
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The 14th edition of the Workshop on Recent Issues in Bioanalysis (14th WRIB) was held virtually on June 15-29, 2020 with an attendance of over 1000 representatives from pharmaceutical/biopharmaceutical companies, biotechnology companies, contract research organizations, and regulatory agencies worldwide. The 14th WRIB included three Main Workshops, seven Specialized Workshops that together spanned 11 days in order to allow exhaustive and thorough coverage of all major issues in bioanalysis, biomarkers, immunogenicity, gene therapy and vaccine. Moreover, a comprehensive vaccine assays track; an enhanced cytometry track and updated Industry/Regulators consensus on BMV of biotherapeutics by LCMS were special features in 2020. As in previous years, this year's WRIB continued to gather a wide diversity of international industry opinion leaders and regulatory authority experts working on both small and large molecules to facilitate sharing and discussions focused on improving quality, increasing regulatory compliance and achieving scientific excellence on bioanalytical issues.
This 2020 White Paper encompasses recommendations emerging from the extensive discussions held during the workshop and is aimed to provide the Global Bioanalytical Community with key information and practical solutions on topics and issues addressed, in an effort to enable advances in scientific excellence, improved quality and better regulatory compliance. Due to its length, the 2020 edition of this comprehensive White Paper has been divided into three parts for editorial reasons.
This publication (Part 3) covers the recommendations on Vaccine, Gene/Cell Therapy, NAb Harmonization and Immunogenicity). Part 1 (Innovation in Small Molecules, Hybrid LBA/LCMS & Regulated Bioanalysis), Part 2A (BAV, PK LBA, Flow Cytometry Validation and Cytometry Innovation) and Part 2B (Regulatory Input) are published in volume 13 of Bioanalysis, issues 4 and 5 (2020), respectively.
| Acronyms | ||
| AAV: | = | Adenovirus-associated virus |
| Ab: | = | Antibody |
| ADA: | = | Anti-drug antibody |
| ADC: | = | Antibody-drug conjugates |
| ADCC: | = | Antibody-dependent cell-mediated cytotoxicity |
| ADHS: | = | Antibody-depleted human serum |
| BAV: | = | Biomarker assay validation |
| BLA: | = | Biologics license application |
| BLAST: | = | Basic Local Alignment Search Tool |
| BMV: | = | Bioanalytical method validation |
| CAR-T: | = | Chimeric antigen receptor T cells are T cells that have been genetically engineered to express one or more receptors targeting specific proteins for use in immunotherapy |
| CBA: | = | Cell-based assays |
| cDNA: | = | Complementary DNA |
| CDR: | = | Complementarity-determining regions |
| CDx: | = | Companion diagnostics |
| cGMP: | = | Current Good Manufacturing Practices |
| CIC: | = | Circulating immune complexes |
| CLBA: | = | Competitive ligand binding assays |
| CLIA: | = | Clinical laboratory improvements amendments |
| Clinically Relevant ADA: | = | ADA impacting PK, PD, efficacy and/or safety of the biotherapeutic in patients |
| CLSI: | = | Clinical and Laboratory Standards Institute |
| CMC: | = | Chemistry, Manufacturing, and Controls |
| COU: | = | Context of use |
| Ct: | = | Threshold cycle |
| CTL: | = | Cytotoxic T lymphocytes |
| CV: | = | Coefficient of variation |
| dPCR: | = | Digital polymerase chain reaction |
| ddPCR: | = | Droplet digital polymerase chain reaction |
| DNA: | = | Deoxyribonucleic acid |
| DoE: | = | Design of experiments |
| Dx: | = | Diagnostic |
| FFP: | = | Fit-for-purpose |
| FIH: | = | First-in-human |
| FPR: | = | False-positive rate |
| GCT: | = | Gene and cell therapy |
| gDNA: | = | Genomic DNA |
| GTx: | = | Gene therapy |
| HDR: | = | Homology directed repair |
| IC: | = | Immune complex |
| IND: | = | Investigational new drug |
| Indel: | = | Insertion–deletion mutations |
| IQR: | = | Inter-quartile range |
| ISI: | = | Integrated Summary of Immunogenicity |
| ISR: | = | Incurred sample reproducibility |
| ITI: | = | Immune tolerance induction |
| KOL: | = | Key opinion leader |
| LBA: | = | Ligand binding assay |
| LCMS: | = | Liquid chromatography mass spectrometry |
| LDT: | = | Laboratory developed test |
| LLOQ: | = | Lower limit of quantitation |
| LOB: | = | Limit of blank |
| LOD: | = | Limit of detection |
| mAb: | = | Monoclonal antibody |
| MDB: | = | Multi-domain biotherapeutics |
| MFI: | = | Mean fluorescent intensity |
| MIQE: | = | Minimum Information for Publication of Quantitative Real-Time PCR Experiments |
| MoA: | = | Mechanism of action |
| MOI: | = | Multiplicity of infection |
| MRD: | = | Minimum required dilution |
| mRNA: | = | Messenger RNA |
| Multiplex: | = | A type of assay that simultaneously measures multiple analytes in a single experiment. |
| NAb: | = | Neutralizing antibody |
| NGS: | = | Next generation sequencing |
| NHEJ: | = | Non-homologous end joining |
| OD: | = | Optical density |
| PBMC: | = | Peripheral blood mononuclear cell |
| PCR: | = | Polymerase chain reaction |
| PD: | = | Pharmacodynamic |
| PK: | = | Pharmacokinetic |
| PMR: | = | Post-marketing request |
| PMT: | = | Photomultiplier tube |
| PVDF : | = | Polyvinylidene difluoride |
| QA: | = | Quality assurance |
| QC: | = | Quality control |
| qPCR: | = | Quantitative (real-time) polymerase chain reaction |
| RNA: | = | Ribonucleic acid |
| RSD: | = | Relative standard deviation |
| RT: | = | Reverse transcription |
| SD: | = | Standard deviation |
| SEC: | = | Size exclusion chromatography |
| SOP: | = | Standard operating procedure |
| ss/ds: | = | Single stranded/double stranded |
| SSM: | = | Spillover spreading matrix |
| TI: | = | Transduction inhibition |
| ULOQ: | = | Upper limit of quantitation |
| WRIB: | = | Workshop on Recent Issues in Bioanalysis |
Index
SECTION 1–Vaccine Clinical Assays and Cell Therapy
Discussions, Consensus and Conclusions
SECTION 2–Gene Therapy, qPCR, NGS and ELISpot Validation
Discussions, Consensus and Conclusions
SECTION 3–NAb Assay Harmonization, Biosimilars and FDA/EMA Guidance/Guideline
Discussions, Consensus and Conclusions
SECTION 4–Immunogenicity Assay Strategies
Introduction
The 14th edition of the Workshop on Recent Issues in Bioanalysis (14th WRIB) was held virtually between 15–29 June 2020 with an attendance of over 1000 representatives from pharmaceutical/biopharmaceutical companies, biotechnology companies, contract research organizations, and regulatory agencies worldwide. The 14th WRIB included three main workshops, seven Specialized Workshops that together spanned 11 days to allow exhaustive and thorough coverage of all major issues in bioanalysis, biomarkers, immunogenicity, gene therapy, cell therapy and vaccine.
Moreover, a comprehensive vaccine assays track; an enhanced cytometry track, and updated Industry/Regulators consensus on bioanalytical method validation (BMV) of biotherapeutics by mass spectrometry (hybrid assays, LCMS and HRMS) were special features in 2020.
As in previous years, this year's WRIB continued to gather a wide diversity of international industry opinion leaders and regulatory authority experts working on both small and large molecules to facilitate sharing, reviewing, discussing and agreeing upon best approaches aimed to achieve scientific excellence and increase regulatory compliance on bioanalytical issues.
The active contributing chairs included Dr. Stephen C Alley (Seattle Genetics), Dr. Anna Edmison (Health Canada), Dr. Chris Evans (GSK), Dr. Christine Fandozzi (Merck), Dr. Sally Fischer (Genentech), Dr. Fabio Garofolo (BRI), Dr. Christine Grimaldi (Boehringer Ingelheim), Dr. Lindsay King (Pfizer), Dr. Rocio Murphy (Merck), Dr. Hendrik Neubert (Pfizer), Dr. Manoj Rajadhyaksha (Regeneron), Dr. Catherine Soo (Health Canada), Dr. Susan Spitz (Incyte), Dr. Roland Staack (Roche), Dr. Scott Summerfield (GSK), Dr. Alessandra Vitaliti (Novartis), Dr. Jan Welink (EU EMA), Dr. Haoheng Yan (US FDA), Dr. Tong-yuan Yang (Janssen), Dr. Hongbin Yu (Boehringer Ingelheim), Dr. Yan Zhang (BMS).
The participation of regulatory agency representatives continued to grow at WRIB [Citation1–25] including the below:
Regulated Bioanalysis and BMV Guidance/Guidelines: Dr. Arindam Dasgupta (US FDA), Dr. Sam Haidar (US FDA), Dr. Mohsen Rajabiabhari (US FDA), Dr. Tahseen Mirza (US FDA), Dr. Nilufer Tampal (US FDA), Dr. Suman Dandamudi (US FDA), Dr. Diaa Shakleya (US FDA), Dr. Jinhui Zhang (US FDA), Dr. Patrick Faustino (US FDA), Dr. Jan Welink (EU EMA), Mr. Stephen Vinter (UK MHRA), Mr. Michael McGuinness (UK MHRA), Dr. Anna Edmison (Health Canada), Dr. Catherine Soo (Health Canada), Dr. Susan Stojdl (Health Canada), Mr. Gustavo Mendes Lima Santos (Brazil ANVISA)
Immunogenicity, Gene Therapy, Cell Therapy and Vaccines: Dr. Susan Kirshner (US FDA), Dr. Daniela Verthelyi (US FDA), Dr. Joao Pedras-Vasconcelos (US FDA), Dr. Haoheng Yan (US FDA), Dr. Meiyu Shen (US FDA), Dr. Mohsen Rajabi Abhari (US FDA), Dr. Isabelle Cludts (UK MHRA), Dr. Elana Cherry (Health Canada), Dr. Lucia Zhang (Health Canada), Dr. Akiko Ishii-Watabe (Japan MHLW), Dr. Sara Gagneten (US FDA), Dr. Andrew Exley (UK MHRA), Dr. Therese Solstad (EU EMA/Norway NoMA), Dr. Richard Siggers (Health Canada)
Biomarkers: Dr. Yow-Ming Wang (US FDA), Dr. Abbas Bandukwala (US FDA), Dr. Kevin Maher (US FDA), Dr. Yoshiro Saito (Japan MHLW)
All the traditional “working dinners” attended by both industry key opinion leaders (KOL) and regulatory representatives were held in a virtual format this year, and the extensive and fruitful discussions from these “working dinners” together with the lectures and open panel discussions amongst the presenters, regulators and attendees culminated in consensus and recommendations on items presented in this White Paper.
A total of 167 recent issues (‘hot’ topics) were addressed and presented in this White Paper, which are the background on each issue, exchanges, consensus and resulting recommendations on these one hundred and sixty-seven topics.
Due to its length, this comprehensive White Paper has been divided into three parts for editorial reasons. This publication covers Part 3 recommendations.
Part 1–Issue 4–February 2021
Hybrid Assays and HRMS
BMV of Biotherapeutics by LCMS and Hybrid Assays: Regulatory Rigor & Acceptance Criteria While Waiting for the ICH M10 Guideline (six topics)
Hybrid Assays for adeno-associated virus (AAV) Gene Therapy & Extracellular Vesicles: Advanced Applications (four topics)
Hybrid Assays for Target Engagement: Novel Applications (three topics)
High Resolution Mass Spectrometry for Protein Therapeutic Bioanalysis: Current Developments (three topics)
Small Molecules Innovation, Peptides and Oligos
Microbiome Contributions to Small Molecule Drug Metabolism and its Impact on Bioanalytical Assays (six topics)
Acoustic-Mass Spectrometry (MS) for Bioanalytical Applications (four topics)
High Resolution Mass Spectrometry (HRMS): Small Molecule Method Development Strategies (four topics)
Design of Experiments for Therapeutic Peptides: Modern Discovery Bioanalytical Laboratories (three topics)
Oligonucleotides and Chain-Shorted Metabolites: Advanced Strategies (four topics)
Regulatory Challenges in Mass Spectrometry
Data Integrity and Regulatory Factors to Consider when Using Cloud Computing (three topics)
Impact of Excipients on Bioanalytical Methods: “What are regulators asking?” (two topics)
Parallelism Evaluation in Small Molecule Endogenous Compounds–New Considerations on ICH M10 Guideline (three topics)
Abnormal Internal Standard Response: Compliance with the 2018 FDA Guidance and ICH M10 Guideline (four topics)
Microsampling in Regulated Bioanalytical Juvenile & Pediatric Studies (three topics)
Part 2A–Issue 5–March 2021
Biomarker Assay Validation (BAV)
Need for a BAV Guidance (three topics)
When Clinical Biomarker Assay Should be Under Clinical Laboratory Improvements Amendments (CLIA)? (three topics)
Current Applications of Context of Use (COU) in Fit-for-Purpose (FFP) BAV (six topics)
Advancements in Extracellular Vesicles (EV) (two topics)
PK LBA Regulated Bioanalysis
Stability Testing of Biotherapeutics: FDA, EMA, & ICH M10 Guidance/Guideline (two topics)
Critical Reagents: Latest Approaches (six topics)
Bispecific Monoclonal Antibodies & Bispecific T-cell Redirectors: Unique Challenges in Pharmacokinetic (PK) Assays (three topics)
Common Issue with Laboratory Information Management System (LIMS) Based Software for Ligand Binding Assay (LBA) Support (two topics)
Parallelism Evaluation in Regulated Bioanalysis for PK LBA: FDA, EMA, & ICH M10 Guidance/Guideline (four topics)
Flow Cytometry Validation
Flow Cytometry Validation: Applicability of Clinical and Laboratory Standards Institute (CLSI) H62 Guideline to Regulated Bioanalysis (four topics)
Flow Cytometry Validation: Target Engagement and Receptor Occupancy (three topics)
Flow Cytometry Validation Strategies for Assays Using Challenging Sample Types (two topics)
Validation Strategies for Image Cytometry Based Assays (three topics)
Validation of Lower Limit of Quantitation (LLOQ) in Flow Cytometry (two topics)
Cytometry Innovation
New Insights in Automated Gating (three topics)
Advantages & Challenges in using Mass Cytometry (CyTOF) (five topics)
High Dimensional/High Parameter Flow Cytometry (five topics)
Part 2B–Issue 5–March 2021
Input from Regulatory Agencies on Bioanalysis & BMV
Input from Regulatory Agencies on Immunogenicity & Biomarkers
Part 3–Issue 6–March 2021
Vaccine Clinical Assays and Cell Therapy
Clinical Vaccine Assay Validation (six topics)
Quality control (QC) Samples in Chimeric Antigen Receptor T Cells (CAR-T) & Vaccine Flow Cytometry Assays: Current Industry Standards (three topics)
Quantitative (real-time) Polymerase Chain Reaction (qPCR) Assays for CAR-T Programs (three topics)
Immunogenicity Strategy for CAR-T Products (three topics)
Gene Therapy, qPCR and ELISpot Validation:
qPCR, Droplet Digital Polymerase Chain Reaction (ddPCR), and Next Generation Sequencing (NGS) Assay Development & Validation: Best Practices (five topics)
AAV Capsid Neutralizing Antibody (NAb) Assays Development and Validation (three topics)
ELISpot & Single Cell Western Blot Assay Validation (three topics)
Application of Current FDA/EMA Immunogenicity Guidance/Guideline to Gene Therapy (two topics)
NAb Assay Harmonization, Biosimilars and FDA/EMA Guidance/Guideline:
Cell-based NAb Assays–Sensitivity and Drug Tolerance and the Relevance for Clinical Outcome (four topics)
NAb Assay Harmonization: Recent Trends and Expectations (four topics)
Biosimilar Immunogenicity: Current Industry Standards (three topics)
The 2019 US FDA Immunogenicity Guidance: Reflections a Year Later (three topics)
Immunogenicity Assay Strategies:
Lessons Learned from Late-Stage Clinical Studies (four topics)
Circulating Immune Complexes (three topics)
Multi-Domain Biotherapeutics: Immunogenicity Assay Strategies (four topics)
Definition of Persistent Anti-Drug Antibody (ADA) Responses and its Clinical Relevance (four topics)
SECTION 1–Vaccine Clinical Assays and Cell Therapy
Bart Corsaro1, Tong-yuan Yang2, Rocio Murphy3, Ivo Sonderegger10, Andrew Exley7, Sylvie Bertholet1, Naveen Dakappagari5, Francis Dessy6, Fabio Garofolo8, Lisa Kierstead9, Holger Koch10, Ghanashyam Sarikonda5, Natasha Savoie11, Richard Siggers12, Therese Solstad13
Authors in Section 1 are presented in alphabetical order of their last name, with the exception of the first five authors who were major contributors.
The affiliations can be found at the beginning of the article
DISCUSSION TOPICS & CONSOLIDATED QUESTIONS COLLECTED FROM THE GLOBAL BIOANALYTICAL COMMUNITY
The topics detailed below were considered as the most relevant ‘hot topics’ based on feedback collected from the 13th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 14th WRIB. The background on each issue, discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
Clinical Vaccine Assay Validation
What validation parameters should be evaluated when transitioning a previously validated vaccine assay into a multiplexed format? How are quality control samples used to monitor assay trending performance? What data is needed when bridging to a new critical reagent in vaccine assays? What are the recommendations on the best practice for standardizing processes for vaccine clinical assay qualification and validation of established immunoassays? How can we overcome linearity issues in vaccine assays, which are sometimes a mixture of different aspects like parallelism, relative accuracy, and dilution? What are strategies for short and long-term stability assessment for vaccine induced antibodies?
Assessment of QC Samples in CAR-T & Vaccine Flow Cytometry Assays: Current Industry Standards
What are the options when using test specific controls? Are they practical and meaningful? Are system level controls routinely used in clinical diagnostic testing applicable for CAR-T monitoring and appropriate for clinical trial submissions? What can CAR-T flow cytometry assays learn from experience with vaccine assay setup?
qPCR Assays for CAR-T Programs
What are considered critical reagents in qPCR assay? How do we perform stability for critical reagents for qPCR? Can we work with WHO to issue a universal human genomic DNA (gDNA) standard so we can use it to calibrate our qPCR assay across the industry by measuring the house keeping gene?
Immunogenicity Strategy for CAR-T Products
Is there agreement that it is straightforward to monitor host humoral responses considering that chronic impact of ADAs on CAR-T persistence is largely unknown or that data have yet to be seen on whether human scFv can result in lower ADAs and better product regarding persistence? Positive host cellular immune responses have been confirmed in treated subjects and seem to be correlated with clearance. Will the use of human scFv as CAR reduce cellular immune response rate? Can lessons on immunogenicity from autologous CAR-T products be applied to allogenic CAR-T products? What are the “right” assay(s) to monitor host cellular immune responses? How do we conduct fit for purpose validation?
DISCUSSIONS, CONSENSUS AND CONCLUSIONS
Clinical Vaccine Assay Validation
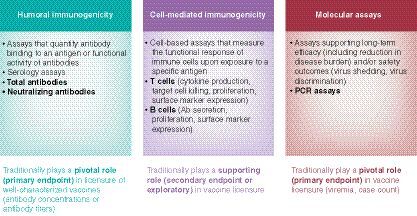
Immunogenicity measurements are key assessments for the clinical development of biotherapeutics and vaccines. While similar assay technologies are applied by the two fields, the goal of the clinical testing and the strategies applied are different. While the immunogenicity assessment for biotherapeutics is focused on the detection of very small amounts of ADA (sensitivity), the immunogenicity assessment for vaccines is more focused on the reproducible quantitation of the antibody response to the vaccine. For this reason, vaccine immunogenicity assessments typically do not use the tiered approach, which is an essential element for biotherapeutic immunogenicity assessments. Many different types of bioanalytical assays are used to assess endpoints in vaccine clinical development (refer to ); this white paper will focus on providing recommendations on serology assays of total antibodies to measure humoral immunogenicity.
Regulatory guidelines and multiple white papers are available to guide the pharmaceutical industry to adequately validate ADA assays [Citation18,Citation21,Citation25–27]. Guidelines and white papers specific for vaccine serology assays, on the other hand, are limited and would be very useful to understand general regulatory expectations regarding assay development, assay validation and clinical immunogenicity testing for vaccine clinical candidates early in development. The 2019 White Paper in Bioanalysis [Citation25] began building the foundation of a framework to define the expectations for vaccine assay qualification, validation, and life cycle management. The 2020 consensus developed the 2019 framework and further focused on providing more detailed and practical recommendations for clinical vaccine assay development and validation. Together, the 2019 & 2020 White Papers were designed to provide a harmonized reference for vaccine assay validation.
