
Abstract
Molecular dynamics is a state of the art computational tool for the investigation of biophysics phenomenon at a molecular scale, as it enables the modeling of dynamic processes, such as conformational motions, molecular solvation and ligand binding. The recent advances in structural biology have led to a bloom in published G-protein-coupled receptor structures, representing a solid and valuable resource for molecular dynamics studies. During the last decade, indeed, a plethora of physiological and pharmacological facets of this membrane protein superfamily have been addressed by means of molecular dynamics simulations, including the activation mechanism, allosterism and, very recently, biased signaling. Here, we try to recapitulate some of the main contributions that molecular dynamics has recently produced in the field.
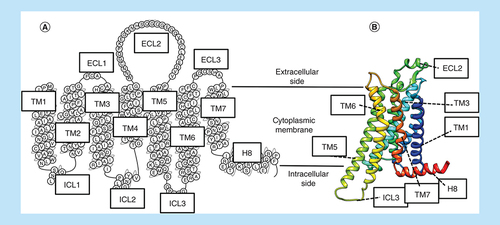
The G-protein-coupled receptors (GPCRs) are a superfamily of integral membrane proteins encoded by more than 800 different human genes [Citation1]. GPCRs transduce extracellular chemical signals into the cellular cytoplasm, where they interact with a set of intracellular effector proteins able to trigger many different physiological effects. GPCRs engage heterotrimeric G proteins, generally able to modulate the cytoplasmic release of calcium [Citation2] and the production of cyclic adenosine monophosphate (cAMP), phosphatidylinositol-4,5-bisphosphate and diacylglycerol [Citation3]; GPCR signaling can also involve arrestins [Citation4], once the receptor C terminus has been phosphorylated by GPCR kinases [Citation5]. GPCRs are structurally characterized by seven transmembrane (TM) helices, interrupted by three flexible extracellular loops (ECLs) and three intracellular loops (ICLs; ) [Citation6,Citation7]. It is the inherent flexibility of GPCRs that make molecular dynamics (MD) such a useful technique for their study.
(A) Snake plot (modified from www.gpcrdb.org) showing the main structural features of a GPCR. The protein single-chain spans the cytoplasmic membrane via seven transmembrane helices (TM1-7), and it is characterized by three ECLs (ECL1-3) and three ICLs (ICL1-3), as well as a helical intracellular C terminus (H8). (B) Ternary organization (ribbon representation) of the structural elements.
ECL: Extracellular loop; GPCR: G-protein-coupled receptor; ICL: Intracellular loop; TM: Transmembrane.
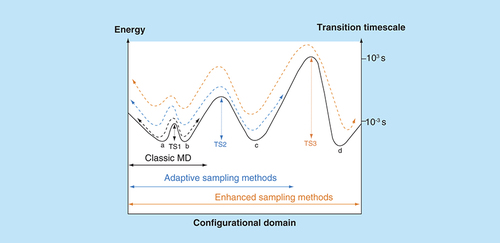
It has been estimated that approximately 34% of approved drugs act on GPCRs [Citation8], making GPCRs the most targeted protein family. Considering that these drugs largely act on a small proportion of the family and that the development of potential drugs for most of the GPCRs currently targeted is still in early or preclinical phases, there is considerable prospect and scope for designing novel therapeutic candidates. In recent years, x-ray crystallography first, and more recently cryo-electron microscopy (cryo-EM) techniques have led to a bloom of published GPCR structures [Citation9], reinforcing the importance of structure-based drug design as a fundamental approach in both academic and industrial research environments. From this point of view, MD simulations represent a helpful computational tool for the rationalization of experimental findings as well as the generation of further working hypothesis at the molecular level. MD algorithms enable a description of the temporal evolution of a system according to its potential energy and Newton’s laws (knowing the acceleration, velocity and position of the atoms at a precise instant in time, t, it is possible to approximate its velocity and position at a successive instant t + Δt). In biological disciplines as well as other fields, computational sciences are taking advantage of the continuous advancement of both hardware and software [Citation10]. It follows that the timescales that MD can cover have leaped from picoseconds, on systems with a few hundred atoms in the 1970s [Citation11], to milliseconds (on purpose-dedicated computers [Citation12]) for systems with hundreds of thousands of atoms. However, the duration of biological phenomena such as conformational activation and ligand unbinding usually go beyond the MD capability [Citation13]. To tackle this issue, many so-called adaptive [Citation14] and enhanced [Citation15] sampling MD protocols have been developed, allowing simulations to overcome energetic barriers between stable basins and, in turn, to sample biologically relevant configurations kinetically inaccessible by means of classic MD simulations ().
