technologies). The TaqMan primer sequences are B2m,mCG11606, Mm00437762_m1 and FG,18S rRNA, 4333760T. qRT-PCR was performed to detect Rpl22 and Rpl22l1 on 25 ng cDNA in a 10 µl reaction using SYBR Green (Biorad cat. 1725271). SYBR Green primer sequences are β-actin (forward) 5′-CGGTTCCGATGCCCTGAGGCTCTT-3’; β-actin (reverse) 5′-CGTCACACTTCATGATGGAATTGA-3′o; Rpl22 (forward) 5′-GTCGCCAACAGCAAAGAGAG-3′; Rpl22 (reverse) 5′-TCCTCGTCTTCCTCCTCCTC-3′; Rpl22l1 (forward) 5′-TGGAGGTTTCATTTGGACCTTAC-3′; Rpl22L1 (reverse) 5′-TTTCCAGTTTTTCCATTGACTTTAAC-3′.
Fat
For UM-HET3 mice, total RNA was extracted from 100 to 110 mg frozen visceral adipose using the miRNeasy Kit (Qiagen), and cDNA was synthesized from 2 µg total RNA using ABI's TaqMan RNA-to-Ct Kit (Life Technologies). qRT-PCR was performed on 25 ng cDNA in a 10 µl reaction (2.5 ng for 18S only) using single tube inventoried TaqMan probes and TaqMan Gene Expression Master Mix (Life Technologies) using the same primer sequences described in colon.
For C57BL6/J fat, total RNA was extracted from ~100 mg frozen visceral adipose tissue using Direct-zol RNA MiniPrep Plus kit (Zymo research, cat. R2070), and cDNA was synthesized from 0.5 µg total RNA using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, cat. 4387406). qRT-PCR was performed on 12.5 ng cDNA in a 10 µl reaction using SsoAdvanced Universal SYBR Green Supermix (Biorad cat. 1725271).
qRT-PCR reactions were carried out on an Applied Biosystems, Inc. 7900HT Fast Real-Time PCR System (Life Technologies) or a CFX384 Real Time System (Bio-Rad). The ΔΔCt (ABS) or ΔΔCq (Bio-Rad) methods were used to calculate fold change for each respective gene. qRT-PCR for Rpl22 and Rpl22l1 was performed on 25 ng cDNA in a 10 µl reaction using SYBR Green (Biorad cat. 1725271) using the same primers as described for colon. Fold changes were graphed and analyzed using an unpaired t-test in Prism 6 (GraphPad Software).
Results and discussion
Local and systemic rapamycin concentrations
We assessed rapamycin concentration in colon, visceral fat, and blood for C57BL/6 female mice (607–627 days old) fed a low (14 ppm) and high (42 ppm) eRapa-containing diet for 42 days and for fat in UM-HET3 mice fed 14 and 42 ppm diets for an average of 5.4 months beginning at an average age of 3.6 months. These diets were shown to extend the life span for Apc Min/ + mice (Citation7) and wild-type UMHET3 mice in a dose-dependent manner (Citation6). The enteric microcapsules used to deliver rapamycin (Eudragit S100) are designed to release the drug in the large bowel in people (Citation30); therefore, we studied eRapa's effect on various tissues to understand local and systemic concentrations. We chose to analyze colon as a follow-up to our previous study of small intestine in C57BL6 mice (Citation7). We also analyzed fat since we previously showed its responsiveness to chronic eRapa (Citation3), and because of the important role it plays in nutrient homeostasis and energy balance (Citation31) and in aging (Citation32).
Average values for rapamycin concentrations in the blood were 37.3±4.7 and 170.2±10 ng/ml, for C57BL/6 mice fed diets containing 14 and 42 ppm rapamycin, respectively (a). In C57BL/6 colon, average levels were 303.5±26.1 and 687.3±77.4 parts per billion (ppb) (b) for the 14 and 42 ppm diets, respectively. Our previous study documented the rapamycin concentration in the proximal and distal small intestine of C57BL/6 mice; these values for the high dose were 266.7±26.35 (n=5) to 1,488±141.6 (n=5) ppb, respectively (Citation7). Collectively, these data indicate that the drug-release profile of Eudragit S100 in mice includes both the small and large intestine, with distal small intestine being the apparent area of highest release.