Regulatory agencies may request the demonstration of assay consistency over the life of the development program from applicants. In response to this regulatory request, it is recommended to use a phased approach to clinical vaccine assay development, separated into 3 distinct phases: 1) assay setup (establish assay format and run parameters), 2) qualification (determine assay performance), and 3) validation (confirm performance in “real life” conditions, with pre-defined acceptance criteria).
Assay Setup
The goal of the initial (set-up) phase of assay development is to identify the format of the assay and potential critical reagents. Understanding the pathogen and the immune response to the vaccine will lead to selection of relevant assays predictive of clinical benefit. The intended use of the assay needs to be clearly defined before selecting and optimizing the methodology. During this phase, development can be limited by sample availability. A barrier to the development of these assays is the lack of appropriate reference standards and positive control samples, particularly those which mimic the matrix and antibody profile of the samples to be tested prior to the first-in-human studies. Serum from pre-clinical studies (e.g., vaccinated laboratory animals) may provide similar breadth of the expected response and high titered serum for understanding the initial assay parameters. However, serum from naturally infected individuals, although not mimicking the antibody profile, may be a better representation of the anticipated human sample matrix. While neither sample is ideal for in-depth clinical assay development, they provide useful and necessary tools. This development phase is fit for a Phase I clinical trial.
Assay Qualification
Qualification is planned and conducted when sufficient human incurred samples are available. During qualification, all relevant assay performance criteria are evaluated. The selection of the qualification parameters is dependent on the use of the assay. These attributes typically include precision, linearity, specificity, accuracy (as applicable), limit of detection (LOD), and analytical range, i.e., LLOQ and upper limit of quantitation (ULOQ). In addition, assay robustness (acceptable variations in incubation times and temperatures) and ruggedness (impact of days, analysts and reagent lots) are ideally evaluated during this phase. Furthermore, sample stability (e.g., short-term stability) may also be evaluated (see analyte stability assessment below).
Linearity
It is recommended to perform the linearity assessment for quantitative vaccine assays. Linearity is a mixture of different aspects like parallelism, relative accuracy, and dilution, depending on the assay format. There is a difference between linearity over the signal range (i.e., from one starting dilution, similar to parallelism) and linearity over the assay concentration range (i.e., from several starting dilutions). To assess dilutional linearity, a dose proportionality approach is recommended. Samples covering the range of interest are serially diluted (independent dilutions) in multiple replicates. For each sample, the dose proportionality is assessed assuming a power model (10αDilutionβ)
Based on a criterion for the ratio between the dilution corrected extremes of the range considered, an acceptance range is computed for the slope in which the 90% confidence interval must fit. The range over which dose proportionality is demonstrated is obtained by a recursive search [Citation28]. Linearity can be determined using 3–5 samples covering the range of interest, serially diluted 4–5 times (independent dilutions) with negative sera. The samples should be run by a minimum of 2 analysts on 2 days at least in duplicate on each day. The expectations for the variability of back calculated titers should be determined.
Specificity
Analytical specificity is the ability of an analytical method to detect the analyte of interest; only the component it purports to measure or the extent to which the assay responds to all subsets of a specified analyte and not to other substances present in the sample. Specificity can be tested with pre-absorption experiments, where the test-serum is pre-incubated with either homologous or heterologous antigen. Initial experiments may use a limited sample set with multiple competitor protein concentrations. Final experiments should use a greater number of samples (5–10 samples) with a single competitor protein concentration. Optical density (OD) or Mean Fluorescent Intensity (MFI) of the sample tested needs to be within the linear range of the assay. The percent inhibition can be used to report specificity results. Multiplexed assays add a second layer of complexity to the specificity testing as the assay must be specific for the homologous protein, but not compete with other measured, heterologous antigens (see multiplex assays).
Limit of Detection
The LOD of an assay is defined as the lowest concentration that has a high probability of producing a response that will be distinguished from the background response (i.e., the response at zero concentration) as determined in a limit of blank (LOB) experiment. It can be determined by spiking 2-fold serial dilutions of a reference standard (where it exists) into negative or antibody-depleted human serum (ADHS). The LOD can also be determined considering the assay precision in the very low concentration range near the LOB.
Precision (Intra-, Inter- and Total Assay Variability)
Intra-assay variability (repeatability) represents within-run variation, while inter-assay variability represents the between-run variation (intermediate precision) attributable to different days, analysts, reagents, etc. The estimate of assay precision may be used to establish acceptance ranges for control samples, and to calculate a statistically meaningful fold-increase in antibody titers for an individual sample. The precision evaluation should test multiple incurred samples a minimum of 6 times.
Analytical Range
The LLOQ and ULOQ define the antibody concentration range over which the assay is acceptably accurate and precisely quantitates samples. LLOQs can be determined by evaluating the precision profile and assay relative accuracy. A recommendation is that at least 80% of the samples within the LLOQs must have variability estimates <20% relative standard deviation (RSD) for a standard antibody binding assay. Ideally, incurred samples are used for the evaluation of precision and relative accuracy because they best reflect the variability of the polyclonal immune response after vaccine administration.
Reference Standards
Assay maintenance activities need to ensure reproducible and precise titer determination over multiple years. A reference standard aligned with an international standard (when available) should be considered. When an international reference standard is unavailable, a high titered pool of samples can be used instead. Arbitrary units to define the standard can be used (e.g., C-value of the 4PL curve). However, it is important that test and reference sera have a parallel dilutional response curve. This parallelism allows sample concentration calculations over the widest range of the standard curve and gives assurance to the relationship of the sample values.
Qualification Reporting and Use
Following the completion of the qualification phase, it is recommended to write a full analytical development report, detailing experiments, experimental design and results (passed and failed). The report should conclude with recommendations for assay validation acceptance ranges. A detailed standard operating procedure (SOP) based on the final procedure used in the assay qualification is required and must be used in an assay validation. A qualified assay is suitable for the analysis of primary and secondary clinical endpoints of Phase I and Phase II clinical trials. Qualified assays may also be used for exploratory clinical endpoints of late-stage trials (Phase III). The interpretation of data from the qualified assay will help establish the design of the late Phase II/Phase III clinical trials.
Assay Validation
If the assay setup and qualification work is completed with a high level of quality, the final development step, assay validation, should be relatively easy and short compared to the initial development phases. Assay validation requires the testing of the assay performance against predefined acceptance criteria optimally using incurred samples spanning the entire analytical range, which are representative of the Phase III program. These criteria are established based on the results from the assay qualification data and the intended use of the assay. Parameters evaluated during validation include: precision, linearity, specificity, accuracy, LOD, quantification range (LLOQ and ULOQ). If any of the validation parameters fail, the validation is considered a failure and the cause of the failure should be reported and investigated. Validated assay support testing for late-stage clinical trials may be reviewed by regulatory agencies prior to Phase III testing.
Regulatory Interactions
Health authorities often request to review validation plans and/or qualification reports before validation and/or to confirm that assays are suitable for testing pivotal clinical study samples. Specific guidance on vaccine immuno-assay validation, issued by regulators, would help decrease the need for such pre-validation regulatory interactions.
Assay Life-Cycle Management
Since data for licensure of vaccines are generated throughout the clinical development program, it is necessary to demonstrate the stability of the assay performance over multiple years. To accomplish this need, many assays will use three quality control systems: implementation of an assay standard, trending, and proficiency panel testing. Assays may develop over time due to changes in conditions or reagents. Therefore, as part of the life cycle management of the assay, the validation should be periodically reassessed to determine if any additional validation work is required. Changes to the assay that may affect assay performance (e.g., new testing laboratories and changes in test samples, new age groups, specific disease populations) may require a partial validation or a full assay revalidation.
Analyte Stability Assessment
Short-term stability experiments assess whether sample handling and storage affect the assay results. The experiments should mimic conditions that are encountered during clinical testing (e.g., freeze/thaw cycles, short term storage at 2–8°C or room-temperature storage). Short-term stability can be assessed early during assay development (e.g., during qualification). If the assay is planned to be further developed, stability may be assessed at a later stage. It has been shown that antibodies are stable in serum or plasma stored at -20°C and -80°C beyond 3–4 years. For this reason, long-term stability studies for frozen matrices may not be required [Citation29,Citation30].
Ideally, samples used for stability experiments would cover the quantifiable range of the assay because incurred samples best reflect the heterogeneity and the matrix of samples used for clinical testing. If insufficient sample volume is available from clinical studies, spiking of negative samples or pooling of positive samples may be considered. When taking the complexity of multiplex assays into account, the number of stability samples per assay subtype might be reduced focusing on concentrations close to the LLOQ of the assay. It is, however, expected that every assay subtype is evaluated as part of the stability study.
Other stability parameters related to the setup and design of the assay should also be considered in a stability program. Critical reagents such as antigen coated plates or beads, and sample predilutions which can be created to enhance assay efficiency must be evaluated to demonstrate that routine experimental practices do not impact assay results.
Long-Term Assay Control
Long-term assay control is important in order to guarantee comparability of test results of long clinical trials and to allow the comparison of data between multiple clinical trials. Two tools may be implemented in order to achieve long-term assay control: assay trending and proficiency panel testing.
Assay trending is performed by analyzing run acceptance QC samples that are measured during routine clinical testing. Trending limits are stricter than run acceptance limits and serve as an early indicator for assay drift. It is recommended to pre-define the trending analysis in a trending plan. This document should describe the trending limits for the QCs, how often the trending is analyzed, the minimal amount of runs per trending-period, and how to react in case of out-of-trend events.
Proficiency panels are additional tools to monitor long-term assay performance. It is recommended to pre-specify the setup, characterization, and the testing of the proficiency panel in a plan. Ideally, a sufficient number of incurred samples that span the quantification range are analyzed multiple times on a regular basis. The volume of samples must be sufficient to run the proficiency panel over multiple years. Results from the panel should be reported in a formal report.
Critical Reagents
Critical reagents are reagents that may impact assay performance. The critical reagents should be identified during assay development and documented as part of the qualification and validation. A process for the bridging of critical reagents prior to use in clinical sample testing is important to guarantee a constant assay performance and avoid drift.
Ideally a new (candidate) critical reagent lot should be qualified by comparing its performance head-to-head to a qualified reagent lot. Minimally, the performance of QC samples should be assessed to qualify a new reagent lot. Optimally a panel of incurred samples is tested over multiple independent runs with the candidate and the qualified reagent lot. In some instances, the qualified lot is not available anymore (e.g., if expired). In this case, a comparison to historically generated data may be considered. The acceptance criteria for the critical reagent qualification should take into consideration the assay performance and the intended use of the assay. Keeping some of the reagents used during qualification/validation can help to assess whether the assay has changed its performance. This is only possible if the critical reagents have a long shelf-life.
Multiplex Testing
Multiplex testing allows the reduction of sample volume. This volume reduction is critical for pediatric studies but may also be beneficial in adult studies. Transitioning from a single-plex to a multiplexed format poses some challenges. Meaningful oversight on robust assay performance and successful critical reagent bridging strategies are more difficult to establish when working with multiplexed assay formats.
It is recommended that all validation parameters are redeveloped and revalidated when transitioning a previously validated vaccine assay into a multiplexed format. Specificity and cross-reactivity will have a major impact on a multiplex format and necessitate re-assessing antigen concentrations and potentially to establish a new reference standard. However, if the change is restricted to the read-out only, then partial validation may be sufficient.
If bridging needs to be established to the previous assay format there should be pre-defined acceptance criteria. Clinical endpoints and data interpretation may be relevant in defining the acceptance criteria of the assay bridging.
A change of platforms is not recommended during Phase III because equivalency may not be possible to demonstrate.
Assessment of QC Samples in CAR-T & Vaccine Flow Cytometry Assays: Current Industry Standards
CAR-T therapies present unprecedented opportunities and challenges for bioanalytical scientists. We are witnessing an explosion in the numbers of next generation CAR-T agents and new clinical trials which is generating an increasing interest in standardizing assays and ensuring quality control. Flow cytometry assays are a key methodology used for monitoring PK/cellular kinetics and efficacy of CAR-T therapies in clinical trials and are also growing in popularity for vaccine trials. Flow cytometry can document vaccine-induced versus natural immunogenicity; vaccine take and response rate (efficacy); support the justification of the final vaccine formulation; and demonstrate non-inferiority versus other vaccines. High parametric flow cytometry can be applied to clinical trial T-cell exploratory endpoints but requires rigorous fit-for-purpose instrument optimization, antibody (Ab) panel design, sample preparation, assay setup and data analysis.
There is currently no finalized regulatory guidance for general flow cytometry assay validation or specifically for the measurement of CAR-T levels by flow cytometry, prompting a consortium of experts from the International Clinical Cytometry society and CLSI to develop guidelines [Citation31]. The main regulatory concern is to demonstrate that the CAR-T assays are actually measuring the desired analyte and QC being used to prove this is representative of samples. To address this, the 2019 White Paper in Bioanalysis recommended the use of QC samples relevant to the cell population of interest such as stabilized whole blood, cryopreserved PBMCs, or “spiked” QCs. Implementation of QCs was recommended to at least periodically track that the assay is performing consistently [Citation25].
Based on real-world experiences encountered with the first approved CAR-T therapy, the 2020 recommendations are focused on describing the practical limitations associated with implementation of traditional QCs, while proposing alternate approaches to ensure the quality of these high complexity flow cytometry assays. These approaches include instrument standardization, appropriate panel design to exclude unwanted cells, personalized gating controls, viability dyes, and bead-based or volumetric approaches for cell counting. Regulatory considerations and future perspectives were also discussed. Two types of controls are currently utilized: test specific controls and system level controls.
Test specific controls are QCs with varying CAR-T levels including transfected cell lines, transduced healthy donor cells, or patient drug product, each with their own benefits and limitations. Transfected cell lines are an excellent option for creating homogenous controls expressing known levels of CAR-T construct. However, these do not contain additional T cell markers required for developing gating approaches for patient samples. While transduced healthy donor T cells do not exhibit light scatter properties of patient specimens exposed to chemotherapy, they are the most practical option for establishing initial gating methods and validation parameters. Patient specific T cells or drug product are ideal controls, but their availability is limited for CAR-T monitoring assays due to their prioritized use in product release assays and treatment of patients. Reliability of test specific controls is based on 3 key factors: (1) instrument, (2) stability of detection reagent and (3) stability of test specific control that should be monitored. A central lab should be used to standardize protocol, using the same instrument type and optical bench configuration, the same cell line, and the same lots of critical reagents to harmonize results across all sites.
System level controls are routinely used in clinical diagnosis and are thus reliable for monitoring CAR-T PK (see ); these approaches have received support from subject matter experts representing FDA and MHRA. Fluorescence Minus One (FMO) controls are used as gating controls to identify the gating boundary for the one antibody that is missing and used to identify background staining due to fluorescence spillover. Isotype controls may be used for identifying nonspecific staining. Instrument QC controls and experimental controls (positive, negative) are important to ensure the assay is performing as expected. Certified participation in a relevant external quality assessment scheme is essential. The use of a single central laboratory is recommended if possible, or evidence to support comparability of results across participating laboratories is required. QCs are not necessary on every sample run but should be used often enough to demonstrate reliable results and should undergo QA oversight. For cocktail antibodies, if they are prepared in-house, stability needs to be demonstrated. If they are sourced from a vendor, the provided stability can be leveraged. In addition, lot to lot bridging study is crucial to qualify the release of new reagent lot.
CAR-T Flow Cytometry assays can also benefit from vaccine experience with instrument and assay setup. It is recommended to use latest peer-reviewed guidelines for the use of flow cytometry and cell sorting in immunological studies to support CAR-T assay development [Citation32]. Instrument setup should be based on the stain index measurement for each detector to determine the best sensitivity and minimize the spillover/spread matrix (SSM). Daily performance checks of instrument precision and sensitivity [Citation33,Citation34] using 3 sets of beads (neutral comp, single peak and rainbow beads) are also needed. It is important to check linearity of the photomultiplier tube (PMT) response. Assay setup defines protocol optimization, background evaluation, gating strategy, specificity, precision, linearity and LOB/LOD. CAR-T detection is usually done together with other cell surface markers in a multi-color, multiplexing fashion. Potential interference from other staining reagents should be investigated thoroughly. In the absence of SRMs for particular flow cytometric applications, it is challenging to perform some performance measures. It is recommended to design appropriate approach and ask regulatory agency for early feedback.
qPCR Assay for CAR-T Programs
Because of its high sensitivity and sampling convenience, qPCR is the most commonly used methodology (even if the use of ddPCR is increasing) for monitoring the fate of CAR-T cells and is especially useful for monitoring low quantities of CAR-T cells as part of long-term studies. Optimal qPCR primers can detect the CAR-T inserted transgene. There is limited regulatory guidance and qPCR method development and validation to support regulated bioanalysis for CAR-T therapies in clinical studies. Discussions built upon the 2019 White Paper recommendations on qPCR validation [Citation25] which recommended following scientifically-led method development and validation strategies, with support from the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) and CLSI guidance [Citation35,Citation36]. A two-phased approach was suggested with initial qualification to determine what performance characteristics can be achieved, followed by validation against pre-defined criteria.
The recommended validation parameters for qPCR assays for CAR-T programs includes sensitivity or limit of quantitation (LLOQ; 50 copies/μg gDNA) and LOD, intra- and inter-assay precision, accuracy, DNA extraction efficiency from tissues, and engineering controls to ensure there is no cross contamination, PCR efficiency, PCR linearity, specificity and selectivity, and robustness. Currently, there is no requirement for performing incurred sample reproducibility (ISR).
For accuracy and precision, QCs can be plasmid spiked into human gDNA for transgene or plasmid only for reference gene or spiked into surrogate nonhuman gDNA. CAR-T from normal donors spiked into diseased whole blood can demonstrate intra- and inter-assay variability while accuracy can be inferred from reference gene measurement from a qualified lot of human gDNA.
Recommendations were also provided for stability assessment of critical reagents which were identified as primers, probes, reference standards, master mix, and positive control (cell lines). To assess their stability, it was recommended to establish a critical reagent qualification program for lot-to-lot bridging, stability, etc.
Finally, the question was raised whether the industry should work with WHO to issue a universal human genomic DNA standard to calibrate qPCR assays across the industry by measuring the house keeping gene. Two options were suggested for these standards. The first was to consider NIBSC wild-type standard 18/164 [widely used as a standard in cancer genome testing] to calibrate CAR-T assays once the copy-number of the reference gene of choice has been characterized. The second proposed the use of WHO 1st International Reference Panel (19/158) for the quantitation of Lentiviral Vector Integration Copy Numbers which will be released shortly.
Immunogenicity Strategy for CAR-T Products
Persistence of CAR-T cells in the subject's circulation plays a critical role in long term efficacy while it could also pose a potential long-term safety risk during treatment and after remission. Immunogenicity to CAR-T products is expected to be more complex when compared to protein biologics. It can be generated from host humoral and cellular responses due to the unique CAR-T product structure and design. Information on immunogenicity to CAR-T products is quite limited due to the fact that most of CAR-T products are still under clinical development [Citation37]. Potential clinical consequences of immunogenicity are largely unknown and are currently still being monitored in clinical studies for each product.
The discussion centered around the bioanalytical strategies and fit for purpose experimental methodologies applied to monitor clinical immunogenicity of CAR-T products based on risk factors and product structure/design. Host humoral immune responses can be measured with LBA-based or cell-based formats [Citation38,Citation39]. FDA still recommends that this assessment be performed for autologous CAR-T cells. Allo-CARs may induce cellular response. There are limited data on the impact of humoral/ cellular response on CAR-T cells safety, efficacy and persistence. Many other transgenes are expressed; humoral responses to all the transgenes are not studied and the impact is unknown. There is a low incidence of cellular immune response against CAR, and no clear relationship between cellular immunity and clinical outcomes exists.
Positive host cellular immune responses have been confirmed in treated subjects and seem to be correlated with clearance [Citation40]. However, supporting data is lacking as many studies do not assess cellular immunogenicity. It was discussed if the use of human scFv as CAR could reduce cellular immune response incidence. It was concluded that CAR can be immunogenic, regardless of the species of origin of the scFv, because foreign sequences are expressed, or novel epitopes are created. Therefore, an immunogenicity assessment is recommended for humanized CAR-T cell therapy. The risk lies in the full length of the CAR construct.