Each state of the system, within all the possible ensemble of configurations (configurational domain), is associated with an energy value. The system can experience a transition from one basin (here indicated as a, b, c and d) to another if it is able to traverse the transition state (TS) between them. By means of classic MD, it is possible to sample minima separated by low-energy TSs (i.e., TS1). By monitoring one or more physical quantities during the simulations (adaptive sampling), it is possible to reduce the time needed to overcome higher energy transition states (TS2). Enhanced sampling methods can enable the system to visit all the states by adding an energetic bias: configurations kinetically unreachable for classic MD and adaptive sampling methods (TS3) can, therefore, be sampled during the simulation. Reported transition timescales are just indicative of the orders of magnitude that enhanced sampling methods allow, compared with classic MD.
MD: Molecular dynamics; TS: Transition state.
In this review, we try to outline and summarize the contribution of atomistic MD simulations to recent advances in the structural comprehension of GPCRs. The main focus will address crucial aspects of ligand binding, the allostery phenomena, solvation effects and the activation mechanism of this captivating receptor family.
Ligand binding
In equilibrium conditions, a binding process can be described as an ensemble of states that are populated with a probability proportional to the energetic stability gained by the ligand (). According to transition state theory (which probably oversimplifies the actual free-energy surface of binding), the probabilities (and the timescales) of transitions between these basins are driven by the energy needed to overcome the saddle points (transition states). It follows that a ligand can be stabilized in several intermediate configurations (metastable states, ) along the path to the final bound state. The energy required for overcoming a transition state and to evolve toward another metastable state is usually due to desolvation phenomena, conformational rearrangements and breaking intermolecular interactions. From this standpoint, MD has proved to be a useful tool for describing the ligand (un)binding mechanism for several families of receptors [Citation16,Citation17].
(A) The centroid of a GPCR ligand (shown as a small red sphere) populates different metastable states during the binding process, before reaching the orthosteric site. Energetically favored transitions from different metastable states describe complete binding paths (in this example, binding path 1 and 2). (B) Centroids corresponding to energetically unfavorable states (white spheres) are widely dispersed at the extracellular vestibule, indicating the receptor/ligand desolvation as a putative kinetic bottleneck, as proposed in [Citation31].
GPCR: G-protein-coupled receptor; ECL: Extracellular loop; TM: Transmembrane.
![Figure 3. Representation of metastable and transition states along the binding pathway of a G-protein-coupled receptor ligand. (A) The centroid of a GPCR ligand (shown as a small red sphere) populates different metastable states during the binding process, before reaching the orthosteric site. Energetically favored transitions from different metastable states describe complete binding paths (in this example, binding path 1 and 2). (B) Centroids corresponding to energetically unfavorable states (white spheres) are widely dispersed at the extracellular vestibule, indicating the receptor/ligand desolvation as a putative kinetic bottleneck, as proposed in [Citation31].GPCR: G-protein-coupled receptor; ECL: Extracellular loop; TM: Transmembrane.](/cms/asset/10dd6e51-dc69-4a09-bc84-27ca564f2a74/ifmc_a_12361681_f0003.jpg)
GPCRs ligands (un)binding: a dynamic perspective
During drug discovery campaigns, it is common to refer to the affinity or potency of a drug candidate, to quantify in vivo activity. However, living organisms are open systems, able to exchange both energy and matter with the surrounding environment. In such physiological conditions, pharmacokinetic phenomena do not allow chemicals to reach a steady state in the proximity of their biological targets after administration. During recent years, there has been a gradual shift from the axiomatic use of the thermodynamic constants KD (dissociation constant) and KA (association constant) to nonequilibrium parameters (i.e., the kinetics constants kon and koff) as ligand-target affinity descriptors. In fact, emerging evidence has pointed out the importance of the binding kinetics on the pharmacology of both endogenous and xenobiotic compounds [Citation18–21]. The dissociation kinetics and therefore the target residence time (RT, defined as the reciprocal of the koff) should be a better predictor of in vivo efficacy in specific cases, as compounds with similar thermodynamic affinity for a biological target can have a very different RT, and therefore a kinetic selectivity (KS). An example of KS is ascribable to tiotropium [Citation22,Citation23], which when dissociating about ten-times slower from the M3-R (muscarinic receptor M3) than the M2-R subtype, is able to trigger the pharmacological effects of the former, limiting the side effects due to the inhibition of the latter one. A further case of how RT can tune in vivo effects could be represented by the atypical antipsychotic drugs [Citation24]. Indeed, it has been proposed that antagonist of the D2-R (dopamine D2 receptor) that are characterized by a faster dissociation rate have reduced on-target extrapyramidal side effects [Citation25].