Fig. 1 Pharmacokinetics of eRapa treatments. (a) Rapamycin blood levels of C57BL/6 mice at 607–627 days of age (42 days on eRapa diets, which averaged 37.3±4.7 and 170.2±10 ng/ml for the 14 and 42 ppm group, respectively). (b) Colon levels of rapamycin averaged 303.5±26.1 and 687.3±77.4 ppb for the 14 and 42 ppm diet-fed groups. In these graphs, tissue data points for 0 ppm rapamycin in diets are shown as zero ppb; most samples were recorded as ≤2.0; however, one assay recorded samples from ≤3 to 11.9 in colon. (c and d) Visceral adipose levels in fat from C57BL/6 mice was 35.12±3.354 ppb for the 42 ppm diet, while they averaged 15.47±3.052 and 25.58±7.011 ppb for the 14 and 42 ppm diets in UM-HET3 mice, respectively.
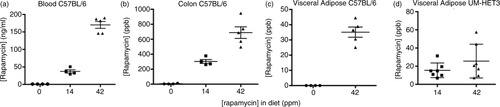
To assess the possibility that eRapa resulted in rapamycin accumulation in internal organs, we measured rapamycin concentration in visceral fat from C57BL/6 mice. We found average rapamycin levels were 35.12±3.354 ppb for the 42 ppm diet (c). Similarly in UM-HET3 mice, we measured 15.47 ±3.052 and 25.58±7.011 ppb for the 14 and 42 ppm diets, respectively (d). Therefore, eRapa clearly delivers rapamycin to internal tissues and organs through a systemic delivery and likely at a lower concentration than that resulting from local release in the intestine.
eRapa's impact on mTORC1
We assayed rapamycin effects on mTORC1 downstream signaling in the colon beginning with levels of rpS6 Ser240/244 phosphorylation. In our mTORC1 tissue immunoblot assays, we interpret the ratio of phosphorylation-dependent anti-rpS6 Ser240/244 antibody integrated intensity quantities (I.I. K counts, see Methods) relative to phosphorylation state-independent counts to reflect changes in the ratio of phosphorylated protein to total at the time of tissue dissection and lysate preparation. We also infer that total protein intensity counts relative to actin antibody intensity counts reflect their ratio in tissues at the same time. a shows representative immunoblot images for the effectors we assayed, along with individual rapamycin concentrations in blood and colon below the blot lanes for each individual mouse assayed. As expected, eRapa represses Ser240/244 phosphorylation in a dose-dependent manner (b) with insignificant effects on rpS6 levels compared to actin (c). In a separate pharmacodynamic experiment, Supplementary Fig. 1 shows immunoblot images with the gel lanes rearranged to reflect increasing rapamycin levels in colon (shown below the blot images). Also shown below are rapamycin blood levels and the ratio of intensity values for rpS6 Ser240/244 phosphorylation signal to total rpS6 intensity values. Note the steep decline in the ratio of phosphorylation-dependent signal to phosphorylation-independent signal (total rpS6) as rapamycin concentration increases up to 86 ppb in colon at which point the ratios level off (graphed in Supplementary Fig. 1). We interpret this to indicate that about 80–100 ppb rapamycin maximally represses the mTORC1→S6K1→rpS6 axis in colon. Note that colon levels of rapamycin do not strictly correlate with blood levels. We also point out that our lysates represent a mixed population of cells, for which there could be different effects on different cell locations and types. Since each lane in this experiment represents the response of an individual mouse, this approach does not take into account variation in individual responses to rapamycin. Because the responses in Ser240/244 phosphorylation in a and b are fairly uniform at colon concentrations of 262 ppb and above (4 mice on mid-dose and 5 on high dose), we feel our interpretation in Supplementary Fig. 1 is valid. Thus, chronic eRapa treatment decreased mTORC1→S6K1→rpS6 signaling in the colon as expected.
Fig. 2 Pharmacodynamics in colon of C57BL/6 mice fed diets containing variable levels of eRapa. (a) Western blot with the indicated antibodies. Below each lane, we show the levels of rapamycin in the colon and blood. (b) Graphs the ratio of intensity values for the phosphorylation state-dependent signal (P(240/244)rpS6) to phosphorylation state-independent (rpS6) signal. (c) The rpS6 to actin antibody signal. (d) Graph showing the ratio of intensity values for the phosphorylation state-dependent signal (Ser209) to phosphorylation-independent signal of eIF4E. (e) Ratio of eIF4E to actin. (f) Ratio of phosphorylation state-dependent signal 4E-BP1 (Thr37/46) to phosphorylation-independent intensity values. (g) Ratios of 4E-BP1 to actin. (h) Ratios of eIF4E to 4E-BP1.