It was also discussed whether lessons on immunogenicity from autologous CAR-T products can be applied to allogenic CAR-T products and how these assays are developed and validated. It was recommended to focus on the clinical problems when developing an assay, such as the context of adverse events or failure of efficacy and how immunogenicity could be implicated in those issues. Generally, immunogenicity concerns are greater for allo-CARs. Therefore, it is recommended to adopt similar approaches to autologous CAR-T cells. For cellular immune response assays (for CAR T and AAV-based gene therapies), both ELISPOT and flow cytometry have been used [Citation41,Citation42].
RECOMMENDATIONS
Below is a summary of the recommendations made during the 14th WRIB:
Clinical Vaccine Assay Validation
Specific Industry/Regulator recommendations for vaccine serology assays would be very helpful to understand general regulatory expectations regarding assay development, assay validation and clinical immunogenicity testing for vaccine clinical candidates early in development
Assay consistency should be demonstrated over the life of the development program
It is recommended to use a phased approach to clinical vaccine assay development, separated into 3 distinct phases: 1) assay setup (establish assay format and run parameters), 2) qualification (determine assay performance), and 3) validation (confirm performance in “real life” conditions with pre-defined acceptance criteria). Refer to for parameters recommended for each phase.
The intended use of the assay needs to be clearly defined before selecting and optimizing the methodology during assay setup.
Assay qualification can only be planned and conducted once sufficient human incurred samples are available.
Perform the linearity assessment for quantitative vaccine assays using 3–5 samples covering the range of interest, serially diluted 4–5 times (independent dilutions) with negative sera. The samples should be run by a minimum of 2 analysts on 2 days at least in duplicate on each day. The expectations for the variability of back calculated titers should be determined.
To assess dilutional linearity, a dose proportionality approach assuming a power model (10αDilutionβ) is recommended.
Test specificity on a minimum of 5–10 samples covering the analytical range in a competition experiment using homologous and heterologous protein. OD or MFI of the sample tested needs to be within the linear range of the assay. The percent inhibition can be used to report specificity results.
LOD is set at the lowest concentration that has a high probability of producing a response that will be distinguished from the background response as determined in a LOB experiment by spiking 2-fold serial dilutions of a reference standard (where it exits) into negative or ADHS or determined considering the assay precision in the very low concentration range near the LOB
LLOQ can be determined by evaluating the precision profile and assay relative accuracy. At least 80% of the samples within the LLOQs must have variability estimates <20% RSD for a standard antibody binding assay.
Precision should test multiple incurred samples a minimum of 6 times.
Write a full analytical development report, detailing experiments, experimental design and results (passed or failed). The report should conclude with recommendations for assay validation acceptance ranges.
A detailed SOP based on the final procedure used in the assay qualification is required and must be used in an assay validation.
Assay validation requires the testing of the assay performance against predefined acceptance criteria optimally using incurred samples spanning the entire analytical range, which are representative of the Phase III program.
If any of the validation parameters fail, the validation is considered a failure and the cause of the failure should be reported and investigated.
Assay validation may periodically be reassessed to determine if any additional validation work is required. Changes to the assay could trigger assay revalidation (e.g., new testing laboratories and changes in test samples).
Standard and test samples need a parallel response from several points on the curve and use of arbitrary units to define the standard can be used (e.g., C-value of the 4PL curve).
Formal trending plans and proficiency panel testing are recommended elements of the serology assay maintenance strategy.
Short-term stability experiments should mimic conditions that are encountered during clinical testing (e.g., freeze/thaw cycles, short term storage at 2–8°C or room-temperature storage).
Long-term stability of antibodies is generally accepted in serum or plasma stored at -20°C and -80°C beyond 3–4 years. For this reason, long-term stability studies for frozen matrices may not be required.
Regulatory agencies may be consulted to confirm agreement on the design and acceptance criteria proposed for validation.
Trending plans
Performed by analyzing run acceptance QC samples
Trending limits are stricter than run acceptance limits
Trending plan defines limits, frequency, and when investigation is required
Proficiency panels
Titers must cover the entire analytical range of the assay.
Volume of samples must be sufficient to perform multiple runs.
Results from the panel should be reported in a formal report.
To define acceptance criteria for bridging critical reagent lots, consider both the intended use of the assay and the assay variability.
Minimally QC samples should be used to qualify a new reagent lot.
Testing should be completed across multiple independent runs with the candidate and qualified lot
If the qualified lot is not available anymore (e.g., if expired), a comparison to historically generated data may be considered
It is recommended that all validation parameters should be redeveloped and revalidated when transitioning a previously validated vaccine assay into a multiplexed format.
Re-assess antigen concentrations and potentially to establish a new reference standard.
If bridging needs to be established to the previous assay format there should be pre-defined acceptance criteria.
A change of platforms is not recommended during Phase III because equivalency may not be possible to demonstrate.
Assessment of QC Samples in CAR-T & Vaccine Flow Cytometry Assays: Current Industry Standards
High parametric flow cytometry can be applied to clinical trial T-cell exploratory endpoints but requires fit-for-purpose albeit rigorous instrument optimization, Ab panel design, sample preparation, assay setup and data analysis.
Two types of controls are currently suggested: test specific controls and system level controls.
Patient specific T cells or drug products are the most ideal option for use as test specific controls but have limited availability.
Reliability of test specific controls is based on 3 key factors: (1) instrument, (2) stability of detection reagent and (3) stability of test specific control that should be monitored.
A central lab should be used to standardize protocol, using same instrument type and optical bench configuration, the same cell line, and the same lots of critical reagents to harmonize results across all sites.
System level controls are reliable for monitoring CAR-T PK/cellular kinetics. Refer to for recommendations.
It is recommended to use latest peer-reviewed guidelines for the use of flow cytometry and cell sorting in immunological studies to support CAR-T assay development.
Instrument setup should be based on the stain index measurement for each detector to determine the best sensitivity and minimize the spillover spread matrix (SSM).
Daily performance checks of precision and sensitivity using 3 sets of beads (neutral comp, single peak and rainbow beads) are needed.
Potential interference from other staining reagents should be investigated thoroughly.
qPCR assay for CAR-T Programs
The recommended validation parameters for qPCR assays for CAR-T programs includes:
Sensitivity or limit of quantitation (LLOQ; 50 copies/μg gDNA) and LOD (range of response),
Intra- and inter-assay precision: CAR-T from normal donors spiked into diseased whole blood can demonstrate intra- and inter-assay variability
Accuracy: can be inferred from reference gene measurement from a qualified lot of human gDNA
DNA extraction efficiency from tissues
Engineering controls to ensure there is no cross contamination
PCR efficiency
PCR linearity
Specificity and selectivity
Robustness
ISR does not need to be performed
Critical reagents were identified as primers, probes, reference standards, master mix, and positive control (cell lines)
It was recommended to establish a critical reagent qualification program for lot-to-lot bridging, stability, etc
NIBSC wild-type standard 18/164 or WHO 1st International Reference Panel (19/158) can be used as a universal human genomic DNA standard to calibrate qPCR assays across the industry by measuring the house keeping gene
Immunogenicity Strategy for CAR-T Products
Host humoral immune responses should be measured with LBA-based or cell-based formats for autologous CAR-T cells
CAR can be immunogenic, regardless of the species of origin of the scFv, because foreign sequences are expressed, or novel epitopes are created. Therefore, an immunogenicity assessment is recommended for humanized CAR-T cell therapy. The risk lies in the full length of the CAR construct
It was recommended to focus on the clinical problems when developing an assay for allogenic CAR-T products, such as the context of adverse events or failure of efficacy and how immunogenicity could be implicated in those issues
Immunogenicity concerns are greater for allo-CARs. Therefore, it is recommended to adopt similar approaches to autologous CAR-T cells; however, additional considerations may be needed for allogeneic cells
SECTION 2–Gene Therapy, qPCR, NGS and ELISpot Validation
Yanmei Lu19, Mark Milton22, Lisa Kierstead9, Jean-Claude Marshall21, Andrew Exley7, Jason DelCarpini14, Fabio Garofolo8, Boris Gorovits17, Swati Gupta15, Lynne Jesaitis16, John Kamerud17, Arno Kromminga18, Anna Ma20, Jim McNally23, Natasha Savoie11, Richard Siggers12, Therese Solstad13
Authors in Section 2 are presented in alphabetical order of their last name, with the exception of the first five authors who were major contributors.
The affiliations can be found at the beginning of the article
DISCUSSION TOPICS & CONSOLIDATED QUESTIONS COLLECTED FROM THE GLOBAL BIOANALYTICAL COMMUNITY
The topics detailed below were considered as the most relevant ‘hot topics’ based on feedback collected from the 13th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 14th WRIB. The background on each issue, discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
qPCR, ddPCR, and NGS Assay Development & Validation: Best Practices
Do we need to update the 2019 recommendations [Citation25] on qPCR validation to further harmonize industry best practice? Are different companies still using different levels of qPCR validation? Are the principles of FFP validation well understood for qPCR or is there still confusion with the assays to be “characterized, qualified or fully validated”? What is required for those validation levels for clinical testing (CLIA laboratory developed tests (LDT) versus Good Clinical Laboratory Practices)? What assay parameters need to be evaluated for NGS and qPCR-based quantitative insertion–deletion mutations (indels) and integration assays? Is there anything we need to add to the 2019 recommendations on assay parameters or generation and characterization of reference standards and QCs for assay development/qualification and assay performance monitoring [Citation25]? NGS computational analysis provides quantitative %indel results based on sequences without the need of using a standard curve, but back calculating %indel against a standard curve increases assay robustness. Should %indel be adjusted using a standard curve? What are the current best practices for managing ddPCR technical challenges? Does guidance on ddPCR need to be implemented? What is the best practice for manually setting the positive/negative threshold when the analysis software is unable to? Frequently the negative control is zero (no positive droplets detected) with multiple independent analyses. If it is zero, what is the best method to establish the LOD? Is less than 90% amplification efficiency acceptable if the assay is quantitative with an otherwise acceptable performance? Would you use ddPCR to determine both biodistribution and shedding of the vector/transgene?
AAV Capsid NAb Assays Development and Validation
Should anti-capsid antibody assays (anti-drug antibody (ADA) and NAb) be used as exclusion criteria for clinical trials of AAV-based gene therapy (GTx)? How is a clinically relevant cut off determined? What regulatory framework is used for these assays: typical ADA guidance or CLIA? Would these assays be considered companion diagnostics (CDx) when the therapeutic is approved? Should cellular immunity assays (such as enzyme-linked immunospot (ELISpot)) for capsid or transgene product be used as exclusion criteria for clinical trials of AAV-based GTx?
ELISpot & Single Cell Western Blot Assay Validation
Do we need to update the 2019 recommendations on ELISpot validation to further harmonize the industry best practice, duplicate or triplicate analysis, cytokine read out, qualification level and definition of positive ELISpot responses [Citation25]? What assay parameters need to be evaluated for ELISpot validation? How does the amount of variability for these assays factor into a potential validation plan? Is there anything we need to add to the 2019 recommendations on assay parameters of precision, assay range (LOQs), ruggedness, linearity [Citation25]? What are the FFP acceptance criteria for Single Cell Western Assays? Are LBA criteria (20%–25%) suitable?
Application of Current FDA/EMA Immunogenicity Guidance/Guideline to Gene Therapy
Are the current Immunogenicity Guidance/Guidelines (FDA/EMA) for biotherapeutics fully applicable to gene and cell therapy? Are we seeing the implementation of clinically relevant immunogenicity strategies via inclusion criteria? Do we have enough data to know what titers are relevant to impact transduction? What is current industry experience?
DISCUSSIONS, CONSENSUS AND CONCLUSIONS
qPCR, ddPCR, and NGS Assay Development and Validation: Best Practices
qPCR Validation
The number of GTx in development has grown significantly in recent years. AAVs have become the most prevalent delivery vector among viral based GTx. Many of these GTx are aimed at treating rare genetic conditions by introducing a functional copy of the gene to restore the function of the protein. Several regulatory guidelines have been published by the EMA [Citation43], FDA [Citation44] and PMDA [Citation45] to provide industry with general considerations related to AAV GTx clinical development, including discussions and recommendations related to the diverse bioanalytical support needed for these unique therapies.
One of the requirements for AAV GTx clinical development is to understand the shedding kinetics and potential infectivity of viral particles by patients after a single dose of AAV GTx. Vector shedding is a concern as exposure to naïve individuals could induce a NAb response rendering future treatment ineffective. Shedding assays are a requirement of EMA, FDA, and PMDA [Citation43–45]. qPCR assays that quantitatively detect the product specific nucleic acid are highly specific, sensitive, reproducible and high throughput. Due to these advantages, qPCR has been the primary assay even though it cannot differentiate intact versus non-infectious or degraded virus. Secondary infectivity assay development is recommended by EMA [Citation43] and PMDA [Citation45] to ensure that rare recombination events do not occur which can enable infection by these vector genome replication incompetent AAVs. The need for the quantification of shed viral vector in a variety of matrix types is clear; however, the level to which these assays should be validated and the specific development criteria for these assays are less clearly defined. Per FDA [Citation44], often an assay with a quantitative readout, like qPCR is used because of the ease of performing and standardizing the assay, high throughput format, rapid turnaround time, and assay sensitivity.
Regulatory agencies require measurement of shed AAV particles from patients in a variety of matrix types, including whole blood, plasma, saliva, urine, semen, stool and potentially tears depending on route of delivery. FDA and EMA have consistently required urine, stool, and saliva [Citation43,Citation44]. Each matrix type can pose its own unique challenge in terms of qPCR and/or ddPCR assay development and should be considered in the overall assay development and validation [Citation46].
Previous White Papers [Citation21,Citation25] have provided the basis on to approach qPCR assay validation in bioanalytical laboratories where the sensitivity requirements may be different in preclinical vs. clinical assays. Similar to LBA validations, assay performance controls and QCs should be established to monitor accuracy, precision, range of quantification, analytical sensitivity and specificity using clinical material if at all possible. Previous discussions have also given strategies for performing qPCR and infectivity assays in challenging matrices such as urine [Citation47] by using a surrogate marker by staining for a late stage viral replication protein in a cell-based assay. Different companies are using different levels of validation, indicating the need for clear industry/regulator recommendations on which parameters should be evaluated. Regulators recommended the application of general principles such as performing FFP validation based upon COU but provided few details. The following 2020 recommendations are aimed to give further practical guidance into the design of qPCR assays for GTx.
Assay Qualification and Validation
qPCR assays should be qualified and validated by the sponsor, with a clear understanding of the assay sensitivity, specificity, reproducibility and variability in each matrix to be tested (). Fit for purpose validation is generally to verify interference and avoid cross-contamination. Spike recovery with qualified internal control in samples is preferable to assess for interference.
For PCR primer probe set efficiencies, most laboratories use criteria of 90–110%, but some go as low as 85% efficiency. Lower than 85% amplification efficiency is not recommended because low efficiency is generally caused by poor primer/probe designs and assay conditions. These are often associated with poor assay performance and may not be suitable for long-term use for sample analysis.
Acceptance Criteria
Acceptance criteria should be established before validation. Specific criteria should be evaluated case by case since specifics will differ with assay and COU. Laboratories should refer to the previously mentioned papers discussing qPCR validation. Most of assays may not meet criteria for accuracy and precision for small molecule assays with %Bias and %CV at 15% (LLOQ at 20%), respectively. Some assays can meet LBA criteria for precision and accuracy criteria with %Bias and %CV at 20% (LLOQ at 25%), respectively but this should be defined on an assay by assay basis and in terms of COU.
Primer/Probe Selection
The primer and probe selection are critical for assay performance [Citation48]. No template controls (NTC) and baseline areas should be clear and clean with proper instrument calibration for the dye. The exponential phase needs to show a strong, straight growth; 100% efficient PCRs show 1 Ct difference between successive 2-fold dilutions with less than 10% CV difference between technical replicates. The plateau should be as close to horizontal as possible (e.g., within +/- 1 Ct difference) and individual replicates should plateau at the same fluorescence level. If the plateau is not horizontal, this indicates that the PCR is not efficient. This can happen if the denaturation midpoint (Tm) of primers in the reaction differs by more than 5°C, especially for primer/probes designed in GC (guanine-cytosine) rich genomic regions. Different concentration levels should plateau at the same level otherwise this indicates reduced assay sensitivity. It was also recommended to design and test multiple oligomer sets per target and perform basic local alignment search tool (BLAST) search to avoid primer dimers for specificity. In addition to a BLAST search, assessment of the primer probe set using gDNA from the same species of intended use to ensure no cross reactivity should also be done during development. Target isolation procedures should be tested for extraction efficiency and potential assay interference using A280/260 or other methods to quantify gDNA/cDNA to normalize results (or use reference targets). Critical reagents (primer, probe, master mix) need to be optimized for assay performance and stability. Each step of thermal cycling should also be optimized for an amplification efficiency above 90% using independently prepared replicate controls and samples. A factorial approach should be used to test factors that impact performance such as primer concentrations, probe concentrations, annealing temperatures, and master mix type. Slope, earliest Ct, and highest fluorescence responses should be assessed for optimization.
Calibrator Material and Assay Controls
Calibrator material should be equivalent or very similar to test samples. If possible, use clinical grade material although it is acknowledged that using clinical grade material may preclude some patients from being treated due to their limited supply. During the design phase, ss/ds (single stranded/double stranded) DNA template can be used (whole or partial). The extractable material for development should demonstrate extraction efficiency with encapsulated ss/ds DNA or cloned/synthesized DNA. If the assay is intended to be used for an extended period of time, it is preferred to use current Good Manufacturing Practices (cGMP) material, but this may not be feasible due to manufacturing constraints. Research lots may be used as long as they are within the same context of use, but bridging to a clinical lot may be required. Long term stability should be performed.
NGS Assay Development and Qualification
Gene editing is advancing rapidly toward clinical applications. For example, AAV-mediated in vivo delivery of gene editing reagents together with a transgene is being developed to introduce a functional gene into the albumin locus of patient hepatocytes. Transgene integration is achieved through engaging homology directed repair (HDR) as well as non-homologous end joining (NHEJ) DNA repair pathways. In addition, the NHEJ DNA repair process can also lead to the introduction of short insertions and deletions (indels) of nucleotides, without transgene integration. Measurement of indels may be used as a surrogate of genome editing efficiency. The evaluation of indels at previously identified off-target sites is, furthermore, essential for monitoring patient safety in clinical trials.
NGS is the technology of choice for the quantification of indels because of its high discovery power for heterogenous indel variants, the ability to multiplex samples and analytes, and the requirement for small DNA input material. For transgene integration, the HDR and NHEJ DNA repair mechanisms generate different DNA sequences at the target integration site, but they produce identical albumin-transgene fusion messenger RNA (mRNA) as a result of pre-mRNA splicing. Reverse transcription (RT) followed by qPCR is a suitable method to quantify transcripts with known sequences.
It was recommended for assay qualification to include precision, accuracy, and sensitivity; reference standards and quality controls were prepared by mixing genomic DNA extracted from a clonal liver cell line carrying 100% indels with wild type unedited genomic DNA at varying ratios. Although next generation sequencing computational analysis pipelines provides quantitative %indels data for patient samples, back calculating %indels against the standard curve makes the assay more robust. This can be especially important in situations where small biopsy samples yield limited amounts of genomic DNA that do not allow for assay failure and repeat sample testing. Discussions uncovered that most sponsors do not adjust %indel using a standard curve but it was agreed that %indel can be adjusted using a standard curve when only limited gDNA material is available.
Regulatory guidance for quantitative molecular assays to monitor editing in patients is lacking. Assay parameters to be evaluated for NGS and qPCR-based quantitative indel and integration assays during validation for intended purpose were discussed. It was recommended that qualification parameters to be evaluated for NGS-based clinical indel assays include gDNA input, specificity of PCR reactions, sequencing coverage, quantifiable range and LOD, precision and accuracy, selectivity, and gDNA stability. In addition, bridging and linearity should be considered when applicable.
It was recommended that in absence of a universal reference standard, a well characterized/qualified reference standard can be used; consideration should be taken for lot-to-lot bridging to ensure consistency. It was also recommended that reference standards and QCs for NGS based clinical indel assays should be generated with different levels of indels such as mixing cell lines of known indel levels with unedited parental cells, followed by gDNA extraction. If possible, the known indel levels would be best confirmed by using an orthogonal method. Stability of reference standards and QCs should be tested, and a qualification/bridging program should be established.