Also the association kinetics is progressively more recognized as a significant factor in determining a ligand’s activity. On-target side effects, in theory, can be reduced by slow-binding ligands, as high target occupancy and activation may be limited. However, a slow binder would not be able to produce a maximal effect in vivo, as the pharmacokinetic elimination may limit the drug tissue concentration and therefore not allow the full occupation of the biological target. Fast binders, on the other hand, are more prone to rebinding phenomenon, which can result in prolonged receptor activity [Citation26]. The necessity of drug candidates with a well-balanced kinetic is increasingly producing efforts on the development of quantitative structure–kinetic relationship protocols, [Citation27–30].
In the GPCR field, the binding to the β2-AR (β2-adrenergic receptor) [Citation31–33] and the M2-R and M3-R [Citation33] were the first to be studied using MD, in 2011. In their pioneering work, Dror et al. [Citation31] simulated several spontaneous binding events of the β2-AR antagonists propranolol, alprenolol, dihydroalprenolol as well as the agonist isoproterenol, observing a common binding path characterized by metastable states in the proximity of extracellular loop 2 (ECL2), extracellular loop 3 (ECL3) and the upper portions of transmembrane helix 6 (TM6) and TM7. Interestingly, the energy barrier of the process was ascribed to the rapid dehydration (within 1 ns) of both the ligand and the receptor in order to establish hydrophobic interactions at the extracellular vestibule. For the M2-R and M3-R [Citation33], simulations highlighted the presence of several metastable states but suggested that conformational rearrangement involving the receptor may act as the rate-limiting step of the overall event. Interestingly, acetylcholine was instead able to bind the M3-R without experiencing any metastable state in concordance with its status as an ultrafast binder [Citation34]. Metastable sites at the protein-solvent interface were also observed during the dissociation paths for β2-AR inverse agonists [Citation32].
The A2A AR (A2A adenosine receptor) is among the most characterized class A GPCRs, as to date more than 40 structures have been published [Citation35]. Several computational studies have addressed the dynamics of this receptor from different points of view. Guo et al. [Citation36] employed temperature-accelerated MD simulations (i.e., a form of enhanced sampling) to sample the dissociation mechanism of the antagonist ZM241385 and identified several amino acids, grouped in two different topological clusters, able to modulate the unbinding kinetics rather than the equilibrium affinity of the ligand. The authors proposed a multistep dissociation, indicating the disruption of the ionic interaction between ECL2 and ECL3 as the early stage of the event. The influence of this intramolecular protein interaction on the koff of different antagonists was addressed in the work of Segala et al. [Citation37], which reported how different ligand substituents can modulate the stability of the salt bridge and, in turn, the ligand RT.
The process of adenosine binding to its A2A AR, both in the intermediate-active [Citation38] and active [Citation39] state, was recently investigated using the supervised MD (SuMD) method, an adaptive method [Citation40,Citation41]. During the simulations the endogenous agonist was able to reach the crystallographic bound conformation only in the presence of the engineered G protein, in accord with the scenario in which receptor pre-coupling with intracellular effectors allosterically influences the association of agonists [Citation42,Citation43]. Very recently, it has been hypothesized that the A2A AR may contemporarily engage two molecules of its natural ligand, leading to a 1:2 complex [Citation44]. This scenario has been partially supported by the experimental observation that adenosine, in presence of the potent agonist CGS-21680, binds to a site alternative to the orthosteric one [Citation45]. SuMD simulations were also combined with a biased approach to characterize putative kinetics bottleneck during the (un)binding of A2A AR antagonists [Citation46], proposing a critical role of the solvation/desolvation of the protein and the ligand in transition states of entropic nature.