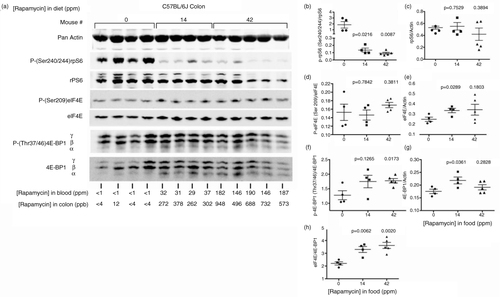
Next, we assessed the status of eIF4E and its repressor, 4E-BP1. Inhibition of mTOR alters the phosphorylation status of the eIF4F components, one of which is eIF4E Ser209 (Citation33). This site is phosphorylated by MNK [MAPK (mitogen-activated protein kinase)-interacting kinases) 1/2, and the Ras/MAPK pathway regulates this catalytic activity. The phosphorylation of eIF4E and MNKs plays an important role in tumorigenesis, most likely by regulating the translation of specific mRNAs (Citation34, Citation35). Interestingly, phosphorylation of MNKs appears necessary for oncogenic transformation but dispensable for development making pharmacologic inhibitors of eIF4E phosphorylation attractive as anti-cancer agents (Citation36, Citation37). Of interest, rapamycin enhanced eIF4E phosphorylation in several cell lines (Citation38–Citation42).
In contrast to the cell culture-based results, we find insignificant changes in eIF4E Ser209 phosphorylation in long-term eRapa-treated colon tissue at the 14 and 42 ppm dose (d). Interestingly, 42 days of eRapa treatment increased eIF4E levels relative to actin at the 14 ppm dose, but was more variable at the 42 ppm dose (e). Since increased levels of eIF4E are considered tumorigenic (Citation23), this suggests a trend toward a pro-growth state in the colon of chronically eRapa-treated mice. Also unexpectedly, we observed an increase in 4E-BP1 Thr37/46 phosphorylation in response to chronic eRapa treatment (f), which would tend to deactivate its repressive potential and promote an increase in eIF4E-sensitive mRNA translation. For resistance to active site inhibitors and clinical response to mTOR inhibition, Alain et al. (Citation43) proposed that the ratio of eIF4E to its repressor, 4E-BP1, is more important than their individual levels or phosphorylation status. g indicates a significant increase in 4E-BP1 levels relative to actin at the 14 ppm dose with the same trend at the 42 ppm dose. However, our results indicate a significant increase in the eIF4E/4E-4E-BP1 ratio in chronically eRapa-treated colon (h). Based on Alain et al.'s model of mTOR inhibitor resistance in transformed MEFs, HeLa, SK-HEP-1, and HepG2 cancer cells (Citation43) and further supported in breast cancer cells (Citation44), these results suggest the possibility of a ‘chronic rapamycin-resistant’ state with respect to the eIF4E/4E-BP1 axis, which might or might not translate to a pro-growth ‘pseudo-anabolic’ state. Regardless, a pro-growth state in rapamycin-treated intestine runs counter to reduced polyp formation we found in Apc Min/ + mice (Citation7).
Rebound activation of oncogenic Akt signaling in response to rapamycin or rapalogs (Citation18, Citation45) represents a significant clinical concern and may be an additional basis for rapamycin resistance (Citation46). Supplementary Fig. 2A shows representative immunoblot images of our assays. In response to long-term eRapa treatment in the colon, quantification intensity data indicate little change in Akt Ser473 phosphorylation (Supplementary Fig. 2B) or levels of Akt except for 3 of the 5 mice fed the 42 ppm diets, which show decreased levels of Akt (Supplementary Fig. 2C). We also observed this pattern in small intestine (Citation7). Therefore, rebound activation of potentially oncogenic Akt signaling does not appear to be an issue with chronic eRapa. Conversely, we do not detect diminution of mTORC2 activity (as indicated by Akt Ser 473 phosphorylation), which might be detrimental according to the model proposed by Lamming et al. (Citation47).
For studies involving intermittent rapamycin treatments (such as NCI-2012-01995, SWOG-S1207 breast cancer trial with a one-year treatment), a withdrawal of an mTOR inhibitor during such a pro-growth state might be tumorigenic. Understanding these adaptive responses to chronic rapamycin exposure in colon (and other organs) clearly requires further studies. In addition, our results suggest that responses to rapamycin in tissue culture do not always translate to in vivo tissue responses, especially with mid- to long-term treatment settings. This difference could reflect the short-term nature of most tissue culture studies or could reflect cell-type-specific responses.