It was recommended that parameters to be evaluated for RT-qPCR gene integration assays include RNA input, specificity of RT-qPCR reactions, quantifiable range and LOD, precision and accuracy, selectivity, tissue storage and shipping conditions to ensure RNA integrity, and QC RNA stability. Reference standards and QCs for RT-qPCR gene integration assay can be generated and characterized by, for example, cloning the fusion cDNA and wild type DNA into a plasmid under the control of a T7 promoter. In vitro transcription can be used to synthesize RNA reference material. The use of A260 to calculate copy number based on size of transcript was recommended. A qualification/bridging program should be established for reference standards and QCs.
ddPCR Assay Development: Best Practices
The efficacy and safety evaluation of gene/cell therapies during development requires measuring target nucleotide sequence levels following their delivery/editing and characterizing their biological distribution and potential to release into the environment. qPCR has historically been the bioanalytical workhorse in studies of nucleic acid quantitation and characterization. More recently a newer technology, droplet digital PCR (ddPCR), has been added to the gene/cell therapy bioanalytical assay toolkit. ddPCR is a dPCR method based on water-oil emulsion partitions or droplets, Similarities between the two PCR methods include the common use of TaqMan chemistry comprised of target-specific primers and fluorescent probes to amplify and detect target sequences in samples. A key difference is the process by which source amplicons are quantitated. In real-time qPCR, the magnitude of fluorescence is measured continuously. The cycle at which the signal is detected above threshold (Ct) or reaches maximal increase in released fluorophore (Cp) is used to determine the concentration of the target sequence by interpolation from a standard calibrator curve. In contrast, dPCR involves the partitioning of target and background sequences to an estimated single copy prior to amplification. End-point reactions are analyzed in thousands of partitions for the presence (positive) and absence (negative) of fluorescence to determine the absolute number of target sequences, without the need for a standard curve and with increased tolerance to variable efficiency of amplification and matrix interference. ddPCR enables specific, accurate quantification of a vector/transgene construct. It readily enables multiplexing, which can be challenging to establish in a qPCR environment where amplicons compete for resources and efficiency close to 2 must be maintained for both amplicons. Multi-color ddPCR facilitates assays that are challenging on the qPCR platforms due to amplicon structure, such as quantitative measurement of AAV vector ITR fusions in cells/tissues over time. It also enables assays that are essentially impossible on the qPCR platform, such as determination of linkage between two different sites on a vector as a measure of vector integrity.
The utility of ddPCR to determine vector shedding and biodistribution as well as transgene biodistribution and expression was discussed. It was concluded that ddPCR can be used for both biodistribution and shedding studies but it was recommended that the same technology be used for both. With respect to both q and ddPCR, it was recommended that a yield threshold be determined for samples with low levels of total DNA (e.g., urine). There is a lack of guidance on collection of spot samples or collection of samples over a defined period of time. Hence, it was recommended for low DNA yield, samples can be stored in single-use aliquots.
Due to the newness of the technology, the current recommendations represent the best practices for managing ddPCR technical challenges. Moreover, another resource is the digital MIQE guidelines: Minimum Information for the Publication of Quantitative Digital PCR Experiments [Citation49].
The 2020 recommendations describe the best practices for manually setting the positive/negative threshold when the analysis software is unable to. It was recommended to use positive controls as guides and/or evaluate 1 or 2D scatter plots and histograms for separation of single from double positives and positives from negatives and to draw appropriate thresholds. The event is documented, verified by a second scientist, and the original and manual raw data and analysis files are saved.
Frequently the negative control is zero (no positive droplets are detected) with multiple independent analyses. The best method to establish the LOD is to determine the lowest concentration at which 95% of the positive samples are detected. In other words, within a group of replicates containing the target at concentrations at the LOD, no more than 5% failed reactions should occur [Citation35]. Alternatives to calculating the LOD include probability analysis or the maximum likelihood estimation method.
If the assay is quantitative with acceptable performance, less than 90% amplification efficiency due to amplicon structure between the primers (e.g., hairpin loops, high GC content), may be acceptable if the assay is FFP and with proper justification supported by other measures (accuracy, precision, linearity). ddPCR is less impacted by amplification efficiency–with reduced efficiency, the droplet is still positive though the signal may be reduced. As long as there is a clear separation of positive/negative, there is no impact to ddPCR quantification. With respect to primer/probe sets, design considerations are similar to those for qPCR and reaction optimization is recommended. Annealing temperature gradients can be used to optimize the separation of positives and negatives. Optimization may include confirmation of quantitative results with an independent nearby primer/probe set [Citation49].
AAV Capsid NAb Assays Development & Validation
Development of GTx utilizing AAV vectors requires a number of unique approaches for bioanalytical and immunogenicity evaluation. Among the most important of these are assays to detect antibodies which may inhibit the ability of the viral capsid to transfect target cells, also known as NAb or transduction inhibition (TI) assays. These cell-based assays are routinely used to measure the ability of patient derived antibodies to block cellular infection. The presence of NAbs has been associated with poor therapeutic efficacy, thus positivity in the assays has sometimes been used as exclusion criteria for clinical trials. Induction of NAb by the initial administration of AAV limits re-administration of the same serotype capsid. Exclusion of anti-AAV NAb seropositive subjects is common in clinical studies [Citation50]. The use of anti-capsid NAb or TI assays and the methodology to identify parameters which may impact performance was discussed. In addition, the regulatory requirements for such testing and the need for developing CDx devices prior to marketing approval of the therapeutic product were discussed.
LBA approaches are used to detect total anti-AAV antibodies and cell-based assays are used for anti-AAV NAb assays. A bridging assay format detecting both non-neutralizing and neutralizing antibodies and using cut point as in other ADA assays gives better analytical sensitivity and specificity than cell-based assays. Cell-based NAb assays measure AAV capsid transduction efficiency in cultured cells using an AAV capsid engineered with reporter gene products. NAb reduces transduction efficiency of AAV capsids and reduces assay signals. There are serum components and compounds other than NAbs present in blood that can inhibit the transduction of cells by AAV capsids. Thus, it is more accurately called transduction inhibition assay although some factors may even enhance transduction. Often a 50% inhibition cut-off has been used to define sample positive/negative score and the reported titer. A statistically defined cut-point value is recommended going forward to align with the approach applied for protein based biotherapeutics. The titer used as threshold for NAb positivity is empirically determined and may differ from one product or assay to the next. Parameters to be optimized include: cell line (HEK 293, HeLa, CHO, Huh-7), cell number, serum (negative control, heat inactivation), multiplicity of infection (MOI) per cell (102 to 105), helper adenovirus or chemicals to enhance AAV transduction and expression, reporter gene (luciferase, GFP, LacZ), incubation time, and cut point assignment (50% inhibition or statistically derived cut point factor) [Citation51].
The regulatory considerations for GTx NAb assays were discussed. During the clinical study, a qualified test for supportive studies (such as Phase I/II) and a validated test for pivotal studies (such as Phase III studies) can be used. During clinical development, these assays may be run in a single lab as LDTs; however, upon marketing of the therapeutic product it may be preferable to have an FDA-approved CDx available. In the EU, country specific regulations or recommendations may apply. Based on current GTx BLA submissions, Phase I/II studies were used as pivotal studies to support approval. Therefore, the fit-for-purpose concept applies to GTx NAb assays. It is recommended to consult regulatory agencies early on for guidance. In other countries, an ISO 15189 accreditation and a diagnostic permit is usually needed. Further discussion on this topic is recommended at next WRIB.
Another concern was the approach to determining exclusion criteria for clinical trials of AAV-based GTx, as discussed in draft guidance provided by the FDA [Citation52]. Options include using anti-capsid antibody assays (ADA and/or NAb) or cellular immunity assays (such as ELISpot) for a capsid or transgene product. The utility of cellular immunity assays for capsid or transgene product as exclusion criteria has not been evaluated by regulators and the current consensus is that this is not required. These assays may be better used as an investigative tool or to guide preventive measures if a problem is suspected. Clinically relevant efficacy and safety issues should be thoroughly investigated, including the use of cellular immunity assays.
When considering the use of anti-capsid antibody assays (ADA and NAb), it was recommended to understand the impact of the presence of anti-capsid antibodies on the clinical outcome. Exclusion criteria for clinical trials of AAV-based GTx require scientific and clinical justification; anti-capsid antibody assays should not be universally used to exclude patients without knowing the impact on safety and efficacy. The precautionary principle is recommended for first-in-human (FIH) studies which may require excluding ADA/NAb positive subjects from AAV-based GTx trials. Where ADA/NAb positive subjects are excluded from pivotal studies, consideration should be given to performing separate studies in these subjects, which might also address the role of immunomodulation including Ab depletion. More data are needed before providing specific guidance. Drug developers should collect data on initial efficacy, duration of efficacy, early adverse events and long-term safety to inform future clinical trials.
ELISpot & Single Cell Western Blot Assay Validation
ELISpot Validation
Detection and monitoring of the immune response triggered by biologic drugs is of paramount importance for the safety and efficacy assessment of large molecule drugs. As the immune responses against these biologics include the induction of CD8-positive cytotoxic T lymphocytes (CTLs), the analysis of activated T-cells is required. Activated CTLs will trigger the killing of the transduced cells, resulting in a decreased efficacy of the gene therapy and in putative organ damage.
ELISpot assays allow the detection of low frequency cells secreting various molecules such as cytokines. ELISpot has recently seen a resurgence and has become the technique of choice for the assessment of the cellular immunity triggered by gene therapies, via the assessment of the activation of cytotoxic T cells. While bioanalytical scientists have gathered extensive expertise with the cell culture, immunoassay and image analysis aspects of ELISpot, the quality of the cells initially used in the assay and their relevant stimulation is one of the key factors impacting the quality and the biological relevance of the results. It is indeed critical that peripheral blood mononuclear cells (PBMCs) are isolated in a timely and consistent manner from patients’ whole blood, across multiple clinical sites and that cell stimulation with peptide pools and positive controls is carried out in a biologically relevant fashion. This need can pose logistical challenges for global studies and may require significant effort to ensure that PBMCs are isolated within a pre-defined time from whole blood collection at a clinical site.
Gene and cell therapy (GCT) programs have unique characteristics that differentiate them from vaccine programs, but the need to identify relevant T-cell immune responses is shared. The experience gleaned from the use of T-cell ELISpot in vaccine clinical trials provides many lessons learned that can be applied to GCT programs.
Within the vaccine industry, T-cell immune responses are induced in humans by vaccination to infectious diseases such as HIV, VZV, HPV and others. However, the determination of a correlate of protection has been elusive for clinical vaccine trials using the ELISpot assay. Several cancer immunotherapy modalities have shown a correlation of IFN-γ T-cell responses with survival however, the studies are very limited in subject number and positive clinical responses making interpretation difficult. Immunogenicity assays used in vaccine clinical trials are expected to be qualified or validated depending upon the phase of the clinical trial. It was agreed that certain parameters used to qualify or validate an antibody binding assay cannot readily be applied to an ELISpot assay. There are key approaches that can be used to optimize the performance of the ELISpot assay so that it can be qualified for clinical application. As an indicator of T-cell immunity, the ELISpot assay is sensitive, relatively robust and can be optimized to reduce variability. Vaccine clinical programs generally run over an extended period of time, resulting in the need for maintaining consistent assay performance. The ELISpot is no exception and brings its own challenges to assay maintenance.
White Papers are available to provide clarity on harmonization of practices and analysis of the quality of ELISpot method results [Citation25,Citation53]. The 2019 White Paper discussed challenges for ELISpot assays including a complex workflow from sample collection to testing, especially for larger multicenter studies.
The current recommendations on ELISpot validation build on the 2019 White Paper in Bioanalysis and ongoing industry experience comprised of performing ELISpot assays according to standard harmonized procedures including automated reading for spot counts [Citation54]. It was recommended that laboratories performing ELISpot assays participate in external QA assessment, for example, T cell ELISpot Proficiency Panels [Citation55].
Critical factors when developing the method are focused on the reagents. High quality frozen peripheral blood mononuclear cell (PBMC) preparations are critical (see below), as are the capture and detection antibody pair. The antigen source can be overlapping synthetic peptides or whole proteins, but purity is a key factor. Filter plates may be polyvinylidene difluoride (PVDF) or some other variety.
Before validation, techniques can be used to optimize the ELISpot process to reduce variability. This optimization includes trained staff, a standardized thaw process, resting of PBMC overnight, and using triplicate vs. duplicate wells. Multifactorial design of experiment (DoE) should be used to assess and optimize the robustness of the critical steps. Automated ELISpot readers are also important tools for optimization. The scan and count function for spot imaging and counting reduces processing time and integrates consistency. Key imager parameters include spot size and shape, which are important for differentiating background from true spots, and spot contrast/intensity. A consistent approach to spot counting through method development and optimization should be established. Definition of a counting template can be useful; counting parameters (e.g., min/max spot size, sensitivity) that have been defined using a set of relevant ELISpot well images would maintain consistency within a study. Maintaining consistency is important for large studies, although there remain some challenges around the correlation of ELISpot positive signals and serious adverse events in patients.
The traditional validation parameters for LBA and vaccine immunogenicity assays can be adapted for ELISpot assay qualification using a sample panel () and assay qualification parameters ().
Assay Suitability Criteria
The assay suitability criteria are derived from the assay qualification and should include QC sample ranges. The negative control has no antigen, mock, and determines the background. The positive control is a mitogen, peptide or responder PBMC. The replicate variability criteria should be <2-fold, and well acceptance criteria should be established.
Defining a Positive Response
Recommendations were provided to define a positive ELISpot response. The critical parameters to assess are ELISpot responses from an existing positive responder population (if available) and a large data set from non-responders. A statistical approach with dual criteria or mean + 2 standard deviations of pre-existing immunity/background is one option that is reasonable. In the absence of an established, clinically relevant positivity criterion for reactivity to peptide antigens, the positivity criteria for a sample must be established based on the level of reactivity that is meaningful above the background reactivity. For example, positive samples are defined by a positive read out 3 times the background when assay variability is twice the background. A different cut-off may be used based on the nature of the assay.
Assay Maintenance
Recommendations were given on how to maintain ELISpot assay performance. A proficiency panel of PBMC samples with expected ELISpot responses (optimally to the antigen of interest) should be established. New analysts should be qualified on the assay and their proficiency should be maintained. Side by side assessment of new reagent lots against qualified lots is necessary for reagent bridging and trending. Both real-time trending of controls during active testing and proficiency panel which monitors assay performance over time were recommended. In the EU, assays need to be confirmed every year. In other countries, the quality control unit of the laboratory will ensure compliance.
Robustness/Ruggedness
Different parameters that can vary over time during routine operation should be tested. The first tier of tests should include detection antibody incubation time, dilutions of conjugated antibodies, and culture period of cells. The second tier of tests can include kit lots (n≥2), and operators (n≥3). Validation experiments should be conducted depending upon COU.
Peripheral Blood Mononuclear Cells
The most critical factor for ELISpot assay performance is PBMC sample quality and availability. It is recommended to use multiple donors (relevant population), and a minimum of 10 vials per donor (1 × 107 PBMC per vial) as part of assay development and validation. Preparation protocols from blood are based on differences in blood cell density: blood cells with round nuclei are lymphocytes (T cells, B cells, natural killer cells) which are the majority of PBMC population, monocytes, and dendritic cells (small percentage). Erythrocytes and platelets contain no nuclei and granulocytes (neutrophils, basophils, and eosinophils) contain multi-lobed nuclei.
It is important to plan PBMC collection in collaboration with the clinical team to mitigate inter-site differences in sample collection and handling. Blood should be collected at clinical sites according to optimized protocol. Central labs should be provided the optimized protocol for isolation, counting, aliquoting, and cryo-preservation. A mock PBMC isolation from blood should be performed to mimic blood storage and shipment delay. After thawing, the bioanalytical lab should assess cell viability (% recovery) and cytokine synthesis. Viability <80% indicates a need to troubleshoot and optimize protocols. If viability ≥80%, cytokine production can be tested.
Prolonged storage of blood (>3h) results in accumulation of granulocytes in the lymphocyte ring of lymphoprep isolation. Increased amounts of granulocyte result in a dramatic dilution of target cells (T cells) causing a high risk to generate false negative ELISpot results. RosetteSep™ granulocyte depletion removes accumulating granulocytes from PBMC preparation and restores T cell frequency.
Cell numbers should be normalized, i.e., PBMCs can be analyzed by flow cytometry and then normalized to the fixed/pre-defined number of T cells (executed at the site of the ELISpot testing). It is acknowledged that limited sample volume, for example, in pediatric samples, might be a limiting factor.
Single Cell Western Blot
Advances in gene therapy are providing benefits to patients by addressing needs that have not been met by traditional biotherapeutics. Expression of the transgene products will be an important pharmacodynamic marker, and new techniques and platforms may need to be employed to measure this analyte in a variety of matrices. For example, a multiplexed western blot assay can measure expression of a transgene product as well as the diseased state biomarker in single red blood cells. The diseased state biomarker has been shown to cause unhealthy cellular morphology in subjects, while the transgene product has been shown to improve cellular morphology. By applying this assay to measure both proteins at the single cell level, a better understanding of the pan-cellularity of the transgene is gained.
Design of experiment studies used to characterize assay variability have been meaningful to establish validation acceptance criteria for this novel approach. In assay development, electrophoresis, cell occupancy, lysis time, antibody selection and concentration, and fluorescence detection instrument settings need to be considered. Electrophoresis optimization will provide better resolution of the analyte, making data analysis easier to automate. Not all fluorophores are created equal–some analytes will be better resolved due to channel and fluorophore selection. In the absence of good control antibody and marker, maintaining consistent PMT voltage becomes critical for tracking assay performance. Analyst technique can impact the number of occupied wells.
The fit-for-purpose validation acceptance criteria for single cell western assays were discussed and whether LBA criteria (20–20%) are sufficient for use. Consensus was reached that FFP validation criteria should be based on context of use and established prior to performing the experiments. Assays should be validated if the data will be used as a crucial parameter for patient safety and efficacy of the treatment. In this case, validation studies should be based on ICH Q2R1 guidance [Citation56]. When only used as supporting data, assay qualification is sufficient.
Application of Current FDA/EMA Immunogenicity Guidance/Guideline to Gene Therapy:
Gene and cell therapies present unique challenges in the assessment of immunogenicity. The potential for host immune responses to impact the efficacy and durability of these therapeutics has led to an emphasis on characterizing the immune response more thoroughly than typically performed for other biotherapeutics. Many of the current principles that are captured in the FDA [Citation26] and EMA [Citation57] guidance documents are applicable to immunogenicity risk assessment and assay development and validation for GTx. However, there are areas where the guidelines do not specifically address the unique concerns surrounding this new area of drug development. Specifically, there are concerns about pre-existing immunity and its impact on transduction of gene therapies, a greater emphasis on cell-based assays, and immunogenicity against transgenes versus the vector.
Unlike the “typical” biotherapeutic which is one molecule, gene and cell therapies have the potential to deliver many foreign proteins into the patient. It is important to consider all foreign proteins that may be introduced/produced. This includes anti-capsid antibodies, anti-transgene antibodies, anti-chimeric antigen receptor (CAR) antibodies, and anti-CAS9 antibodies, etc. Performing an early risk assessment is essential if only to get started on the different assays that may be needed. A typical screen, confirm, titer approach for testing can be used. Screening and NAb assays are likely to have significant numbers of baseline positives which will require far more individuals than typical for cut point setting. It may be difficult in rare disease or pediatric populations to obtain sufficient number of individuals and sample volumes. Significant geographic differences have also been observed [Citation58–60]. Cell based assays should be implemented earlier than typically seen for biotherapeutics including transduction-based neutralization measures and determination of cellular immunity to AAV as inclusion/exclusion criteria.
It was agreed that the current immunogenicity guidance documents for biotherapeutics [Citation26,Citation57] are broadly applicable to gene/cell therapy [Citation51], even though the FDA guidance specifically states that it does not apply to GTx. Although neither the FDA nor EMA have issued detailed guidance on the immunogenicity assessment of gene/cell therapy, consideration should be given to developing immunogenicity assays for, for example, anti-vector, anti-transgene product, etc. with timely measurements. Risks associated with gene therapies are not clear, so a product specific risk assessment should be considered.
Consensus was reached to base immunogenicity assessments on several general principles. First, initiate an immunogenicity risk assessment and update as needed during development along with justification of the details of the assessment. Given significant uncertainty, store samples in order to perform additional analyses later on, if needed. Ideally, execute the immunogenicity assay at a single, central laboratory. Investigate the impact of immunogenicity on clinically relevant issues such as lack of efficacy, loss of efficacy, and adverse events.
Current data do not support a general recommendation on what pre-existing titers are relevant in terms of clinical safety and efficacy. For protein-based biologics, it has been proposed that pre-existing antibodies may, but do not necessarily lead to, boosted immune responses. A clinically relevant cut point needs to be determined, anticipating that high titers may have a higher clinical relevance. Other factors like serotype, dose, product type, intended target organ, etc. may impact inclusion criteria. Due to challenges in comparing assays across sponsors, it is impossible to select a titer for exclusion/inclusion for all serotypes.