Besides the bulk solvent, the lipid environment also allows the diffusion of (lipophilic) ligands towards GPCR orthosteric sites. From this standpoint, MD simulations have shed light on the binding processes to the P2Y1 R (purinergic P2Y1 receptor) [Citation47], the CB2R (cannabinoid receptor 2) [Citation48], and the S1PR (sphingosine-1-phosphate receptor 1) [Citation49]. Interestingly, the latter study reported ionic interactions at the protein surface in contact with lipid environment receptor membrane vestibule as well as the desolvation required for the ligand entrance to the binding site as kinetics bottlenecks of the event. Temperature-accelerated MD was used to study the unbinding paths of the PAR1 (protease-activated receptor 1) antagonist vorapaxar [Citation50,Citation51], proposing the putative orthosteric access of the ligand through the cleft between TM6 and TM7.
MD is increasingly employed as a tool for the challenging direct estimation of binding kinetics parameters [Citation52]. Besides the construction of Markov state models [Citation53] (which are usually based on long timescale-unbiased simulations [Citation54,Citation55]), other computational approaches allow simulating GPCR (un)binding events by keeping track of the energy needed to achieve the transition between basins. Bortolato et al. [Citation56] ranked a set of CRF1R (corticotropin-releasing factor 1 receptor) ligands on the basis of the maximum energy required for simulating the dissociation, gaining good agreement with the experimental RTs. In a subsequent work [Citation57], the same authors proposed that a binding pathway from the membrane bilayer, between TM5 and TM6, was the energetically favored pathway for the allosteric modulator CP-376395.
GPCR activation & signaling
In order to accomplish their membrane transducer functions [Citation58,Citation59], GPCRs undergo structural transitions capable of transferring the extracellular chemical information (i.e., the agonist binding) to the intracellular side of the cytoplasmic bilayer, where cognate proteins bind. A few decades after the ternary model was formulated [Citation60]), structural biology and related disciplines [Citation3] are now increasingly shedding light on GPCR activation mechanisms [Citation7,Citation61], highlighting an intrinsic dynamicity of this receptor superfamily [Citation62] and common structural hallmarks among different GPCR classes [Citation6,Citation9,Citation62–66]. As shown in , the principal structural change involves the intracellular side of the receptor, where TM6 kinks and swings as a result of well-placed conserved proline residues (additionally for class B, conserved glycine residues), moving away (6–14 Å) from the TM bundle and shaping the G-protein-binding site. Also TM5, TM2 and TM7 slightly rearrange, in order to accommodate the intracellular effector, for example, the C-terminal helix of a G protein.
The most striking structural change is the outward movement of TM6 (about 6–14 Å) from the TM bundle that allows the intracellular effector to bind. Further conformational switches (ribbon representations) involve the ‘ionic lock’ between R3.50 and D/E6.30, the NPxxY (N7.49, P7.50 and Y7.53) motif, and the highly conserved W6.48. The residue numbering follows the Ballesteros–Weinstein scheme [Citation92].
ECL: extracellular loop; TM: Transmembrane.
![Figure 4. Structural hallmarks of a class A G-protein-coupled receptor activation mechanism.The most striking structural change is the outward movement of TM6 (about 6–14 Å) from the TM bundle that allows the intracellular effector to bind. Further conformational switches (ribbon representations) involve the ‘ionic lock’ between R3.50 and D/E6.30, the NPxxY (N7.49, P7.50 and Y7.53) motif, and the highly conserved W6.48. The residue numbering follows the Ballesteros–Weinstein scheme [Citation92].ECL: extracellular loop; TM: Transmembrane.](/cms/asset/a459307d-e28c-4650-91ba-a51e2affa372/ifmc_a_12361681_f0004.jpg)
The first direct computational insights into the β2-AR dynamic activation mechanism were proposed in the work by Dror et al. [Citation67], following the first x-ray crystal structure of an active (G-protein or G-protein mimic-bound) GPCR [Citation68,Citation69]. Starting from the agonist-bound active state, with the intracellular G-protein or G-protein mimetic nanobody removed, the authors performed extensive simulations observing several spontaneous transitions toward the experimentally determined inactive conformation. The receptor experienced numerous intermediate conformations along the deactivation pathway, but without a strong coupling mechanism between the orthosteric and G-protein-binding sites, giving a putative rationale for the experimental difficulties in fully stabilizing the β2-AR in the presence of an agonist [Citation70]. In contrast, other MD studies have shown an allosteric communication between the orthosteric site and a conserved polar network of the μOR (opioid receptor μ) [Citation71,Citation72].