eRapa's impact on RiBi
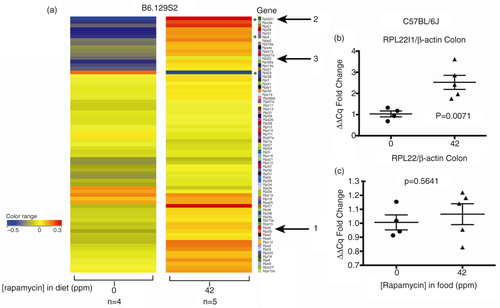
To expand our understanding of the effects of chronic rapamycin in colon, we performed RNA profiling experiments. Based on the significant reduction of rpS6 240/244 phosphorylation by eRapa (b) and the demonstration by Chauvin et al. (Citation26) that S6K1→rpS6 controls the ribosome biogenesis transcriptional program, we first analyzed the profiles of mRNAs encoding ribosomal protein genes (RPGs). shows an unfiltered heat map for RPG mRNA levels in the colon in response to high-dose rapamycin. One gene that did not appreciably change is Rps6 (arrow 1), in agreement with protein levels in b. However, note the striking upregulation in the upper and lower quadrants of the heat map. An interesting example in the top group is ribosome protein L22 like 1 (Rpl22l1) mRNA (arrow 2), which encodes a paralog of Rpl22 (arrow 3). These mRNAs and their products are of interest since studies identified specific functions of Rpl22 in T- and B-cell development in mice (Citation48, Citation49). Additionally, Zhang et al.'s (Citation50) studies indicated that Rpl22 and Rpl22l1 are critical for T-cell development in zebrafish and are functionally antagonistic in the regulation of hematopoietic stem cells. Additionally, depletion of Rpl22 was documented in cancer (Citation51). qRT-PCR validated the increase in the Rpl22l1 mRNA (b). Based on our array data, Rpl22 mRNA levels increase marginally (arrow 3), also quantified by qRT-PCR (c). Interestingly, O'Leary et al. (Citation48) proposed that Rpl22 regulates the expression level of paralog, Rpl22l1, by binding to a hairpin in exon 2 of its mRNA. They also showed incorporation of both paralogs into ribosomes. Our data suggest that one adaptation to chronic rapamycin by colon may include an altered large subunit ribosome composition, in addition to an overall increase in mRNAs encoding ribosomal proteins.
Fig. 3 Rapamycin effects on ribosomal protein genes (RPGs) in colon of eRapa-fed mice. (a) Unfiltered heat maps comparing mRNA levels in mice fed 0 or 42 ppm diets. The arrow labeled 1 shows rpS6, arrow 2 indicates the upregulation of rpl22l1 and rpl22, all of which we discuss in the text. We interpret these data to indicate a trend toward overall upregulation of these mRNAs. Asterisks indicate canonical pathway genes identified by IPA analysis and are discussed in the text. Visualized data are baselined to the median and log 2 transformed. From 0 to −0.5 would be a 1.5-fold change. (b) and (c) Graphs showing qRT-PCR results normalized to (actin) of Rpl22l1 and Rpl22, respectively.
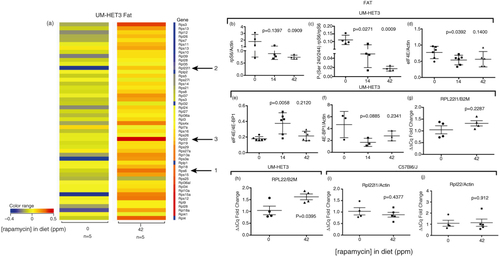
We next asked if RPG upregulation by FKBP-rapamycin is an intestine-specific response by examining their profile obtained from visceral adipose in mice fed 42 ppm eRapa diets. The heat map in a shows a general upregulation of most RPG in response to chronic eRapa treatment in fat, including rpS6 (a, arrow 1). This does not agree with rpS6 protein levels which tend to decline in fat (b), but which is accompanied by a significant dose-dependent inhibition of rpS6 phosphorylation relative to total protein (c). Opposite to colon, we observe a decrease in eIF4E relative to actin at the 14 ppm dose and the same trend at the higher dose (d). However, at the lower dose, we observe a significant increase in the ratio of eIF4E to 4E-BP1 with the same trend seen at the higher dose (e). One potential reason for the increase in eIF4E:4E-BP1 is a tendency toward a decrease in 4E-BP1 relative to actin at the 14 and 42 ppm doses (f). Curiously in this setting, we observe that the level of the rpl22 mRNA relative B2M (a, arrow 3) appears to be higher than rpl22l1 (a, arrow 2), which is opposite to the response to colon. qRT-PCR validated these results in UM-HET3 mice (g and h), but not in C57BL6/J mice (i and j), which had higher tissue levels of rapamycin (c compared to d). A caveat of our fat RNA genomic and other data is that we acquired it using UM-HET3 mice as opposed to the C57BL/6 mice used to collect tissue data. Thus, strain-specific differences appear be an element in responses to chronic rapamycin.