RECOMMENDATIONS
Below is a summary of the recommendations made during the 14th WRIB:
qPCR, ddPCR, and NGS Assay Development and Validation: Best Practices
qPCR Validation
qPCR assays should be qualified and validated by the sponsor, with a clear understanding of the assay sensitivity, reproducibility and variability in each matrix to be tested. Parameters to consider include: standard calibrators, accuracy, precision, LLOQ and LOD, stability, amplification efficiency and factorial optimization. Refer to
Acceptance criteria should be established before validation. Specific criteria should be evaluated case base case since specifics will differ with assay and COU
Primer/probe selection:
NTCs and baseline areas should be clear and clean with proper instrument calibration for the dye
The exponential phase needs to show a strong, straight growth (target 1 Ct difference between successive 2-fold dilutions)
The plateau should be as horizontal as possible and individual replicates should plateau at the same fluorescence level
Different concentration levels should plateau at the same level otherwise this indicates reduced assay sensitivity
Design and test multiple oligomer sets per target and perform BLAST search to avoid primer dimers and for specificity
Target isolation procedures should be tested for extraction efficiency and potential assay interference using A280/260 or other methods to quantify gDNA/cDNA to normalize results (or use reference targets)
Calibrator material should be equivalent or very similar to test samples. If possible, use the clinical material
For long term calibrators, it is preferred to use cGMP material but research lots may be used with bridging
Extraction efficiency can be evaluated with encapsulated ss/ds DNA or cloned/synthesized DNA
NGS indel assays and RT-qPCR integration assays
Measurement of indels may be used as a surrogate of genome editing efficiency.
Evaluation of indels at previously identified off-target sites is essential for monitoring patient safety in clinical trials
NGS is the technology of choice for the quantification of indels because of its high discovery power for heterogenous indel variants, the ability to multiplex samples and analytes, and the requirement for small DNA input material
Reverse transcription followed by qPCR is a suitable method to quantify transcripts with known sequences
In absence of a universal reference standard, a well characterized/qualified reference standard can be used; consideration should be taken for lot-to-lot bridging to ensure consistency
Reference standards and QCs for NGS based clinical indel assays
Should be generated with different levels of indels such as mixing cell lines of known indel levels with unedited parental cells, followed by gDNA extraction
If possible, the known indel levels would be best confirmed by using an orthogonal method.
Stability should be tested
Qualification/bridging program should be established
Parameters to be evaluated for NGS based clinical indel assays include:
gDNA input
Specificity of PCR reactions
Sequencing coverage
Quantifiable range, linearity and LOD
Precision and accuracy
Selectivity
gDNA stability
Bridging and linearity should be considered when applicable
Reference standards and QCs for RT-qPCR integration assays
Clone the fusion cDNA and wild-type DNA into plasmids under the control of a T7 promoter
In vitro transcription can be used to synthesize RNA reference material
Copy number of purified RNA can be calculated using A260 and size of transcript
Parameters to be evaluated for RT-qPCR gene integration assays include:
RNA input
Specificity of RT-qPCR reactions
Quantifiable range
Precision and accuracy
Sensitivity and LOD
Selectivity
Tissue storage and shipping conditions to ensure RNA integrity
QC RNA stability
Qualification/bridging program should be established for reference standards and QCs
Back calculating %indel against a standard curve increases assay robustness when only limited gDNA material is available
ddPCR Assay Development: Best Practices
ddPCR enables specific, accurate quantification of a transgene construct and readily enables multiplexing
ddPCR can be used for both biodistribution and shedding studies but it was recommended that the same technology be used for both
For low DNA yield, samples can be stored in single-use aliquots
Best practices for manually setting the positive/negative threshold when the analysis software is unable to:
Use positive controls as guide
Evaluate 1 or 2D plots and histograms for separation of positive and negative (single/double positives)
Draw threshold
Document event and second scientist confirms
Save original and manual raw data and analysis files
Establish the LOD by determining the lowest concentration at which 95% of the positive samples are detected. Within a group of replicates containing the target at concentrations at the LOD, no more than 5% failed reactions should occur
If the assay is quantitative with acceptable performance, less than 90% amplification efficiency due to amplicon structure between the primers (e.g., hairpin loops, high GC content), may be acceptable if the assay is FFP and with proper justification supported by other measures (accuracy, precision, linearity)
AAV Capsid NAb Assays: Development & Validation
LBA approaches are used to detect total anti-AAV antibodies and cell-based assays are used for anti-AAV NAb assays.
A bridging assay format detects both non-neutralizing and neutralizing antibodies and uses cut point as in other ADA assays giving better assay sensitivity and specificity than cell-based assays.
Parameters to be optimized include:
Cell line (HEK 293, HeLa, CHO, Huh-7)
Cell number
Serum (negative control, heat inactivation)
MOI per cell (102 to 105)
Helper adenovirus or chemicals to enhance AAV transduction and expression
Reporter gene (luciferase, GFP, LacZ)
Incubation time
Cut point assignment (50% inhibition or statistically derived cut point factor)
During the clinical study, a qualified test for supportive studies (such as Phase I/II) and a validated test for pivotal studies (such as Phase III studies) can be used
For commercial products, an ADA/NAb assay may be considered a CDx and should be performed in CLIA-certified lab in US. In the EU, country specific regulations or recommendations may apply. In other countries, an ISO 15189 accreditation and a diagnostic permit is usually needed
When considering the use of anti-capsid antibody assays (ADA/NAb)
Understand the impact of the presence of anti-capsid antibodies on the clinical outcome
Exclusion criteria require scientific and clinical justification
Anti-capsid antibody assays should not be universally used to exclude patients without knowing the impact on safety and efficacy
The precautionary principle is recommended for FIH studies which may require excluding ADA/NAb positive subjects from AAV-based GTx trials
Where ADA/NAb positive subjects are excluded from pivotal studies, consideration should be given to performing separate studies in these subjects, which might also address the role of immunomodulation including corticosteroids and Ab depletion
The utility of cellular immunity assays for capsid or transgene product as exclusion criteria has not been evaluated by regulators. Consensus from industry is that that this is not required. These assays may be better used as an investigative tool or to guide preventive measures if a problem is suspected
ELISpot & Single Cell Western Blot Assay validation
ELISpot Validation
Multifactorial DoE can be used to assess and optimize the robustness of the critical steps
Reagents:
High quality frozen PBMC preparations are critical (see below)
Capture and detection antibody pair
Antigen source can be overlapping synthetic peptides or whole proteins
Filter plates may be PVDF or other variety
Automated ELISpot readers:
Scan and count function for spot imaging and counting reduces processing time and integrates consistency
Key imager parameters include spot size and shape, and spot contrast/intensity
A consistent approach to spot counting through method development and optimization should be established. Definition of a counting template can be useful
Validation experiments should be conducted depending upon COU
Laboratories performing ELISpot assays participate in external QA assessment
Assay parameters for an ELISpot validation:
Precision, assay range (LLOQs), ruggedness, linearity and specificity
Use high purity grade peptides
PBMC preparation harmonization between sites is recommended
Do not to use cells with a viability <80%.
Cells can be normalized, i.e., PBMCs need to be analyzed by flow cytometry and then the PBMC should be adjusted to the fixed/pre-defined number of T-cells (executed at the site of the ELISpot analysis)
Different parameters that can vary over time during routine operation should be tested to determine ruggedness
Assay suitability criteria are derived from the assay qualification and should include QC sample ranges. The replicate variability criteria should be <2-fold, and well acceptance criteria should be established
Positive Response:
Critical parameters are the existing positive responder population (if available) and a large data set from non-responders
A statistical approach, for example, with dual criteria or mean + 2 standard deviations of pre-existing immunity/background can be used
The positivity criteria for a sample must be established based on the level of reactivity that is meaningful above the background reactivity
for example, a positive read out equals 3 times the background when assay variability is twice the background. A different cut off may be used based on the nature of the assay
Assay Maintenance:
A proficiency panel of PBMC samples with expected ELISpot responses (optimally to the antigen of interest) should be established
New analysts should be qualified, and their proficiency maintained.
Side by side assessment of new reagent lots against qualified lots is necessary for reagent bridging and trending
Real-time trending of controls during active testing and proficiency panel, which monitors assay performance over time, were recommended for trending
In the EU, assays need to be confirmed every year. In other countries, the quality control unit of the laboratory ensures compliance
PBMC:
Use multiple donors (relevant population), and a minimum of 10 vials per donor (1 × 107 PBMC per vial)
Blood should be collected at clinical sites according to optimized protocol. Central labs should be provided the optimized protocol for isolation, counting, aliquoting, and cryo-preservation
After thawing, the bioanalytical lab should assess cell viability (% recovery) and cytokine synthesis
RosetteSep™ granulocyte depletion removes accumulating granulocytes from PBMC preparation and restores target cell (T cell) frequency
PBMCs can be analyzed by flow cytometry and then the PBMC adjusted to the fixed/pre-defined number of T cells
Single Cell Western Blot
Design of experiment studies used to characterize assay variability have been meaningful to establish validation acceptance criteria for this novel approach
In assay development, electrophoresis, cell occupancy, lysis time, antibody selection and concentration, and fluorescence detection instrument settings need to be considered
In the absence of good control antibody and marker, maintaining consistent PMT voltage becomes critical for tracking assay performance
FFP validation criteria should be based on context of use and established prior to performing the experiments
Assays should be validated if the data will be used as a crucial parameter for patient safety and efficacy of the treatment. In this case, validation studies should be based on ICH Q2R2 guidance
When only used as supporting data, assay qualification is sufficient
Application of Current FDA/EMA Immunogenicity Guidance/Guideline to Gene Therapy
Current Immunogenicity Guidance/Guideline (FDA/EMA) for biotherapeutics [Citation26,Citation57] are broadly applicable to gene/cell therapy.
All foreign proteins that may be introduced/produced by the patient should be considered. This includes anti-capsid antibodies, anti-transgene antibodies, and anti-CAS9 antibodies
Perform an early risk assessment
A typical screen, confirm, titer approach for testing can be used
Screening and NAb assays are likely to have significant numbers of baseline positives which will require far more individuals than typical for cut point setting
Cell based assays should be implemented earlier than typically seen for biotherapeutics including transduction-based neutralization measures and determination of cellular immunity to AAV as inclusion/exclusion criteria
Consideration should be given to developing immunogenicity assays for example, anti-vector, anti-transgene product, etc. with timely measurements
Risks associated with gene therapies are unclear, so a product-specific risk assessment should be considered
Base immunogenicity assessments on several general principles:
Initiate an immunogenicity risk assessment and update as needed during development
Justify the details of the risk assessment
Store samples in order to perform additional analyses later on, if needed
Ideally, execute the immunogenicity assay at a single, central laboratory
Investigate the impact of immunogenicity on clinically relevant issues such as lack of efficacy, loss of efficacy, and adverse events
A clinically relevant cut point needs to be determined, expecting that high titers may have a higher clinical relevance
SECTION 3–NAb Assay Harmonization, Biosimilars and FDA/EMA Guidance/Guideline
Haoheng Yan4, Bonnie Wu2, Daniela Verthelyi4, Susan Kirshner4, Joao Pedras-Vasconcelos4, Manoj Rajadhyaksha24, Roland F. Staack25, Elana Cherry12, Isabelle Cludts26, Madeleine Dahlbäck27, George R. Gunn28, Akiko Ishii-Watabe29, Vibha Jawa30, Robert Kubiak31, Michael Partridge24, Marco Petrillo32, Samuel O. Pine33, Johann Poetzl34, Sam Song35, Chris Stebbins36, Yuling Wu31, Lucia Zhang12
Authors in Section 3 are presented in alphabetical order of their last name, with the exception of the first seven authors who were major contributors.
The affiliations can be found at the beginning of the article
DISCUSSION TOPICS & CONSOLIDATED QUESTIONS COLLECTED FROM THE GLOBAL BIOANALYTICAL COMMUNITY
The topics detailed below were considered as the most relevant ‘hot topics’ based on feedback collected from the 13th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 14th WRIB. The background on each issue, discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
Cell-Based NAb Assays–Sensitivity and Drug Tolerance and the Relevance for Clinical Outcome
When do cell-based NAb assays add value if ADAs do not have an apparent effect on clinical outcome? When do they add value to measure in vitro neutralization in samples during treatment if the drug is not of high immunogenicity risk and NAb results may not be needed during treatment to ensure patient safety? Would regulators expect two NAb assay platforms (one NAb assay during treatment, and one NAb assay for follow up samples) when high drug trough levels are impacting assay sensitivity? What are regulatory and industry opinions on biotherapeutic mechanism of action (MoA) as the primary driving consideration when deciding if a functional cell-based NAb assay is needed?
NAb Assay Harmonization: Recent Trends and Expectations
Are NAb assays required for low immunogenic compounds known to only elicit very low titer ADA responses? Do we need to test NAbs for baseline ADA positive samples, placebo ADA positive samples, or samples with low risk molecules which were demonstrated to contain low-level ADA incidence in Phase I/II studies? What additional value do NAbs provide, given the availability of ADA status, titer, transient/persistent, PK, pharmacodynamics (PD), and efficacy? For bi- or multi-specific biotherapeutics, when should the NAb assay be implemented and what are the viable assay platforms? What is the risk level of immunogenicity for the T-cell redirected cancer cell killing biotherapeutics? When should NAb assay be implemented for this category of drug products?
Biosimilar Immunogenicity: Current Industry Standards
Are cell-based functional assays sensitive and drug tolerant enough to detect potential differences in the immunogenic profile of a proposed biosimilar compared to its reference? Regarding anti-IgG Fc response, how likely is the development of an ADA response towards this highly conserved region of antibody drug molecules? With 8 mg/mL IgG present in blood circulation, would assays be able to detect anti-IgG Fc ADAs specific for the drug? Should the interpretation of immunogenicity results in light of the assay used be included when presenting or publishing immunogenicity data?
The 2019 US FDA Immunogenicity Guidance: Reflections a Year Later
A relatively high false positive rate (FPR) of 5% in the ADA screening assays is expected to ensure high sensitivity. According to industry experience, assay sensitivity is dictated by the choice of analytical platform and not by artificially inflating the FPR. Is there a scientific reason to continue using a 5% FPR instead of a lower value (e.g., 1% or 0.1%)? The 2019 FDA guidance [Citation26] requires 1% of false positives in the ADA confirmatory assays while the 2017 EMA guidance [Citation57] states that ADA confirmatory assays should eliminate any false positives. Do 1% and 0% FPR, respectively, refer to the confirmatory assay only or to the final data set? There are disadvantages to using box-plots with 1.5- or 3-inter-quartile range (IQR) to identify outliers in cut point data. What alternative approaches could be used to ensure conservative cut point values without unnecessarily excluding ADA-negative samples and preserving normal biological variability within the subject population? What comprises a good risk-based strategy that can be written as part of the Investigational new drug (IND)? What would be needed to perform an ongoing risk assessment that could then be summarized as part of Integrated Summary of Immunogenicity (ISI) for the BLA submissions?
DISCUSSIONS, CONSENSUS AND CONCLUSIONS
Immunogenicity assessments are an essential element of safety and efficacy evaluations in the product development of biopharmaceuticals. NAb assays characterize detected ADAs for their neutralizing activities as a component of an immunogenicity testing strategy. Analysis of ADA and NAbs can contribute to the understanding of drug PK, PD, efficacy, and safety observed during product development and patient treatment.
Cell-Based NAb Assays–Sensitivity and Drug Tolerance and the Relevance for Clinical Outcome
Assays used during clinical development to detect NAbs typically fall into one of two categories, either cell-based assays (CBA) or competitive ligand binding assays (CLBA). Assay formats are typically selected in alignment with drug MoA, assay performance, and the immunogenicity risk assessment; cell-based assay formats being generally preferred by most regulatory agencies for complex MoAs. Ideally, cell-based bioassays provide experimental systems that reflect all or a key portion of the pharmacologic activity of a biotherapeutic. Cell-based NAb assays often provide more appropriate platforms for assessment of ADA-mediated neutralization for agonists and many biotherapeutics targeting cell-surface receptors. However, they are generally more complex and challenging to develop. CLBAs may be more robust, sensitive and drug tolerant when used to support antagonistic biotherapeutics or receptor-Fc fusion proteins. The sensitivity of a CLBA typically reaches the 100–200 ng/mL range for the positive control detection, while CBAs may be less sensitive. In addition, CBAs can be adversely impacted by sample pre-treatment procedures such as those aiming to improve drug tolerance. Consequently, CBAs can have poorer drug tolerance than CLBAs [Citation61,Citation62].
EMA and FDA immunogenicity guidelines [Citation26,Citation57] present the current regulatory views on NAb assay format. The EMA recommends developing a NAb assay which responds well to the biotherapeutic and is sufficiently drug tolerant. The risk of NAb incidence and potential clinical consequences should also be considered. The EMA is open to the use of CLBA for therapeutics that exert their activity only through direct binding to their target (e.g., antagonistic mAbs), as long as the assay “reflects neutralizing capacity/potential in an appropriate manner” [Citation57]. The FDA guideline indicates a preference for CBAs; however, there are bioanalytical considerations such as assay sensitivity, drug tolerance and selectivity that can lead to the use of CLBAs.
USP and industry consensus of opinion is that the biotherapeutic MoA is a primary consideration when deciding whether a functional cell-based NAb assay is to be used. According to Wu, et al., [Citation63] and USP-NF <1106.1> [Citation64], the CBA NAb assay format is preferred for agonist and antibody-drug conjugates (ADC). For an agonist or enzyme-replacement therapy, a CBA or an enzyme activity assay can be used; USP also allows for a CLBA. For an antagonistic MoA, there is disagreement on the preferred approach. Wu et al stated that CLBA is generally preferred, but CBAs can be useful for some biotherapeutics targeting cellular receptors, while USP advocates the use of CBA for soluble receptor biotherapeutics and for antibody-dependent cell-mediated cytotoxicity (ADCC)+ modalities.
Recommendations on the timing of NAb assays in clinical programs were provided in the 2013 White Paper in Bioanalysis [Citation6], which advised taking a risk-based approach to decide when a NAb assay should be implemented, a recommendation that is still supported. In 2017, this recommendation was expanded to state that NAb assays should be included at all clinical phases for high risk molecules [Citation18]. This year, the discussion focused on products of low immunogenicity risk and the possibility of using alternative measures such as PD markers to assess neutralizing activity of samples during treatment when drug interference presents a problem. Indeed, a PD marker or biomarker that correlates with clinical outcome could be part of an integrated approach that uses PK, ADA, and PD data to assess NAb activity over time during treatment. The success of an integrated approach would rely on the data quality from PK, ADA, and PD assays. Similar positions have previously been recommended [Citation15,Citation18,Citation25].
For products where NAb assessment needs to be conducted during treatment but drug interferes with the sensitivity, it could be possible to use two different kinds of NAB assays, although this is not expected by the regulators: one NAb assay with a higher drug tolerance but lower sensitivity during treatment and one NAb assay with lower drug tolerance but higher sensitivity for the follow-up samples in the wash-out period. However, the two methods would need to be bridged for use in the same study and the rationale for using two assay platforms should be clearly described. As it is difficult to compare results from different NAb assays, the use of two methods in the same study is not common and would need to be discussed with regulators prior to implementation.
Since NAb positive controls are only surrogates and do not represent NAbs in clinical samples, a NAb assay with drug tolerance lower than the trough concentration may still detect NAbs in clinical samples. From an industry perspective, sponsors could use the actual drug concentration in NAb positive samples to reassess drug tolerance to help justify the adequacy of the assay; these results can be provided as supplemental data to support the validation report. Regardless of the assay format, NAb assays should be developed to characterize the neutralizing capacity of ADA in the samples collected, being mindful of potential drug and matrix interference. Hence, a certain degree of pragmatism is appropriate when selecting the NAb assay format.
NAb Assay Harmonization: Recent Trends and Expectations
Nab activity assessment for low Immunogenicity risk molecules
Regulatory agencies recommend immunogenicity assessment be conducted for all classes of biotherapeutics. NAb assays would characterize the neutralizing capacity of ADAs and may correlate with clinical outcomes. Some products can be considered to be of low immunogenicity risk, if they are not endogenous mimics, are administered to immunosuppressed patients, or have been previously shown to have low immunogenicity risk. Therefore, it was discussed whether it was possible to reduce the scope of NAb testing for low risk drug molecules. It is important to point out that a low risk molecule could attain a higher risk profile when patient population changes or the drug has a critical quality attribute that could make it more immunogenic. The industry perspective regarding the need for NAb testing for low risk molecules was varied. While some considered that testing for NAb places an unnecessary burden on drug development of low risk biotherapeutics, others voiced that the immunogenicity risk, Ab titers, and the potential clinical consequence of developing NAbs inform the immunogenicity risk; currently they are not sufficient to predict neutralizing activity and thus assessment of NAb activity is needed. Regarding the timing of NAb assay implementation, the need for development, validation and implementation of ADA and NAb assays early in product development is dictated by the risk of the product. To avoid holding up the drug development program, for low-risk products, NAb assays can be developed in parallel with early studies and implemented based on the evolving incidence and titer of ADA.