The GPCRs’ conformational heterogeneity, observed upon agonist binding, may be the basis of the different coupling possibilities with intracellular counterparts (i.e., stimulatory G protein – Gs, inhibitory G protein – Gi, and arrestin) [Citation73]. Recently, Kohlhoff et al. [Citation74] performed milliseconds timescale MD simulations of the β2-AR on the Google’s Exacycle cloud, obtaining a detailed description of the overall receptor activation mechanism. Even though different pathways were observed, the model that the author proposed confirmed the displacement of TM6 away from TM3, either before or concurrently with conformational changes at the NPxxY motif () and the connector region (a TM cluster of residues located between the orthosteric site and the G-protein-binding sites).
Li et al. [Citation75] proposed that the direct contacts between the A2A AR and the agonist could induce a side-chain rearrangement in residues located at the bottom of the orthosteric site, leading to a loss of hydrophobic packing in the core of the receptor. This, in turn, allows TM6 to move away from TM3 due to the break of the ‘ionic lock’ (). Further molecular switches of the A2A AR have been reported in the work by Lee et al. [Citation76].
In the M2-R, enhanced sampling MD simulations of the activation mechanism highlighted the concomitant formation of polar interactions between TM5 and TM7, and the outward movement of the intracellular portion of TM6 [Citation77], while TM3 was confirmed as fundamental for the functionality of the receptor [Citation63]. Very recently, a study on the M2-R deactivation (simulated by replacing an agonist with an inverse agonist in the active conformation of the receptor [Citation78] deprived of the G-protein mimetic nanobody [Citation79]), suggested that the opening of the extracellular side of the receptor may occur before the intracellular relaxation of TM6.
About 15 years ago, the concept of biased signaling was first introduced in the GPCR field [Citation80]. According to this novel view, an agonist-bound GPCR can preferentially couple to either one or more of a number of G-proteins or a β-arrestin, generating a particular intracellular signal that can be triggered by any of the three components of the ternary complex (e.g., the agonist, the receptor or the transducers) [Citation81,Citation82]. Even though the structural determinants of the biased signaling are still poorly understood, it is well accepted that small variations in the conformational energy landscape of the receptor lead to different intracellular signaling pathways, in other words, different GPCR active conformations preferentially bind to particular effectors [Citation83,Citation84].
Understanding agonist bias offers the opportunity to develop drugs with an enhanced therapeutic profile, and reduced on-target side effects [Citation84]. Recently, ligand RT and biased agonism have been described as inter-related in the ‘kinetic contest’ [Citation85], indicating that the overall binding kinetics can modulate the nature of the receptor signaling [Citation86]. In theory, interactions at any site of the receptor surface that recognize a ligand can shift the conformational equilibrium toward a specific conformation, therefore triggering a particular physiological effect [Citation82]. From this standpoint, MD may represent a useful tool for the investigations of the GPCR conformational landscape, not only in their canonical orthosteric-bound state but also when potential metastable sites are engaged. Schneider et al. [Citation87] investigated the association mechanism and the orthosteric behavior of the μOR agonist oliceridine (OLINVO™, TRV-130), the first biased opioid to enter Phase III clinical trials [Citation88]. Simulations highlighted marked differences in the structural communication, determined by complex networks of both polar and nonpolar intramolecular interactions between the extracellular side and the intracellular portion of the receptor, depending on whether TRV-130 or the reference unbiased agonist morphine was bound to the orthosteric pocket (mainly at the level of TM3 and TM6). The significance of this resides in the reduced side effects reported for oliceridine in Phase III clinical trials for moderate-to-severe acute pain as a result of reduced β-arrestin signaling (www.trevena.com).