Fig. 4 Rapamycin effects on RPGs in visceral adipose of eRapa-fed mice. (a) Unfiltered heat maps comparing mRNA levels in mice fed 0 and 42 ppm diets. The arrows point to the same genes shown in . Visualized data are baselined to the median and log 2 transformed. From 0 to −0.5 would be a 1.5-fold change. (b) Graph of rpS6 levels mice fed 0, 14, and 42 ppm diets, relative to actin. (c) Graph of phosphorylation-dependent (Ser240/244) signals in mice fed 0, 14, and 42 ppm diets, respectively. (d–f) Graphs of the following ratios: eIF4E:actin; eIF4E:4E-BP1; 4E-BP1:actin, respectively. (g) and (h) Graphs of qRT-PCR results compared to (B2M) for Rpl22l1 and Rpl22, respectively.
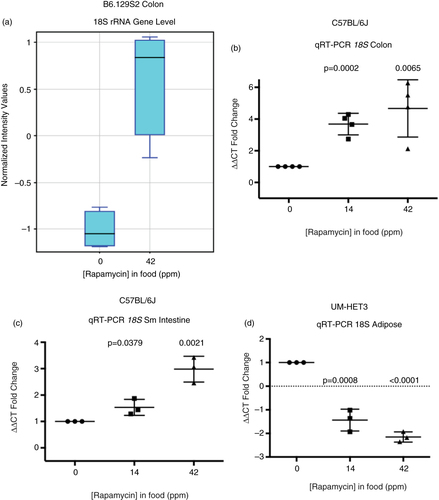
Unexpectedly, microarray data indicated that 18S rRNA levels increased in response to 42 ppm eRapa treatment (a), which we also validated by qRT-PCR in colon (b) and distal small intestine (c). Thus, in colon (and likely small intestine), the overall increase in RPG mRNAs aligns with an increase in 18S rRNA. Our ribosomal RNA result runs counter to the prevailing notion that rapamycin inhibition of mTORC1 leads to lower 18S rRNA (Citation27, Citation28). However, in fat, we observed the expected decline in 18S rRNA in response to chronic rapamycin (d), consistent with the difference in Rpl22l1 and Rpl22 responses in b. Thus, colon responses included an increase in both RPG mRNA and 18S rRNA levels, while in fat we observed an increase in some RPGs, but a decrease in 18S rRNA. The reason we see a more coordinated response in colon, but a less coordinated response in fat is not understood, although fat has a 27-fold lower rapamycin tissue level than colon (). Different mouse strains (C57BL/6 vs. UM-HET3) could also be an explanation. At this stage of our studies, we cannot distinguish between the possibilities that the overall increase in RPG mRNAs could represent an increase in RNA Pol II transcription rate or a decrease in mRNA turnover (Citation52). The changes in 18S rRNA could represent an increase (colon) or decrease (fat) in Pol I transcription, an alteration of 18S processing (Citation53) or altered half-life (Citation54).
Fig. 5 Rapamycin effects on 18S ribosomal RNA in colon of eRapa-fed C57BL/6 mice. (a) Graphic comparison of the normalized intensity values for 18S rRNA as quantified by microarray analysis. (b–d) Graphs showing fold changes in rRNA by qRT-PCR normalized to B2M mRNA in response to 14 and 42 ppm diets in colon (b), small intestine (c), and adipose (d).
eRapa's impact on canonical pathways
An IPA® analysis of all genes in the colonic response to chronic rapamycin identified several canonical pathways including decreases in phosphatidylcholine, phosphatidylethanolamine, and cholesterol biosynthesis I, II, and III. Of interest to the present discussion, IPA ® identified an upregulation of eIF2 signaling [−log(p-value) = 1.12E00 9.95E-03]. eIF2 promotes recruitment of the initiator methionyl-tRNA to the 40S ribosome subunit, which it requires for locating the start codon. Cellular responses to an insufficiency of amino acids are regulated via eiF2/eIF2B [reviewed by Proud (Citation20)]. Affected RiBi molecules in colon identified by IPA® include Rn18s (18S rRNA, a–c), Rpl22l1, and Rpl4 and striking downregulation of Rpl23 (, asterisks). Interestingly, an IPA analysis of liver from 14 ppm eRapa-fed C57BL/6 mice by Karunadharma et al. (Citation55) also identified the eIF2 as an affected pathway, and heat maps showed a general upregulation of RPBs in young and older mice, in basic agreement with our data. The downregulation of Rpl22l1 was a notable exception in their study, but in agreement with our analysis of kidney (data not shown).