NAb assessment for multi-domain biotherapeutics
Bispecific and multi-domain biotherapeutics could elicit immune responses with multiple specificities to different domains. Risk factors derived from the drug product of such molecules could include novel amino acid sequences or epitopes in each functional domain, neo-antigens in the linker sequences or in the interface between domains, and post-translational modifications, for example, glycosylation, oxidation, deamidation. This increased complexity presents unique challenges to method development and characterization for immunogenicity. As with other therapeutics, the timing for implementing a NAb assay would depend on the risk of the molecule. The NAb assay should capture all the activities of the product which might be accomplished by single or multiple assays.
Biosimilar Immunogenicity: Current Industry Standards
A biosimilar is a biological product that is highly similar to and has no clinically meaningful differences with an approved reference product. Immunogenicity data for the proposed biosimilar product and the reference product is essential to support a demonstration of biosimilarity or interchangeability for a proposed biosimilar product.
Regulatory feedback on the bioanalytical strategy and method requirements to evaluate immunogenicity of biosimilars has evolved in the last few years, and the current thoughts are described in FDA guidance to industry [Citation65,Citation66] and method validation guidelines [Citation26,Citation57,Citation67].
Clinical Immunogenicity Considerations for Biosimilar and Interchangeable Insulin Products
Most recently in 2019, FDA proposed an exception for the insulin program, which is discussed in the draft FDA guidance for industry “Clinical Immunogenicity Considerations for Biosimilar and Interchangeable Insulin Products” [Citation68]. This guidance states that “…generally, if a comparative analytical assessment based on state-of-the-art technology supports a demonstration of high similarity for a proposed biosimilar or interchangeable insulin product, there would be little or no residual uncertainty regarding immunogenicity; in such instances, a comparative clinical immunogenicity study generally would be unnecessary to support a demonstration of biosimilarity or interchangeability”. The recommendation for insulin products is based on a number of factors, including “the relatively small, structurally uncomplicated and well-characterized nature of insulin products”; “extensive experience and literature survey that confirm minimal or no clinical relevance of immunogenicity with insulin product use”; and “scientific thinking on the lack of clinical impact of immunogenicity with insulin product use”, as reflected from multiple sources detailed in the guidance. In conclusion, “current analytical tools used to evaluate quality attributes for insulin products can support a comprehensive analytical comparison thorough enough to support a conclusion that a particular proposed biosimilar insulin product that is “highly similar” to its reference product generally would have little or no residual uncertainty regarding immunogenicity and would be expected, like the reference product, to have minimal or no risk of clinical impact from immunogenicity. In such cases, a comparative clinical immunogenicity study would generally not be necessary to support licensure of a proposed biosimilar or interchangeable product [Citation68]”.
In practice, FDA recommends that a 351(k) BLA for a biosimilar or interchangeable insulin product covered by the guidance contains, among other things: adequate Chemistry, Manufacturing, and Controls (CMC) information, a comprehensive and robust comparative analytical assessment, a comparative clinical pharmacology study, and an immunogenicity assessment justifying why a comparative immunogenicity clinical study is not necessary. A comparative clinical immunogenicity study may be needed in some circumstances where there is residual uncertainty about immunogenicity, such as differences in certain impurities giving rise to questions related to immunogenicity.
Immunogenicity Assays for Biosimilar Programs
A one-assay approach for ADA assays, recommended in the FDA guidance [Citation66] and 2016 White Paper in Bioanalysis [Citation15], is widely used for the detection and characterization of ADAs in comparative clinical trials.
Immunogenicity assay validation parameters are the same for innovator and biosimilar programs, and include assay cut points, sensitivity, drug tolerance, specificity, selectivity, precision and reproducibility, robustness and reagent stability and system suitability controls. However, when the one-assay approach is used in a biosimilar program, additional data are needed to demonstrate the assay performs similarly with both products. This was exemplified in a talk by FDA with case studies, including the assay parameters from 5 approved biosimilars to US-licensed Humira. For ADA binding assays, all programs used the one-assay approach. Similarly, the one-assay approach was used for NAb assays, regardless of assay format being CBAs or CLBAs.
It is possible that differences in assay performance result in higher rates of ADAs or NAbs in the comparative immunogenicity testing when compared to the published data. Therefore, immunogenicity data from biosimilar studies need to be assessed using parallel arm studies and be interpreted in the context of the assays used and the totality of the data, including the clinical relevance of the detected ADAs. Although the ADA incidence for a compound may be increased when using highly sensitive ADA assays compared to historical data, the immunogenicity risk class most likely has not changed. While health authorities are aware of this, attention needs to be paid when discussing immunogenicity data in the literature as patients or physicians may not properly interpret the varying immunogenicity rates for the same originator product.
The concept for biosimilar immunogenicity evaluation is tailored for biosimilar development. Industry representatives presented their considerations specific for biosimilar immunogenicity assessment: the potential immunogenicity of the innovator product and published data are evaluated. Since the proposed biosimilar should match the reference product, the risk for new and not already described immune reactions is low. Depending on the immunogenicity risk assessment and product class (e.g., monoclonal antibodies or fusion proteins with human IgG Fc), alternative assay strategies to assess immunogenicity should be considered. In order to allow a sensitive and meaningful comparison of the immunogenic profile of a proposed biosimilar to its reference product, highly sensitive assays are required, including NAb assays. CLBAs are considered as powerful tools in biosimilar immunogenicity assessments due to their high sensitivity, while CBAs usually generate lower sensitivity and drug tolerance. In case anti-drug Fc antibodies may be of interest and potentially neutralize the efficacy of the drug, high levels of endogenous IgG Fc (approximately 8 mg/mL) in circulation, which compete for binding to anti-drug Fc antibodies, need to be factored in for assay development and data interpretation. Furthermore, the drug Fc region is highly conserved and unlikely to elicit ADA responses on its own. NAbs are often specific to the CDR region. For mAb biotherapeutics with point substitutions on the Fc region (e.g., to enhance effector function), a NAb assay can be developed to assess antibodies specific to such mutations. However, non-neutralizing polyclonal antibodies to an Fc mutation have been reported. Use of anti-Fc positive controls may be challenging in the presence of high concentrations of endogenous IgG Fc in the sample matrix. Depending on the immunogenicity risk assessment, a NAb assay focusing on the antigen binding region of the biosimilar molecule could be sufficient to assess neutralizing capacity of a proposed biosimilar irrespective of its MoA.
2019 US FDA Immunogenicity Guidance: Reflections a Year Later
Building on the 2019 White Paper recommendations [Citation25] on the interpretation and implementation of the 2019 US FDA Immunogenicity Guidance [Citation26], further discussions were held on the impact of those recommendations on current industry practice.
FDA guidance on immunogenicity assay development and validation stresses the importance of selecting appropriate minimum required dilution (MRD) for the assay, but there is no industry perspective put forth in the literature. Z’ factor was proposed for MRD selection but its utility is not ideal [Citation64,Citation69]. There is agreement on the importance of MRD but not on the definition of what constitutes an “appropriate” MRD per guidance.
Current EMA [Citation57] and FDA [Citation26] guidance documents display a fair degree of overlap in terms of scientific approach for immunogenicity testing [Citation70]. Both agencies recommend using a tiered strategy for sample testing with sequential screening and confirmatory assays followed by semi-quantitation and characterization of ADA. Assessment of false negative (sensitivity) and false positive (specificity) rates are recommended by both EMA and FDA. Both agencies agree that 5% of false positives in the screening assay is recommended to minimize false negatives. Some concern was expressed regarding an interpretation of EMA guideline as recommending avoiding any false negatives; this would suggest that 5% of false positives can ensure not only high but infinite sensitivity. Industry representative discussed the value of 5% screening FPR vs. lower values (e.g. 1% or 0.1%). According to industry experience, assay sensitivity is dictated by the choice of analytical platform and not by increasing FPR. The consensus from both regulators and industry representatives was that the 5% FPR is reasonable for minimizing false negative and it allows for an initial assessment of clinical samples.
EMA guideline expects the confirmatory tier to eliminate any false positives while FDA guidance recommends 1% of false positive classifications in the confirmatory assay. It was unclear if the 0% and 1% FPR, respectively, referred to the confirmatory assay only or to the final data set. Industry consensus indicated that these FPR refer to the confirmatory cut point determination, which includes all samples. Confirmatory assays aim to identify true ADA positives; however, it is challenging to eliminate all false positives in the confirmatory assay. An FPR lower than 1% could be implemented if the assay demonstrates high sensitivity, specificity, and selectivity, and the data suggest the assay allows for a meaningful interpretation of the clinical data. The regulators recommend the industry consult with the regulatory agencies when alternative FPRs are desired.
FDA 2019 guidance suggests a 1% false positive rate for NAb assays performed on confirmed ADA positive samples. A confirmatory step for NAb assays is not routinely performed. However, there are cases when screening and confirmatory steps are needed for NAb assays. In that situation, the cut points for screening and confirmatory NAb assays should be determined case-by-case.
Industry representatives stated that ideal immunogenicity assays where both false negative and positive rates equal zero are impractical and not necessary and state-of-the-art immunogenicity assays can deliver reasonably high sensitivity without adjusting false positive rates. At the same time, immunogenicity assays can sometimes generate significantly more than 1% of false positives when applied to clinical samples. This could be caused by conservative outlier exclusion during determination of assay cut points [Citation71] and failure to differentiate between analytical and biological variability [Citation72]. The use of non-orthogonal confirmatory assays can also contribute to excessive false positives [Citation73,Citation74].
Regarding the impact of outliers, outlier removal is typically based on Tukey's box-plot [Citation27], but there are disadvantages to using a box-plot with 1.5- or 3-IQR to identify outliers in cut point data. Industry representative stated that the box plot outlier removal assumes the distribution of assay responses for baseline samples is symmetrical, which is often not the case. A box-plot suggests which results should undergo more rigorous statistical analysis, but it cannot easily distinguish between analytical and biological outliers. It has been shown that eliminating outliers using a box-plot can result in cut points that are not appropriate for testing subjects from different study populations [Citation71]. The regulators highlighted that box plot is the most common outlier removal methods seen in the current submissions and appropriately justified in most cases. When in doubt, industry is encouraged to discuss statistical approaches for outlier removal and cut point determination with the regulatory agencies.
Regarding immunogenicity risk assessments, the FDA guidance [Citation26] suggests performing a risk assessment during early development of the therapeutic drug. This could be initiated early, and include data collected during lead selection as well as rank ordering of the multiple variants of the candidate to bring the least risky sequence forward for further development. To accomplish a comprehensive risk assessment, appropriate tools and methods are needed to identify, characterize and mitigate the risks. A sound risk-based strategy requires a risk versus benefit analysis. A comprehensive end to end risk assessment requires an evaluation of both intrinsic and extrinsic risks spanning the life cycle of the therapeutic protein and includes product, process, patient and disease/treatment related factors [Citation75]. Several tools have been developed and qualified for their ability to identify risks related to the therapeutic protein. Multiple algorithms that can evaluate the different aspects of antigen uptake, processing and presentation can be implemented early on to develop the ability to identify the intrinsic sequence-based risks. Additionally, the post translational modifications and other extrinsic risks related to the process, e.g., changes in product quality attributes (PQAs) that are noted during the later stage of process development, can be assessed using specialized human immune cell-based assays. Such assays can also capture risk of impurities like host cell protein related contaminants, excipient interaction with the therapeutic protein, and any immune modulatory target engagement related liability [Citation76].
Additionally, the preclinical and clinical experience related to exposure, efficacy and safety can be captured through the course of development and the risk designation identified as low, moderate or high. The bioanalytical strategy for clinical trials can also be streamlined based on the identified risk and provided in the IND. An ongoing effort by industry and regulators for further harmonization of the risk assessment tools and their outputs will help support the adoption of these strategies. The risk assessment activities can be performed at key touch points in pipeline development and the assessments should be summarized in the form of key deliverables at various stages of development (IND, Clinical study reports, ISI/BLA etc.).
RECOMMENDATIONS
Below is a summary of the recommendations made during the 14th WRIB:
Cell-Based NAb Assays–Sensitivity and Drug Tolerance and the Relevance for Clinical Outcome
It is recommended to use the biotherapeutic MoA as the primary consideration when deciding whether a functional cell-based NAb assay is used
Cell-based NAb assay are considered more reflective of mechanism action for assessment of ADA-mediated neutralization for agonists and many biologic therapeutics targeting cell-surface receptors. In these cases, CBA formats are generally preferred by most regulatory agencies
CLBAs may be more robust, sensitive and drug tolerant compared to cell based NAb assay, and can be used to support antagonistic biotherapeutics or receptor-Fc fusion proteins, when appropriately justified
For low risk molecules:
A PD marker or biomarker that correlates with clinical outcome could be part of an integrated approach that uses PK, ADA, and PD data to assess NAb activity over time during treatment. This would rely on quality data from PK, ADA, and PD assays
For NAb assays with good drug tolerance but compromised sensitivity:
Two NAb assay platforms can be used: one NAb assay with a higher drug tolerance and low sensitivity during treatment and one NAb assay with a higher sensitivity and lower drug tolerance for the follow-up samples during the wash-out period
The two methods must be bridged for use in the same study and the rationale for using two assay platforms should be clearly described
Sponsors could use actual drug concentrations in NAb positive clinical samples to reassess assay drug tolerance in the study phase to help justify the adequacy of the assay; these results can be provided as supplemental data to support the validation report
NAb Assay Harmonization: Recent Trends and Expectations
Immunogenicity risk, ADA titers and incidence, and the potential clinical consequence of NAb response are not currently sufficient to predict neutralizing activity and thus assessment of NAb activity may be needed
The need for development, validation and implementation of ADA and NAb assays early in product development is dictated by the risk of the product
To avoid holding up the drug development program, for low-risk products, NAb assays can be developed in parallel with early studies and a decision to implement made later based on the results on incidence and titer of ADA
Biosimilar Immunogenicity: Current Industry Standards
“If a comparative analytical assessment based on state-of-the-art technology supports a demonstration of “high similarity” for a proposed biosimilar or interchangeable insulin product, there would be little or no residual uncertainty regarding immunogenicity. In such instances, a comparative clinical immunogenicity study would generally not be necessary to support licensure of a proposed biosimilar or interchangeable insulin product [Citation68]”.
A one-assay approach for ADA binding assays and NAb assays is widely used for the detection and characterization of ADAs in comparative clinical studies.
If this approach is used instead of the two-assay approach additional data are needed to demonstrate the assay performs similarly with both products (antigenic equivalence)
The generated immunogenicity data from biosimilar studies need to be interpreted in the context of the assays used and the totality of the data, including the clinical relevance of the detected ADAs.
Attention needs to be paid when discussing immunogenicity data in the literature as patients or physicians may not properly interpret the varying immunogenicity rates for the same originator product
The 2019 US FDA Immunogenicity Guidance: Reflections a Year Later
Assay sensitivity is influenced by the choice and performance of analytical platform
Adjusting FPR should not be used to manipulate sensitivity
The 5% FPR is widely accepted by industry and the regulatory agencies. It is a reasonable choice for ADA assays and allows for an initial assessment of clinical samples
An FPR lower than 1% for the confirmatory assay could be implemented if the assay demonstrates high sensitivity, specificity, and selectivity and allows for a meaningful clinical immunogenicity data interpretation.
In rare cases when there is a need for separate screening and confirmatory steps for NAb assay. The cut points for screening and confirmatory NAb assays should be determined case by case
A risk assessment strategy starting from early discovery and progressing through the entire life cycle of the product development is preferred. The risk assessment can be adjusted based on knowledge obtained, summarized at development milestones and submitted through key regulatory submissions. It is recommended that the entire risk assessment be provided as part of integrated summary of immunogenicity in the marketing application
SECTION 4–Immunogenicity Assay Strategies
Roland F. Staack25, Bonnie Wu2, Haoheng Yan4, Daniela Verthelyi4, Susan Kirshner4, Joao Pedras-Vasconcelos4, Elana Cherry12, Isabelle Cludts26, Boris Gorovits17, Sumit Kar11, Meina Liang37, Mohsen Rajabi Abhari4, Manoj Rajadhyaksha24, Becky Schweighardt16, Kay Stubenrauch25, Yuanxin Xu38, Lucia Zhang12
Authors in Section 4 are presented in alphabetical order of their last name, with the exception of the first six authors who were major contributors. Author affiliations can be found at the beginning of the article.
The affiliations can be found at the beginning of the article
DISCUSSION TOPICS & CONSOLIDATED QUESTIONS COLLECTED FROM THE GLOBAL BIOANALYTICAL COMMUNITY
The topics detailed below were considered as the most relevant ‘hot topics’ based on feedback collected from the 13th WRIB attendees. They were reviewed and consolidated by globally recognized opinion leaders before being submitted for discussion during the 14th WRIB. The background on each issue, discussions, consensus and conclusions are in the next section and a summary of the key recommendations is provided in the final section of this manuscript.
Lessons Learned from Late Stage Clinical Studies
How long should immunogenicity responses be monitored in the clinic until considered fully characterized? If anti-drug immune responses are not associated with clinical impact in the trials, should immunogenicity monitoring be required in the post-marketing setting? If so, how long should it be? Should clinical monitoring be emphasized, rather than immunogenicity monitoring, in the post-approval setting? Should immunogenicity testing only be triggered post-approval in the event of a new safety signal, reduced efficacy, or altered PD marker? What type of immunogenicity monitoring is relevant in the post-market setting versus being unwarranted and of limited clinical value?
Circulating Immune Complexes
Circulating Immune Complexes (CIC) can be formed upon drug administration due to immunogenicity/ADA formation and presence of soluble (multimeric) ligands. What is the industry best practice in bioanalysis for better understanding of CICs? Is size-specific bioanalysis of CIC possible by size exclusion chromatography (SEC)-LBA? Faster clearance of immune complexes (ICs) is mainly driven by high molecular weight CICs. Do you agree that the clearing property of ADA is a function of the size of the formed CICs? What is the industry experience in biodistribution regarding the clearance pathway of the CICs, impact of IgG subclasses/Fc variants on CIC PK, impact of CIC formation on PK, impact of CIC formation on immunogenicity, and CIC-complement complexes?
Multi-Domain Biotherapeutics: Immunogenicity Assay Strategies
For low risk multi-domain biotherapeutics (MDB) with low ADA incidence, is it necessary to evaluate ADA domain specificity? If so, would implementation of such an assessment only in Phase III be sufficient? If low ADA incidence does not allow impact assessments of domain specific ADA on the clinical outcome, how will the data be used during BLA review? For MDBs that have domains for function (e.g., ADC Ab and toxin), structure (e.g., ADC linker) or PK (e.g. Fc modification), is it sufficient to assess the domain specificity for only the functional domains and not for all domains? When will epitope mapping be needed instead of domain specificity? Will multiplex domain specificity assays be acceptable? Has this assay approach been implemented in drugs that have successfully received market approval?
Definition of Persistent ADA Response and its Clinical Relevance
Should an ADA response be characterized as “persistent” if only the last immunogenicity time point tested ADA positive? What if earlier time points fluctuate between positive and negative? Should a persistent response be defined as consistently positive over a certain period of time, such as 16 weeks? Does “less than 16 weeks before an ADA-negative last sample” truly represent a persistent response? Can persistence coupled with other characteristics (NAb, high titer) be better predictors of impact of immunogenicity? Should the term “persistent” only be used when there is an association with altered exposure, safety or efficacy?
DISCUSSIONS, CONSENSUS AND CONCLUSIONS
Lessons Learned from Late Stage Clinical Studies
There is continued concern that the immunogenicity risks identified in clinical trials will not accurately reflect the real-world clinical experience due to the limited numbers of patients in clinical trials, overly-restrictive eligibility criteria, assays with low sensitivity and drug tolerance, and relatively short-term follow-up after initial dosing in the clinical trial setting. As a result, the clinical relevance of the ADA responses may not be fully evaluated and unexpected immunogenicity-related safety events or reduced efficacy may occur once more patients are assessed for longer periods of time. This concern has been realized in rare examples where newly emergent immunogenicity-related issues have arisen in the commercial setting. For this reason, comprehensive long-term immunogenicity monitoring is often required and included in pharmacovigilance and risk management plans in the post-approval setting.