Very recently, the dynamic recognition process between a GPCR and an intracellular binding partner was reconstructed by means of MD. Miao et al. [Citation89] simulated the intracellular association between the M2-R and a G-protein mimetic nanobody. In concordance with mutagenesis experiments, the authors proposed that ICLs play a fundamental role by stabilizing the complex by establishing both hydrophobic and hydrophilic contacts. Moreover, the cytoplasmic end of TM6 favors the association by becoming transiently more disordered as the nanobody approached the receptor. At the extracellular side of the system, the binding pocket evolved from an open conformation (observed in the inactive state) to a closed one (), in concordance with the allosteric coupling proposed also for the β2-AR [Citation43]. On the other hand, the arrestin activation mechanism was extensively studied by means of simulations in two parallel works by Eichel et al. [Citation90] and Latorraca et al. [Citation91]. Latorraca et al., in particular, rationalized an astonishing amount of previous experimental data. They highlighted three independent sets of GPCR–arrestin interactions that could trigger effector activation, all be it with different affinities and propensities. These are: interactions mediated by both the receptor core and the phosphorylated cytoplasmic tail, interactions involving the phosphorylated cytoplasmic tail only and interactions involving the receptor core only. Importantly, the capability of the receptor core to induce arrestin stimulation was linked mainly to intracellular loop 2 (ICL2) and ICL3. From this standpoint, compared with the relatively low TM domain flexibility, the higher conformational space explored by ICLs can putatively provide a set of interactions able to engage the effector in a wider number of subtly different conformations, leading to the fine-tuning characteristic of GPCR signaling.
How does molecular dynamics address GPCRs allosterism?
While the GPCR transduction of chemical signals from the extracellular medium to the intracellular one can be considered unidirectional (i.e., external stimuli trigger an intracellular response), the structural mechanism through which this is realized is finely tuned in a bidirectional way. Indeed, the agonist and the intracellular effector reciprocally enhance their stability (and therefore affinity) for the receptor when bound to it [Citation93]. Moreover, as integral membrane proteins, GPCRs communicate with the aqueous and the lipid environments, both contributing to the overall regulation and hetero/homodimerization in a number of different ways through the binding of lipid, cations and metabolites [Citation93–95]. One of the most characterized GPCR endogenous allosteric mechanisms is exerted by sodium binding to a specific site, located in the core of the class A TM bundle below the orthosteric site, where the ion coordinates the side chains of conserved residues, including the Asp of the TM2 LXXXD motif [Citation96], and exerts a negative allosteric effect by stabilizing inactive conformations [Citation97]. Following the first structure resolved with the sodium-bound (the A2A AR [Citation97]), MD simulations were extensively employed with the aim of uncovering the mechanism underlying the sodium allosteric effect [Citation98–104]. Both the active and inactive conformations of the A2A AR were simulated in the presence and absence of bound cation [Citation101]; the outcomes showed that the receptor active state is not able to stabilize the sodium, and that the cation’s first and second sphere of solvation heavily contribute to the overall structural effect by stabilizing the inactive conformation of the receptor. In a very recent work, Selvam et al. [Citation105] computed the free energy profiles of sodium association to a set of 18 GPCRs. By means of extensive MD simulations, they revealed beside a conserved mechanism of binding, the possibility for the ion to access the allosteric site also from the intracellular vestibule of the receptors, at least for the purinoceptor P2Y12 and PAR1. Interestingly, during simulations on the class B GPCR glucagon receptor the sodium visited a putative binding site. However, the short RT observed is in accordance with the lack of experimental evidence (and structural motif) linking this class of GPCRs and ion-mediated allosteric effects. Indeed, while class B GPCRs share a number of structural motifs with class A [Citation106], they lack the LXXXD motif.
Besides the sodium, it is well known that bivalent cations are able to modulate the binding of GPCRs. A positive allosteric effect has been observed for the ORs (opioid receptors) [Citation107], the M2-R [Citation108] and the A2A AR [Citation109,Citation110]. This latter was investigated in depth by Ye et al. [Citation111], employing MD simulations to rationalize experimental data. Several putative binding sites for cations (Na+, K+, Ca2+, Mg2+ and Zn2+) were located by considering acidic residues at the extracellular vestibule as well as residues involved in the switch from the active to the inactive state (like the class A GPCR ‘ionic lock’ – ).
Increasing attention has been focused on the development of GPCR allosteric modulators as potential therapeutics [Citation112]. The reasons for this lie in the possibility of enhancing the response of an orthosteric drug or the natural ligand by selectively targeting the usually less conserved allosteric sites, and of reducing on-target side effects, by altering the signal bias [Citation95,Citation113]. Understanding the structural basis of allosteric modulation may enable the rational design of new agents. From this standpoint, the work by Dror et al. [Citation114] represents a milestone. The authors reconstructed the putative binding of both positive allosteric modulators (PAMs) and negative allosteric modulators of the M2-R, showing that despite the structural diversity, the ligands were able to form similar interactions at the same site on the extracellular vestibule of the receptor. The occupation of this spot affected the shape of the orthosteric site and vice versa, thus explaining the cooperative behavior observed experimentally.