Our studies raised many questions, for example, what cell types are responsible for our observations in colon and fat, and could this explain the difference in responses to rapamycin? What, if any, changes in the ribosome profile does FKBP-rapamycin effect in these tissues? And, how does rapamycin regulate mRNA levels for Rpl22l1 and Rpl22, and does this translate to the protein/ribosome level? How do our observations translate to prevention of polyp formation and a normal life span in Apc Min/ + mice? And, are intestinal crypt stem cells a target for this effect, as the results of Yilmaz et al. (Citation56) might suggest? Finally, are patients on rapamycin/rapalog therapy at risk for cancer development and progression when taken off the drug?
An important question raised during the review of this manuscript is ‘which strain might provide better translational data depending on the experimental objectives?’ Miller et al. (Citation57) provided a compelling rationale for use of UM-HET3 rather than inbred mice to screen for agents that extend life span studies conducted by the Intervention Testing Program (ITP). They argued that the use of genetically heterogeneous mice would help ‘to avoid missing true positive effects of agents that might fail to work in a specific inbred genotype, and to reduce the chance of giving undue emphasis to an agent that might work only in the specific inbred stock selected’. To this end, the ITP has been very successful having identified several drugs that extend life span, including acarbose (Citation58) and the eRapa used in this study. Flurkey et al. (Citation59) compared diet restriction of UM-HET3 with a standard F1 hybrid (CByB6F1/J) and reported the same increases in mean and maximum life span in both. These authors stated that their results ‘support the use of a genetically mixed model, with reproducible allelic composition, for studies in aging research’. Understanding that no genetic composition is perfect, if the experimental objective is to test for an intervention in the aging process, UM-HET3 mice would appear to be a good choice.
If the experimental objective is uncovering the mechanism(s) of action of rapamycin as an anti-aging agent, inbred strains (and their cells) amenable to genetic manipulation might be preferable. This would be especially important in studies of specific processes and diseases associated with aging. An ability to make specific genetic alternations in these pathways in cells (and then mice) studied under defined conditions will be useful in understanding rapamycin effects on life span. Similar approaches in other important pathways might lead to new targets for novel agents for testing in UM-HET3. Thus, we believe that testing in multiple genetic backgrounds will lead to a clearer picture of aging and how to ameliorate its effects.
We and others are actively pursuing these and other questions in the fast expanding field of mTOR biology to identify additional anti-aging and anti-cancer targets, which should then be tested in multiple genetic environments.
Conflict of interest and funding
Under a licensing agreement between Rapamycin Holdings, Inc. and the University of Texas Health Science Center San Antonio, Z.D. Sharp, P. Hasty, R. Strong, and C.B. Livi, the university is entitled to milestone payments and royalty on sales of the rapamycin formulation used in this paper. This work was supported by NIH grants (RC2AG036613, Project 1, ZDS and PH, and P01AG017242, Core B and Project 3, PH; R01 CA193835-01A, ZDS) and Pilot Funds from the Nathan Shock Center of Excellence in the Biology of Aging, ZDS and PH; P30AG013319 and U01AG022307, RS.
Supplementary Material
Download PDF (1 MB)Acknowledgements
Animals were under the expert care of Vivian Diaz and her team in the Nathan Shock Aging Animal and Longevity Core directed by Dr Jim Nelson. The Genomics Shared Resource provided valuable assistance and reagents with the RNA profiling experiments. Nameer Kirma provided critical advice for all qRT-PCR analysis and their interpretation.
Notes
To access the supplementary material for this article, please see Supplementary files under ‘Article Tools’
Related Research Data
References
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, etal. Rapamycin slows aging in mice. Aging Cell. 2012; 11: 675–82.
- Zhang Y, Bokov A, Gelfond J, Soto V, Ikeno Y, Hubbard G, etal. Rapamycin extends life and health in C57BL/6 mice. J Gerontol A Biol Sci Med Sci. 2014; 69(2): 119–30.
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, etal. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460: 392–5. [PubMed Abstract] [PubMed CentralFull Text].
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, De Cabo R, etal. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66(2): 191–201.
- Neff F, Flores-dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, etal. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. 2013; 123(8): 1–2.
- Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, etal. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. 2014; 13(3): 468–77.
- Hasty P, Livi CB, Dodds SG, Jones D, Strong R, Javors M, etal. ERapa restores a normal life span in a FAP mouse model. Cancer Prev Res. 2014; 7: 169–78.
- Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, etal. Rapamycin extends life span of Rb1 + /( mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013; 5(2): 100–10.