The goal of post-approval monitoring of immunogenicity is to detect signals of clinically relevant ADA. These signals should be relatively apparent, such as a negative impact on PK, identification of a subset of patients losing efficacy or PD response over time, hypersensitivity responses occurring after repeat dosing, a safety signal associated with neutralization of an endogenous counterpart, and additional clinical outcomes (acute or chronic) that are suggestive of immunogenicity related mechanisms.
The best strategy to adopt should be science-based, data- and purpose-driven. Literature has no shortage of reports that provide industry experience from protein therapeutics [Citation21,Citation25,Citation77]. Case studies like those described below demonstrate that the immunogenicity section of product inserts contains useful information as well. Finally, regulatory guidelines are invaluable [Citation26,Citation57,Citation78].
Despite the perceived need to continue vigilant post-approval immunogenicity monitoring, many cases have demonstrated that data obtained from late-stage clinical trials have accurately predicted the real-world immunogenicity experience. It was agreed that there are several case studies within the industry experience in which more sensitive bioanalytical assays and re-analysis of pivotal clinical trial data were performed due to a post-marketing request (PMR) yet similar immunogenicity findings were found. Relevant, meaningful and quality clinical trial data could predict patients’ immunogenicity in the post-approval setting.
One case regarded an approved intravenous administered enzyme replacement therapy for which the FDA requested the optimization of the NAb assay and the inclusion of a titer-based step, as well as re-optimization of the drug specific IgE assay in the post-marketing setting. Following re-optimization of the assays, the sponsor was asked to re-test all the clinical specimens from the pivotal clinical trial with the new assays, as well as investigate a prophylactic immune tolerance regimen in a cohort of patients treated with the biotherapeutic. Low-level sporadic IgE positivity was detected in a newly developed ImmunoCAP assay but these results were not associated with or predictive of hypersensitivity, similar to the results obtained with the original IgE assay in the clinical trials. Likewise, the newly optimized NAb assay demonstrated comparable results to those derived from the original assay, demonstrating no association with efficacy endpoints and similarly sustained PD effect across all NAb quartiles [Citation79]. These results supported the release from the immune tolerance PMR 5 years after drug approval without the need for performing an immune tolerance induction (ITI) regimen.
Conversely, clinically relevant ADA was detected for bacterially derived enzyme replacement therapy where all patients developed a biphasic immune response [Citation80]. As a result of the wide immunogenicity monitoring, information was incorporated into the label instructing physicians to titrate the dosage in a step-wise manner based on tolerability to achieve an effective maintenance dosage and consider increasing the dosage in patients who have not achieved phenylalanine reduction. Extensive immunogenicity assay optimization and re-testing was requested in the post-approval setting, despite the data not being used to inform clinical decision making. In addition, the sponsor received a PMR to perform ITI in an attempt to reduce the overall immunogenicity against the biotherapeutic.
Based on industry experience with assessing risks and benefits of biotherapeutics in the post-market setting, recommendations were provided on how to decide what is justifiable immunogenicity monitoring and what is unwarranted and of limited clinical value. Sufficient ADA data to understand clinical response and impact should be available at the time of submission. Monitoring should be based on scope of available data, clinical signals (safety and efficacy related observations), and the overall clinical impact of the immunogenicity data. In cases where the ADA analysis from the clinical trials demonstrates a lack of clinical impact on PK, PD, and/or safety profiles, continued immunogenicity monitoring may be unwarranted in the post-marketing setting and should be discussed with the Agency.
Clinical impact (triggers), for example, a decrease in efficacy or an emergent safety finding, should be defined by the sponsor on a case-by-case basis depending on the drug mode of action. Establishing clinical triggers for immunogenicity monitoring is more relevant than defining titer-based threshold criteria for long-term testing.
For chronic multi-dose treatments, a time frame of a minimum of 1–2 years of total clinical experience per patient was proposed. One year is typically sufficient for a single dose therapy while 2 years may be needed for multi-dose product depending on dosing intervals. Clinical experience may include the pivotal study period and post-market monitoring. For high immunogenicity risk products, active discussions with the relevant regulatory agencies are encouraged throughout the product life-cycle management. Acute treatments may need special considerations based on the dosing regimen and clinical risk.
If PMR and/or PMC is warranted, to determine when enough post-market immunogenicity monitoring has been completed and the anti-drug immune response has been sufficiency evaluated, an endpoint should be defined in the clinical protocol based on clinical triggers. If, within the 1 to 2F-year monitoring, there is no observed immunogenicity-related clinical signals (loss of efficacy or immunogenicity related safety findings), cessation of immunogenicity monitoring should be considered. PK and PD data are usually not required as part of PMR and/or PMC although that information is valuable for understanding the correlation of immunogenicity and clinical signals.
Further industry and health authority discussions are needed to determine when there is value in continuing to monitor immunogenicity, develop more sensitive bioanalytical assays, and re-analyze immunogenicity data in the post-market setting.
Circulating Immune Complexes
Administration of therapeutic proteins might evoke the formation of immune complexes. The occurrence of an immune response with the formation of ADA which bind the therapeutic protein is one potential mechanism leading to the formation of immune complexes. CICs can influence drug activity, PK and safety. CIC formation can reduce the clearance of the target and/or drug [Citation81], while CIC clearance via FcγR and/or complement can increase drug clearance [Citation82,Citation83]. Clearance of CIC depends on the size and structure (e.g., lattice) [Citation84] and immunoglobulin class within the complex. CIC can also provoke adverse events, like hypersensitivity reactions [Citation80,Citation85,Citation86]. A detailed understanding of the immune complex formation, structure, and PK could improve drug development and interpretation of preclinical/clinical data.
Methodologies for measuring CICs are evolving. SEC and asymmetric flow field flow fractionation (A4F) can detect CICs but provide no actual size information if not coupled to specific detectors such as Multi Angle Light Scattering (MALS). Furthermore, application of these technologies to analyze biological samples is challenging with regard to sensitivity and specificity [Citation87,Citation88]. There is a need to develop methods to better characterize CIC, particularly in biological samples, in terms of their size and to better understand their time of onset, structure, and their impact on PK. Detailed knowledge in this field is of increasing complexity and growing importance with the increasing complexity of newly-designed biotherapeutics. Major bioanalytical challenges include how to monitor and evaluate CIC in the product life cycle. However, progress has been made for accurate size-specific CIC bioanalysis by use of SEC-LBA [Citation89]. These methods are best suited for research purposes to better understand drug characteristics and to design improved therapies with the desired PK and clearance profiles. Decisions regarding clinical monitoring of CIC needs to be based on potential for clinically-relevant outcomes.
Recommendations were given on the industry best practice in bioanalysis for better understanding of CIC and its role in clinical studies. First, a consistent definition of the term CIC is required. The term CIC could describe different complexes such as drug-ADA, drug-target, or complement complexes. In addition, a better understanding of in vivo significance of CICs is needed. There is a differentiation between clinical relevance and research relevance (e.g., clinical/safety events vs. PK). Safety and impact-based monitoring was recommended for CIC. If there is a safety signal in non-clinical or early phase studies, then relevant CIC (e.g., complement complexes) measurement could be added to the protocol [Citation80]. The aim would be to understand whether CIC is involved in the mechanism of the clinical/safety events.
So far, commercial generic functional CIC assays based on complement activation have been used, which are not drug- and size-specific but more practical in a clinical setting and likely demonstrate association with some of the safety related signals. Whether drug- and size-specific analysis adds clinical value needs further discussion and investigation. Immunoglobulin isotype (such as IgM) and IgG subclass of ADA may determine the ability to impact drug PK profile and safety more than the CIC size. In addition, very limited data is available on whether CIC size specific analysis is feasible [Citation90]. Given the concentration of drug and ADA in each patient varies over time, the size (based on molar ratio of each component) of the CIC is expected to vary from patient to patient. Size specific analysis is technically challenging, and more investigation is necessary to understand the relevance to CIC clearance. At present, these topics are considered highly relevant for research purposes to understand product attributes but only limited data is available. Further discussions at future WRIB meetings are needed to update and expand the recommendations based on newly acquired industry experience. For now, clinical safety signals matter more than CIC size measurement, because the correlation between the two is unknown.
Multi-Domain Biotherapeutics: Immunogenicity Assay Strategies
With recent advances in technology, biologics became more complex with the emergence of multi-domain modalities. Multi-domain biotherapeutics (MDBs) contain complex structures multiple functional domains, and, sometimes, a multi-step pharmacological MoA. Examples include ADC, Fc-fusion proteins, PEGylated proteins, and bi/multi-specifics [Citation91–93].
Like other biotherapeutic molecules, the PK/PD and exposure-response correlation for MDBs rely heavily on the bioanalytical strategy employed for their characterization. However, the multi-functional domains and dynamic nature of these complex biological entities require additional scientific considerations for selecting an appropriate bioanalytical strategy. MDBs can induce immune response to various domains, which may have different effects on the PK, PD, safety and efficacy of the molecule. Therefore, characterization of ADA domain specificity has become a regulatory expectation for this class of molecule.
The conventional approach to assess domain specificity uses competitive inhibition with domain-containing molecules. Although it is advantageous to employ a single assay, this type of method often has a unique bioanalytical challenge for detection of domain specific ADA subtypes, leading to false negative classification and thus an inaccurate immunogenicity assessment. An alternative approach to overcoming this challenge is to determine various ADA subtypes with multiple assays. However, it is resource intensive to develop multiple assays and challenging to compare results across assays.
Other unique bioanalytical challenges for MDBs include the need to better characterize in vivo biotransformation and/or structural instability, generation of multiple domain-specific critical reagents, evaluation of potential assay interference(s) due to multiple circulating targets and/or pre-existing/treatment-emergent ADA against individual functional domains, quantitation of multiple analytes, requirement of high assay sensitivity and alternate analytical platforms owing to the low clinical doses, and assay continuity across different development stages of the program.
Since there is no single bioanalytical strategy that works for all MDBs, the strategy will need to be adapted on a case-by-case basis depending on the MDB structure, its MoA and target biology, and the PK questions that need to be addressed at a specific development stage of the program.
The 2016 and 2018 White Papers in Bioanalysis [Citation15,Citation21] recommended domain specificity for MDBs at the confirmatory assay tier to characterize immune dominant regions. When developing an assay, an approach based on the complexity of therapeutic MoA was recommended. Discussions on the use of streamlined competitive binding assays using components of MDBs versus more demanding assays for individual domains were inconclusive. Sponsors were recommended to consult with the corresponding regulatory agencies. However, two years of additional experience with MDBs has allowed the initial recommendations on the need for domain specificity characterization and the best bioanalytical approaches to be updated in this White Paper.
The need for evaluating ADA domain specificity should be based on a risk assessment that evaluates the clinical and safety risk instead of a business risk. A high risk MDB has domains with high homology to endogenous counterparts whereas low risk MDBs refer to lack of structural homology and low risk of cytokine release syndrome [Citation94]. The best approach is to first monitor total ADA. If total ADA is positive with impact on PK/PD and safety, then domain specificity should be analyzed. ADA domain specificity for the relevant domains as part of MDBs is not considered equivalent to ADA antigenic epitope mapping. B cell epitopes to proteins could be either linear amino acid based and/or 3D-structure based conformational epitopes given the nature of a polyclonal antibody response. Epitope mapping is part of the tier 3 immunogenicity testing as part of ADA characterization if patient ADA showed negative impact on PK, PD, and/or safety.
Characterization of ADA domain specificity is recommended (tier 3 of immunogenicity testing) and should assess responses to all functional domains that play a role in mechanism of action of the drug. In certain cases where characterization information from phase I and II is sufficient to support low clinical risk, further analysis should be discussed with regulatory authorities. However, most often the data from phase I & II studies is insufficient to make a determination.
For MDBs that have domains for function (e.g., ADC Ab and toxin) and for structure (e.g., ADC linker) or PK (e.g., Fc modification), it was discussed whether it is sufficient to assess the domain specificity for the functional domains only. From a regulatory perspective, data are only necessary for the functional domains but additional data may be helpful to sponsors for follow-up molecules. For example, Fc domain specificity characterization is relevant if it has a pharmacological function or is modified/engineered. This can be determined before phase III and sufficient sample volume should be collected to perform retrospective domain analysis if needed. Furthermore, although only functional domain information is recommended, epitope mapping may be useful to improve a platform or molecule for follow-up generations.
Characterization of ADA domain specificity presents technical challenges, particularly for low titer total ADA samples, and several methodologies exist [Citation95,Citation96]. It was determined that multiplex domain specific assays are acceptable, but experience is very limited. Recently, an approach has been published to overcome technical challenges of the competitive domain specificity method [Citation97]. A review of the different approaches, technical aspects and challenges for MDB assays was recommended as a topic for a future WRIB.
Definition of Persistent ADA Response and its Clinical Relevance
There is an acknowledged lack of relationship between the reported ADA incidence and associated clinical parameters for example, PK, efficacy, safety [Citation15]. Magnitude (titer) and duration (persistence) of the ADA response may correlate better with clinical impact of ADA, rather than the ADA incidence [Citation26,Citation57]. Although methods for objective assessment of the duration of ADA responses have been proposed, colloquial classifications into transient and persistent responses are more prevalent. Industry and regulatory harmonization efforts have provided some basis for the definition of “persistent” ADA response. However, use of these definitions with some modifications is observed in practice. The impact of these modifications on the correlation between duration of ADA response and clinical safety and efficacy was discussed.
The current definition of a persistent ADA response, as outlined by Shankar et al. [Citation98], is a “treatment-induced ADA detected at two or more sampling time points during the treatment (including follow-up period if any), where the first and last ADA-positive samples (irrespective of any negative samples in between) are separated by a period of 16 weeks or longer, or treatment-induced ADA incidence only in the last sampling time point of the treatment study period or at a sampling time point with less than 16 weeks before an ADA-negative last sample”.
Since there is no FDA guidance for the nomenclature and the EMA guideline references the Shankar paper, the possibility of employing an alternative definition for “persistence” was discussed. It was discussed that low-titer ADA positive results near the assay cut point that do not show upward trending in titer and/or consistent positivity over time are unlikely to be clinically meaningful and should be distinguished from a clinically meaningful treatment-induced persistent ADA response.
The impact of the following definition was evaluated in a presented case study: “Treatment-emergent ADA positive response with two or more consecutive ADA positive sampling time points, separated by at least 12/16-week period (based on nominal sampling time), with no ADA negative samples in between, regardless of any missing samples”. Using this definition, three categories were possible: persistent (potential impact on drug exposure; may or may not translate to impact on efficacy), transient (no impact on drug exposure, efficacy or patient safety), and indeterminate (generally no impact on drug exposure and/or efficacy; no impact on patient safety). An analysis of clinical study data resulted in some patients identified as persistent by the working definition “switch” to the transient or indeterminate category using the modified definition. Preliminary analysis suggested that patients identified as “persistent” by the working definition may have drug concentration profiles that appear to be similar to ADA negative patients. Considering inter-patient variability, patients identified as “ADA persistent” by the modified definition exhibited a noticeable impact on drug concentration profile. Analysis is underway in other studies to assess if these observations are consistent across studies.
This topic was extensively discussed in the 2016 White Paper in Bioanalysis [Citation15]. The recommendations stated:
“To properly classify the ADA duration data, at least 1 year of immunogenicity data were recommended. A 16- or 12-week duration were both deemed acceptable to characterize the patient as having an ADA as persistent response and an adequate sampling schedule should be selected”
“The duration of follow-up testing for ADA-positive patients should be data driven and should not default to continuous monitoring until subjects become baseline negative”
These variations led to discussion to bring consistency to the definition of “persistent”. It was recommended that the existing white paper defining transient versus persistent responses is sufficient [Citation98]. This information is rarely placed on labels but if it is, the sponsor definition should be stated. If the sponsor deviates from the working definition for example, using the 12 weeks criteria instead of the 16 weeks, it is recommended that this be clearly stated in the dossier. However, there are challenges to interpretation of low-level positive responses near to the assay cut point, potential false positives particularly if no clinical relevance is associated. Therefore, discussions on the term “persistent” may be required with other stakeholders for example, physicians, because of the potential for misinterpretations with clinical relevance.
RECOMMENDATIONS
Below is a summary of the recommendations made during the 14th WRIB:
Lessons Learned from Late Stage Clinical Studies
Clinically relevant ADA signals should be relatively apparent in the form of negatively affected PK after repeat dosing, a subset of patients losing efficacy or PD response over time, occurrence of hypersensitivity responses, or a safety signal associated with neutralization of an endogenous counterpart
The best strategy for post-approval monitoring of immunogenicity should be science-based and data-driven. Published data, case studies, labels, and guidelines should be used to inform decisions
Sufficient immunogenicity data to understand clinical impact should be collected and available at the time of approval
The need for immunogenicity monitoring should be based on safety signals and clinical impact
High ADA titers alone may have little clinical relevance and monitoring until samples become negative may be challenging and of limited clinical value
Clinical triggers for post-marketing immunogenicity monitoring should be defined by the sponsor on a case-by-case basis depending on the mode of action of the biotherapeutic. Establishing clinical triggers for immunogenicity monitoring is more relevant than defining a titer threshold
For chronic treatments, a time frame of a minimum of 1–2 years of total clinical experience per patient was proposed, depending on the immunogenicity risk categorization of the drug. Clinical experience may include the pivotal study period and post-marketing monitoring, if warranted
For high risk products, active discussions with the relevant regulatory agencies are encouraged throughout the product lifecycle. Acute treatments may need special considerations based on the regimen and clinical risk.
An endpoint should be defined based on clinical triggers to determine when enough post-marketing immunogenicity monitoring has been collected and the anti-drug immune response has been sufficiently assessed
PK and PD data are valuable for understanding the correlation of immunogenicity and clinical consequence but not required
Circulating Immune Complexes
A consistent definition of the term CIC needs to be established for a better understanding of the in vivo significance of CICs
It is important to continue with the collective effort from industry to understand the immune complex formation, structures, and impact on PK to improve drug development and interpretation of preclinical/clinical data
It was agreed that additional studies are needed to better characterize CIC in terms of their size and to better understand their formation, structure, PK, and clinical relevance
Safety and impact-based monitoring in patients was recommended for CIC. If a CIC-related clinical signal is identified in non-clinical or early phase studies, then inclusion of CIC measurement in the phase III clinical protocol should be considered
Functional CIC based on complement activation may be more suited for clinical impact driven analysis, rather than size-based CIC analysis
Multi-Domain Biotherapeutics: Immunogenicity Assay Strategies
It was agreed that characterization of ADA domain specificity has become a regulatory expectation for multi-domain biotherapeutics
The conventional approach to assess domain specificity uses competitive inhibition with domain-containing molecules
An alternative approach to overcoming the challenge of detection of low level ADA subtypes is to determine various ADA subtypes with multiple domain detection assays
Domain specificity is not considered equivalent to epitope mapping
As such there is no single bioanalytical strategy that works for all MDBs; rather the strategy will need to be adapted on a case-by-case basis depending on the MDB structure and its MoA, target biology, and the PK question that needs to be addressed at a specific development stage of the program
The need for evaluating ADA domain specificity should be based on a risk assessment that evaluates the clinical and safety risk instead of a business risk. The best approach is to monitor total ADA, and if ADA is positive with impact on PK/PD and safety, then domain specificity can be analyzed
Characterization of ADA domain specificity is recommended (tier 3 of immunogenicity testing), especially for low risk molecules, to help elucidate ADA domain binding specificity and its impact if there is a clinical signal. If characterization information from phase I and II indicates low clinical risk, this immunogenicity data may be sufficient to streamline Phase III monitoring
Data are only necessary for the functional domains, but evaluation of other domains, including epitope mapping, may be useful for the design of a novel drug with lower immunogenicity. Multiplex domain specific assays are acceptable, but experience is very limited due to technical challenges
Definition of Persistent ADA Response and its Clinical Relevance
It was confirmed that magnitude (titer) and duration (persistence) of the ADA response may correlate better with clinical impact of ADA than only considering incidence alone
Consensus was achieved that the existing Shankar white paper definition of transient versus persistent responses is still adequate [Citation98]
If information on ADA persistence is included on the label, the sponsor-specific definition of persistence should be provided in the label, particularly if the sponsor deviates from the standard definition (e.g., using the 12 weeks criteria instead of the 16 weeks)
Table 1. Parameters for vaccine serology assay qualification and validation.
Table 2. System Level QC Practices for CAR-T Monitoring Assays.
Table 3. qPCR Assay Validation Considerations for Cell and Gene Therapy.
Table 4. Sample Panel for ELISpot Assay Qualification.
Table 5. ELISpot Assay Qualification Parameters.
Acknowledgements
The US FDA, Europe EMA, UK MHRA, Health Canada, Norway NoMA, Brazil ANVISA, Japan MHLW, and for supporting this workshop
Dr. Steven Alley (Seattle Genetics), Dr. Anna Edmison (Health Canada), Dr. Chris Evans (GSK), Dr. Christine Fandozzi (Merck), Dr. Sally Fischer (Genentech), Dr. Fabio Garofolo (BRI), Dr. Christine Grimaldi (Boehringer Ingelheim), Dr. Lindsay King (Pfizer), Dr. Rocio Murphy (Merck), Dr. Hendrik Neubert (Pfizer), Dr. Manoj Rajadhyaksha (Regeneron), Dr. Catherine Soo (Health Canada), Dr. Susan Spitz (Incyte), Dr. Roland F. Staack (Roche), Dr. Scott Summerfield (GSK), Dr. Alessandra Vitaliti (Novartis), Drs. Jan Welink (EMA), Dr. Haoheng Yan (US FDA), Dr. Tong-yuan Yang (Janssen), Dr. Hongbin Yu (Boehringer Ingelheim), Dr. Yan Zhang (BMS) for chairing the workshop and/or the White Paper discussions
All the workshop attendees and members of the Global Bioanalytical Community who have sent comments and suggestions to the workshop to complete this White Paper
Future Science Group as a trusted partner
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
References
- Savoie N , BoothBP, BradleyTet al. 2008 White Paper: The 2nd Calibration and Validation Group Workshop on Recent Issues in Good Laboratory Practice Bioanalysis. Bioanalysis1(1), 19–30 (2009).
- Savoie N , GarofoloF, van AmsterdamPet al. 2009 White Paper on Recent Issues in Regulated Bioanalysis from the 3rd Calibration and Validation Group Workshop. Bioanalysis2(1), 53–68 (2010).
- Savoie N , GarofoloF, van AmsterdamPet al. 2010 White Paper on Recent Issues in Regulated Bioanalysis and Global Harmonization of Bioanalytical Guidance. Bioanalysis2(12), 1945–1960 (2010).
- Garofolo F , RocciM, DumontIet al. 2011 White Paper on Recent Issues in Bioanalysis and Regulatory Findings from Audits and Inspections. Bioanalysis3(18), 2081–2096 (2011).
- DeSilva B , GarofoloF, RocciMet al. 2012 White Paper on Recent Issues in Bioanalysis and Alignment of Multiple Guidelines. Bioanalysis4(18), 2213–2226 (2012).
- Stevenson L , RocciM, GarofoloFet al. 2013 White Paper on Recent Issues in Bioanalysis: “Hybrid”–the best of LBA & LC/MS. Bioanalysis5(23), 2903–2918 (2013).
- Fluhler E , HayesR, GarofoloFet al. 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 1–small molecules by LCMS). Bioanalysis6(22), 3039–3049 (2014).
- Dufield D , NeubertH, GarofoloFet al. 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 2–hybrid LBA/LCMS, ELN & regulatory agencies’ input). Bioanalysis6(23), 3237–3249 (2014).
- Stevenson L , AmaravadiL, MylerHet al. 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 3–LBA and immunogenicity). Bioanalysis6(24), 3355–3368 (2014).
- Welink J , FluhlerE, HughesNet al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 1–small molecules by LCMS). Bioanalysis7(22), 2913–2925 (2015).
- Ackermann B , NeubertH, HughesNet al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 2–hybrid LBA/LCMS and input from regulatory agencies). Bioanalysis7(23), 3019–3034 (2015).
- Amaravadi L , SongA, MylerHet al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 3–LBA, biomarkers and immunogenicity). Bioanalysis7(24), 3107–3124 (2015).
- Yang E , WelinkJ, CapeSet al. 2016 White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV) (Part 1–small molecules, peptides and small molecule biomarkers by LCMS). Bioanalysis8(22), 2363–2378 (2016).
- Song A , LeeA, GarofoloFet al. 2016 White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV): (Part 2–Hybrid LBA/LCMS and input from regulatory agencies). Bioanalysis8(23), 2457–2474 (2016).
- Richards S , AmaravadiL, PillutlaRet al. 2016 White Paper on recent issues in bioanalysis: focus on biomarker assay validation (BAV): (Part 3–LBA, biomarkers and immunogenicity). Bioanalysis8(23), 2475–2496 (2016).
- Welink J , YangE, HughesNet al. 2017 White Paper on recent issues in bioanalysis: aren't BMV guidance/guidelines ‘Scientific’? (Part 1–LCMS: small molecules, peptides and small molecule biomarkers). Bioanalysis9(22), 1807–1825 (2017).
- Neubert H , SongA, LeeAet al. 2017 White Paper on recent issues in bioanalysis: rise of hybrid LBA/LCMS immunogenicity assays (Part 2: hybrid LBA/LCMS biotherapeutics, biomarkers & immunogenicity assays and regulatory agencies’ inputs). Bioanalysis9(23), 1895–1912 (2017).
- Gupta S , RichardsS, AmaravadiLet al. 2017 White Paper on recent issues in bioanalysis: a global perspective on immunogenicity guidelines & biomarker assay performance (Part 3–LBA: immunogenicity, biomarkers and PK assays). Bioanalysis9(24), 1967–1996 (2017).
- Welink J , XuY, YangEet al. 2018 White Paper on Recent Issues in Bioanalysis: ‘A global bioanalytical community perspective on last decade of incurred samples reanalysis (ISR)’ (Part 1–small molecule regulated bioanalysis, small molecule biomarkers, peptides & oligonucleotide bioanalysis). Bioanalysis10(22), 1781–1801 (2018).
- Neubert H , OlahT, LeeAet al. 2018 White Paper on Recent Issues in Bioanalysis: focus on immunogenicity assays by hybrid LBA/LCMS and regulatory feedback (Part 2–PK, PD & ADA assays by hybrid LBA/LCMS & regulatory agencies’ inputs on bioanalysis, biomarkers and immunogenicity). Bioanalysis10(23), 1897–1917 (2018).
- Stevenson L , RichardsS, PillutlaRet al. 2018 White Paper on Recent Issues in Bioanalysis: focus on flow cytometry, gene therapy, cut points and key clarifications on BAV (Part 3–LBA/cell-based assays: immunogenicity, biomarkers and PK assays). Bioanalysis10(24), 1973–2001 (2018).
- Garofolo W , SavoieN. The Decennial Index of the White Papers in Bioanalysis: ‘A Decade of Recommendations (2007–2016)’. Bioanalysis9(21), 1681–1704 (2017).
- Fandozzi C , EvansC, WilsonAet al. 2019 White Paper on Recent Issues in Bioanalysis: Chromatographic Assays (Part 1–Innovation in Small Molecules and Oligonucleotides & Mass Spectrometric Method Development Strategies for Large Molecule Bioanalysis). Bioanalysis11(22), 2029–2048 (2019).
- Booth B , StevensonL, PillutlaRet al. 2019 White Paper On Recent Issues in Bioanalysis: FDA BMV Guidance, ICH M10 BMV Guideline and Regulatory Inputs (Part 2–Recommendations on 2018 FDA BMV Guidance, 2019 ICH M10 BMV Draft Guideline and Regulatory Agencies’ Input on Bioanalysis, Biomarkers and Immunogenicity). Bioanalysis11(23), 2099–2132 (2019).
- Piccoli S , MehtaD, VitalitiAet al. 2019 White Paper on Recent Issues in Bioanalysis: FDA Immunogenicity Guidance, Gene Therapy, Critical Reagents, Biomarkers and Flow Cytometry Validation (Part 3–Recommendations on 2019 FDA Immunogenicity Guidance, Gene Therapy Bioanalytical Challenges, Strategies for Critical Reagent Management, Biomarker Assay Validation, Flow Cytometry Validation & CLSI H62). Bioanalysis11(24), 2207–2244 (2019).
- US FDA, Center for Drug Evaluation and Research . Guidance for Industry: Immunogenicity Testing of Therapeutic Proteins Products–Developing and Validating Assays for Anti-Drug Antibody Detection (2019). https://www.fda.gov/media/119788/download
- Shankar G , DevanarayanV, AmaravadiLet al. Recommendation for the validation of immunoassay used for detection of host antibodies against bioanalytical products. J. Pharm. Biomed. Anal.48, 1267–1281 (2008).
- Smith BP , VandenhendeFR, DeSanteKAet al. Confidence interval criteria for assessment of dose proportionality. Pharm. Res.17(10), 1278–1283 (2000).
- Hendriks J , StalsC, VersteilenAet al. Stability studies of binding and functional anti-vaccine antibodies. Bioanalysis6(10), 1385–1393 (2014).
- Michaut L , LaurentN, KentschKet al. Stability of anti-immunotherapeutic antibodies in frozen human serum samples. Bioanalysis6(10), 1395–1407 (2014).
- CLSI . CLSI Guideline H62: Validation of Assays Performed by Flow Cytometry (2019). https://www.wrib.org/PDFs/Flow_H62_Proposed_Draft-190919.pdf
- Cossarizza A , ChangH-D, RadbruchAet al. Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). European Journal of Immunology49(10), 1457–1973 (2019).
- Perfetto SP , AmbrozakD, NguyenRet al. Quality assurance for polychromatic flow cytometry. Nat. Protoc.1(3), 1522–1530 (2006).
- Perfetto SP , AmbrozakD, NguyenRet al. Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nature Protocols7, 2067–2079 (2012).
- Bustin SA , BenesV, GarsonJAet al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem.55(4), 611–622 (2009).
- CLSI . CLSI.org
- Gorovits B , KorenE. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs33(3), 275–284 (2019).
- Potthoff B , McBlaneF, SpindeldreherSet al. A cell-based immunogenicity assay to detect antibodies against chimeric antigen receptor expressed by tisagenlecleucel. J. Imm. Meth.476, 112692 (2020). 10.1016/j.jim.2019.112692
- EMA 2018 . Assessment Report: Kymriah (Tisagenleucel) (2018). https://www.ema.europa.eu/en/documents/assessment-report/kymriah-epar-public-assessment-report_en.pdf
- Lamers CHJ , KlaverY, GratamaJWet al. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem. Soc. Trans.44(3), 951–959 (2016).
- Annika CK et al. Comparison of the ELISPOT and cytokine flow cytometry assays for the enumeration of antigen-specific T cells. J.Immunol. Methods283(1-2), 141–153 (2003).
- Theresa LW et al. Enzyme-linked immunospot, cytokine flow cytometry, and tetramers in the detection of T-cell responses to a dendritic cell-based multipeptide vaccine in patients with melanoma. Clin. Cancer Res.9(2), 641–649 (2003).
- European Medicines Agency . EMA General Principles to Address Virus and Vector Shedding (EMEA/CHMP/ICH/449035/2009) (2009). https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-10.pdf
- US FDA, Center for biologics evaluation and research . Guidance for industry: Design and Analysis of Shedding Studies for Virus or Bacteria-Based Gene Therapy and Oncolytic Products (2015). www.fda.gov/media/89036/download
- Pharmaceuticals and Medical Devices Agency . PMDA Japan Guidance on ensuring the quality and safety of products for gene therapy (2019). https://www.pmda.go.jp/files/000235607.pdf
- Rangarajan S , WalshL, LesterWet al. AAV5-Factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med.377(26), 2519–2530 (2017).
- Zen Z , EspinozaY, BleuT, SommerJM, WrightJF. Infectious titer assay for adeno-associated virus vectors with sensitivity sufficient to detect single infectious events. Hum. Gene Ther.15(7), 709–715 (2004).
- Bustin S , HuggettJ. qPCR primer design revisited. Biomol. Detect. Quantif.14, 19–28 (2017).
- Huggett JF et al. The Digital MIQE Guidelines: minimum information for publication of quantitative digital PCR experiments. Clin. Chem.59(6), 892–902 (2013).
- Wang D , TaiPWL, GaoG. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discovery18, 358–378 (2019).
- Gorovits B , FiscellaM, HavertMet al. Recommendations for the Development of Anti-Viral Vector Neutralizing Antibody Assays. AAPS J.22(2), 24 (2020).
- US FDA, Center for Biologics Evaluation and Research . Guidance for Industry: Human Gene therapy for Rare Diseases (2020). https://www.fda.gov/media/113807/download
- Janetzki S , PanageasKS, Ben-PoratLet al. Results and harmonization guidelines from two large-scale international Elispot proficiency panels conducted by the Cancer Vaccine Consortium (CVC/SVI). Cancer Immunol. Immunother.57(3), 303–315 (2008).
- Janetzki S , PriceL, SchroederHet al. Guidelines for the automated evaluation of Elispot assays. Nat. Protoc.10(7), 1098–1115 (2015).
- Immudex . Proficiency Panels:How Well Do You Perform T-cell Immune Monitoring?https://www.immudex.com/proficiency-panels.aspx
- ICH . ICH Q2R1: Validation of Analytical Procedures: Text and Methodology (2005). https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf
- European Medicines Agency . EMA Guideline on Immunogenicity Assessment of Therapeutic Proteins.EMEA/CHMP/BMWP/14327/2006 Rev 1 (2017). https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf
- Calcedo R , MorizonoH, WangLet al. Adeno-associated virus antibody profiles in newborns, children, and adolescents. Clin. Vaccine Immunol.18, 1586–1588 (2011).
- Mingozzi F , ChenY, EdmonsonSCet al. Prevalence and pharmacological modulation of humoral immunity to AAV vectors in gene transfer to synovial tissue. Gene Ther.20, 417–424 (2013).
- Calcedo R , VandenbergheLH, GaoGet al. Worldwide epidemiology of neutralizing to adenoassociated viruses. J. Infect. Dis.199, 381–390 (2009).
- Finco D , BaltrukonisD, Clemens-EganAet al. Comparison of competitive ligand-binding assay and bioassay formats for the measurement of neutralizing antibodies to protein therapeutics. J. Pharm. Biomed. Analysis54(2), 351–358 (2011).
- Wu Y , AkhgarA, LiJJet al. Selection of a ligand-binding neutralizing antibody assay for benralizumab: comparison with an antibody-dependent cell-mediated cytotoxicity (ADCC) cell-based assay. AAPS J.20(3), 49 (2018).
- Wu B , ChungS, JiangXRet al. Strategies to determine assay format for the assessment of neutralizing antibody responses to biotherapeutics. AAPS J.18(6), 1335–1350 (2016).
- USP-NF <1106.1> Immunogenicity Assays–Design and Validation of Assays to Detect Anti-Drug Neutralizing Antibody (2015).
- US FDA, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) . Guidance for Industry: Clinical Pharmacology Data to Support a Demonstration of Biosimilarity to a Reference Product (2016). https://www.fda.gov/media/88622/download
- US FDA, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER) . Guidance for Industry: Scientific Considerations in Demonstrating Biosimilarity to a Reference Product (2015). https://www.fda.gov/media/82647/download
- US FDA, Center for Drug Evaluation and Research, Center for Veterinary Medicine . Guidance for Industry, Bioanalytical Method Validation (2018). https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf
- US FDA, Center for Drug Evaluation and Research . Draft Guidance for Industry: Clinical Immunogenicity Considerations for Biosimilar and Interchangeable Insulin Products (2019). https://www.fda.gov/media/133014/download
- Devanarayan V , ToveyMG. Cut point and performance characteristics for anti-drug antibody assays. In: Detection and Quantification of Antibodies to Biopharmaceuticals.MichaelG ( Ed.). Tovey, Wiley (2011).
- Kurki P . Compatibility of immunogenicity guidance by the EMA and the US FDA. Bioanalysis11(17), 1619–1629 (2019).
- Kubiak RJ , ZhangJ, RenP, YangH, RoskosLK. Excessive outlier removal may result in cut points that are not suitable for immunogenicity assessments. J. Imm. Meth.463, 105–111 (2018).
- Zhang J , ArendsRHGP, KubiakRJ, RoskoLK, LiangM, LeeN, ChenC-K, YangH. A new method for identification of outliers in immunogenicity assay cut point data. J. Imm. Meth.484–485, 112817 (2020).
- Kubiak RJ , ZhangJ, ZhangJ, ZhuYet al. Correlation of screening and confirmatory results in tiered Immunogenicity testing by solution-phase bridging assays. J. Pharm. Biomed. Anal.74, 235–245 (2013).
- Kubiak RJ , ArendsRGHP, LeeN, LiangM, ZhangJ, RoskosLK. Confirmatory cut point has limited ability to make accurate classifications in Immunogenicity assays. Bioanalysis12(4), 246–256 (2020).
- Tovey MG , LallemandC. Immunogenicity and other problems associated with the use of biopharmaceuticals. Ther. Adv. Drug Safety2(3), 113–128 (2011).
- Jawa V , TerryF, GokemeijerJet al. T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation–updated consensus and review. Front. Immunol.11, 1301 (2020).
- Rosenberg AS , SaunaZE. Immunogenicity assessment during the development of protein therapeutic. JPP70(5), 584–594 (2018).
- US FDA, Center for Biologics Evaluation and Research . Guidance for Industry: Human Gene Therapy for Rare Diseases (2020). https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-rare-diseases
- Andrew CM et al. Antibodies that neutralize cellular uptake of elosulfase alfa are not associated with reduced efficacy or pharmacodynamic effect in individuals with Morquio A syndrome. J. Immunol. Meth.440, 41–51 (2017).
- Gupta S et al. Association of immune response with efficacy and safety outcomes in adults with phenylketonuria administered pegvaliase in phase 3 clinical trials. EBioMedicine37, 366–373 (2018).
- Bruce NR , MooJC. Antibodies as carrier proteins. Pharm. Res.15(11), 1652–1656 (1998).
- Hogarth PM . Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr. Opin. Immunol.14(6), 798–802 (2002).
- Chauhan AK , MooreTL. T cell activation by terminal complex of complement and immune complexes. J. Biol. Chem.286(44), 38627–38637 (2011).
- Rojas JR , TaylorRP, CunninghamMRet al. Formation, distribution, and elimination of infliximab and anti-infliximab immune complexes in cynomolgus monkeys. J. Pharm. Exp. Ther.313(2), 578–585 (2005).
- van Schouwenburg PA et al. Adalimumab elicits a restricted anti-idiotypic antibody response in autoimmune patients resulting in functional neutralisation. Ann. Rheum. Dis.72(1), 104–109 (2013).
- Krishna M , StevenGN. Immunogenicity to biotherapeutics–the role of anti-drug immune complexes. Front. Immunol.7, 21 (2016).
- Hoffmann E et al. Generation, characterization, and quantitative bioanalysis of drug/anti-drug antibody immune complexes to facilitate dedicated in vivo studies. Pharm. Res.36(9), 129 (2019).
- van Schie KA et al. Restricted immune activation and internalisation of anti-idiotype complexes between drug and antidrug antibodies. Ann. Rheum. Dis.77(10), 1471–1479 (2018).
- Regenass-Lechner F et al. Immunogenicity, inflammation, and lipid accumulation in Cynomolgus monkeys infused with a Lipidated Tetranectin-ApoA-I fusion protein. Toxicol. Sci.150(2), 378–389 (2016).
- Tada M , TakuoS, AkikoI-W. Development and characterization of an anti-rituximab monoclonal antibody panel. MAbs10(3), 370–379 (2018).
- Labrijn AF et al. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov.18(8), 585–608 (2019).
- Hoofring SA , LopezR, HockMBet al. Immunogenicity testing strategy and bioanalytical assays for antibody-drug conjugates. Bioanalysis5(9), 1041–1055 (2013).
- Stubenrauch K , KunzelC, VogelR, TuerckD, SchickE, HeinrichJ. Epitope characterization of the ADA response directed against a targeted immunocytokine. J. Pharm. Biomed. Anal.114, 296–304 (2015).
- Gorovits B , PengK, KrommingaA. Current Considerations on Characterization of Immune Response to Multi-Domain Biotherapeutics. BioDrugs34(1), 39–54 (2020).
- Gorovits B et al. Recommendations for the characterization of immunogenicity response to multiple domain biotherapeutics. J. Immunol. Methods408, 1–12 (2014).
- Broders O , WesselsU, ZadakM, BeckmannR, StubenrauchK. Novel bioanalytical method for the characterization of the immune response directed against a bispecific F(ab) fragment. Bioanalysis12(8), 509–517 (2020).
- Vainshtein I , SunB, RoskosLK, LiangM. A novel approach to assess domain specificity of anti-drug antibodies to moxetumomab pasudotox, an immunotoxin with two functional domains. J. Immunol. Methods477, 112688 (2020).
- Shankar G , ArkinS, CoceaLet al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J.16(4), 658–673 (2014).