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, etal. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176(5): 2092–7.
- Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, etal. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011; 10(24): 4230–6.
- Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, etal. New nanoformulation of rapamycin rapatar extends lifespan in homozygous p53 − /− mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4(10): 715–22.
- Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, etal. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/−mice. Aging (Albany NY). 2012; 4(10): 709–14.
- Dao V, Pandeswara S, Liu Y, Hurez V, Dodds S, Callaway D, etal. Prevention of carcinogen and inflammation-induced dermal cancer by oral rapamycin includes reducing genetic damage. Cancer Prev Res (Phila). 2015; 8(5): 400–9.
- Rho O, Kiguchi K, Jiang G, Digiovanni J. Impact of mTORC1 inhibition on keratinocyte proliferation during skin tumor promotion in wild-type and BK5.AktWT mice. Mol Carcinog. 2013; 12: 1–12.
- Saha A, Blando J, Tremmel L, DiGiovanni J. Effect of metformin, rapamycin, and their combination on growth and progression of prostate tumors in HiMyc mice. Cancer Prev Res (Phila). 2015; 8(7): 597–606.
- Fujishita T, Aoki K, Lane HA, Aoki M, Taketo MM. Inhibition of the mTORC1 pathway suppresses intestinal polyp formation and reduces mortality in ApcDelta716 mice. Proc Natl Acad Sci USA. 2008; 105: 13544–9.
- Piselli P, Serraino D, Segoloni GP, Sandrini S, Piredda GB, Scolari MP, etal. Risk of de novo cancers after transplantation: results from a cohort of 7217 kidney transplant recipients, Italy 1997–2009. Eur J Cancer. 2013; 49: 336–44.
- Houghton PJ. Everolimus. Clin Cancer Res. 2010; 16: 1368–72.
- Squillace RM, Miller D, Wardwell SD, Wang F, Clackson T, Rivera VM. Synergistic activity of the mTOR inhibitor ridaforolimus and the antiandrogen bicalutamide in prostate cancer models. Int J Oncol. 2012; 41(2): 425–32. [PubMed Abstract] [PubMed CentralFull Text].
- Proud CG. Control of the translational machinery by amino acids. Am J Clin Nutr. 2014; 99: 231–6.
- Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the Human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010; 38: 768–74.
- Aylett CHS, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, etal. Architecture of human mTOR complex 1. Science. 2016; 351(6268): 48–52.
- Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N. eIF4E – from translation to transformation. Oncogene. 2004; 23: 3172–9.
- Alain T, Sonenberg N, Topisirovic I. mTOR inhibitor efficacy is determined by the eIF4E/4E-BP ratio. Oncotarget. 2012; 3: 1491–2.
- Gentilella A, Kozma SC, Thomas G. A liaison between mTOR signaling, ribosome biogenesis and cancer. Biochim Biophys Acta Gene Regul Mech. 2015; 1849(7): 812–20.
- Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau-Aveilla C, Dreazen A, etal. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene. 2014; 33(4): 474–83.
- Iadevaia V, Liu R, Proud CG. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin Cell Dev Biol. 2014; 36: 1–8.
- Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006; 25: 6384–91.
- Meyuhas O, Dreazen A. Ribosomal protein S6 kinase from TOP mRNAs to cell size. Prog Mol Biol Transl Sci. 2009; 90: 109–53. [PubMed Abstract].
- Khan MZI, Prebeg Ž, Kurjaković NA. pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers. I. Manipulation of drug release using Eudragit® L100-55 and Eudragit® S100 combinations. J Control Release. 1999; 58: 215–22.
- Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. 2015; 33: 55–66.
- Liao CY, Rikke BA, Johnson TE, Gelfond JAL, Diaz V, Nelson JF. Fat maintenance is a predictor of the murine lifespan response to dietary restriction. Aging Cell. 2011; 10: 629–39.
- Thoreen CC. Many roads from mTOR to eIF4F. Biochem Soc Trans. 2013; 41: 913–16.
- Furic L, Rong L, Larsson O, Koumakpayi IH, Yoshida K, Brueschke A, etal. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci USA. 2010; 107(32): 14134–9.
- Ueda T, Sasaki M, Elia AJ, Chio IIC, Hamada K, Fukunaga R, etal. Combined deficiency for MAP kinase-interacting kinase 1 and 2 (Mnk1 and Mnk2) delays tumor development. Proc Natl Acad Sci USA. 2010; 107: 13984–90.
- Hou J, Kam F, Proud CG, Wang S, Lam F, Proud CG, etal. Targeting Mnks for cancer therapy. Oncotarget. 2012; 3(2): 118–31.
- Proud CG. Mnks, eIF4E phosphorylation and cancer. Biochim Biophys Acta. 2015; 1849(7): 766–73.
- Bianchini A, Loiarro M, Bielli P, Busà R, Paronetto MP, Loreni F, etal. Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. Carcinogenesis. 2008; 29(12): 2279–88.
- Marzec M, Liu X, Wysocka M, Rook AH, Odum N, Wasik MA. Simultaneous inhibition of mTOR-containing complex 1 (mTORC1) and MNK induces apoptosis of cutaneous T-cell Lymphoma (CTCL) cells. PLoS One. 2011; 6(9): 2–9.
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, etal. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005; 65: 7052–8.
- Wang X, Yue P, Chan C-B, Ye K, Ueda T, Watanabe-Fukunaga R, etal. Inhibition of mammalian target of rapamycin induces phosphatidylinositol 3-kinase-dependent and Mnk-mediated eukaryotic translation initiation factor 4E phosphorylation. Mol Cell Biol. 2007; 27(21): 7405–13.
- Zang C, Eucker J, Liu H, Müller A, Possinger K, Scholz CW. Concurrent inhibition of PI3-kinase and mTOR induces cell death in diffuse large B cell lymphomas, a mechanism involving down regulation of Mcl-1. Cancer Lett. 2013; 339(2): 288–97.
- Alain T, Morita M, Fonseca BD, Yanagiya A, Siddiqui N, Bhat M, etal. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer Res. 2012; 72(24): 6468–76.
- Decarlo L, Mestel C, Barcellos-Hoff M-H, Schneider RJ. eIF4E is a Feed-forward translational coactivator of TGFβ early pro-transforming events in breast epithelial cells. Mol Cell Biol. 2015
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, etal. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006; 66(3): 1500–8.
- Efeyan A, Sabatini DM. MTOR and cancer: many loops in one pathway. Curr Opin Cell Biol. 2010; 22(2): 169–76.
- Lamming DW, Mihaylova MM, Katajisto P, Baar EL, Yilmaz OH, Hutchins A, etal. Depletion of Rictor, an essential protein component of mTORC2, decreases male lifespan. Aging Cell. 2014; 13: 911–17.
- O'Leary MN, Schreiber KH, Zhang Y, Duc ACE, Rao S, Hale JS, etal. The ribosomal protein Rpl22 controls ribosome composition by directly repressing expression of its own paralog, Rpl22l1. PLoS Genet. 2013; 9(8): e1003708.
- Anderson SJ, Lauritsen JPH, Hartman MG, Foushee AMD, Lefebvre JM, Shinton SA, etal. Ablation of ribosomal protein L22 selectively impairs alphabeta T cell development by activation of a p53-dependent checkpoint. Immunity. 2007; 26(6): 759–72.
- Zhang Y, Duc A-CE, Rao S, Sun X-L, Bilbee AN, Rhodes M, etal. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev Cell. United States. 2013; 24(4): 411–25.
- Rao S, Lee S-Y, Gutierrez A, Perrigoue J, Thapa RJ, Tu Z, etal. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood. 2012; 120(18): 3764–73.
- Pérez-Ortín JE, Alepuz P, Chávez S, Choder M. Eukaryotic mRNA decay: methodologies, pathways, and links to other stages of gene expression. J Mol Biol. 2013; 425(20): 3750–75.
- Henras AK, Plisson-Chastang C, O'Donohue M-F, Chakraborty A, Gleizes P-E. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015; 6: 225–42.
- Nwagwu M, Nana M. Ribonucleic acid synthesis in embryonic chick muscle, rates of synthesis and half-lives of transfer and ribosomal RNA species. J Embryol Exp Morphol. 1980; 56: 253–67. [PubMed Abstract].
- Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, etal. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell. 2014; 13: 529–39.
- Yilmaz ÖH, Katajisto P, Lamming DW, Gültekin Y, Bauer-Rowe KE, Sengupta S, etal. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature. 2012; 486(7404): 490–5. [PubMed Abstract] [PubMed CentralFull Text].
- Miller RA, Harrison DE, Astle CM, Floyd RA, Flurkey K, Hensley KL, etal. An aging interventions testing program: study design and interim report. Aging Cell. 2007; 6: 565–75.
- Harrison DE, Strong R, Allison DB, Ames BN, Astle CM, Atamna H, etal. Acarbose, 17-(-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell. 2014; 13(2): 273–82.
- Flurkey K, Astle CM, Harrison DE. Life extension by diet restriction and N-acetyl-L-cysteine in genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2010; 65(12): 1275–84.