Allosteric sites were proposed by means of MD studies in the proximity of other GPCR orthosteric sites, as in the δOR (the PAM BMS-986187 putatively binds in a spot delimited by TM2, TM6 and TM7 [Citation115]), or overlapping the canonical orthosteric one. According to this latter hypothesis, some PAMs could reach the orthosteric site and interact with the bound agonist further stabilizing it, as proposed for the A3 AR (adenosine receptor A3) [Citation116] and, more recently, for the CB1R (cannabinoid receptor 1) [Citation117].
MD simulations represent a powerful tool for investigating allosteric communications and how structural information propagates from allosteric sites to the interfaces with intracellular GPCR effectors [Citation118]. From this standpoint, some recent approaches focused on correlated residue motions [Citation119,Citation120] and mutual information metrics [Citation121]. In the GPCR field, Bhattacharya et al. mapped the allosteric communication pipelines responsible for the transmission of the activation signal from the orthosteric site to the intracellular interface of the β2-AR [Citation122]. Interestingly, results showed a higher dynamicity of the agonist-bound intermediate state of the receptor compared with the inactive or fully active one (the agonist-bound intermediate state is a conformation that, despite a TM structural organization resembling the inactive state, is characterized by side-chain conformational rearrangements observable in the fully active conformation). Moreover, upon binding of an agonist, the extracellular side of the receptor experienced a decoupling from the intracellular one. A further computational approach, able to detect protein pockets and the communication between adjacent pockets during MD simulations, has been proposed by La Sala et al. [Citation123] and applied to the A2A AR. Their findings showed dynamic communication, between the orthosteric site and the sodium allosteric binding site, ascribable to residues located at the interface between the two cavities, able to exchange from one site to the other one.
Solvation patterns in GPCRs
The presence of conserved hydrated patterns along the GPCR TM domain should suggest a major role for water molecules in promoting conformational transitions. Active and inactive states are characterized by different spatial organizations of residues located in the core of the receptors and, in turns, by different stable networks of water-mediated interactions [Citation124]. In this scenario, the binding of the orthosteric ligand and the intracellular effector may affect the water molecules accessibility to the GPCR core, besides triggering direct conformational changes responsible for activation/deactivation. It has been proposed that water molecules, flowing in the GPCR TM core from both membrane sides, contribute to activation of the κOR (κ opioid receptor) [Citation125], the A2A AR [Citation126,Citation127], the β2-AR [Citation126] and Rho (rhodopsin) [Citation126]. Indeed, changes in the rotameric state of conserved amino acids upon activation putatively allow the formation of a hydrated channel that spans and stabilizes the receptors.
In drug design, the role played by water molecules in the whole ligand-receptor recognition event is increasingly considered. Different computational tools based on MD simulations have recently been developed with the aim of characterizing hydrated spots in druggable protein-binding sites [Citation128–133]. WaterMap [Citation128] was employed by Higgs et al. [Citation134] for estimating the free energy profile of water molecules in the unoccupied orthosteric site of the A2A AR. This allowed the authors to infer a link between the structure–activity relationships (SARs) of the triazolylpurine antagonists and the extent to which the antagonists overlap several hydrated spots that were energetically classified as unfavorable. Among these, two sites occupied by ligand atoms were directly involved in ligand–protein hydrogen bonds. Effects on the solvation of the A2A AR binding site, produced upon binding, were investigated also for a set of triazine inhibitors [Citation135]. In this study, Bortolato et al. proposed that the RT could be increased by displacing unstable water molecules that are trapped in the first hydration shell of the bound ligand. The same triazine – A2A AR complexes were also employed for the validation of AquaMMapS [Citation132], a recent method developed to locate hydration spots in protein cavities by monitoring the solvent kinetic state [Citation132]. The AquaMMapS protocol detects protein sites more prone to be occupied by water molecules with low root-mean-square fluctuation, distinguishing volumes that can be favorably occupied by a ligand’s hydrophilic moieties from the volumes where water molecules can be favorably displaced by hydrophobic substituents.
Besides the hydration patterns, the membrane environment also has an active role in the dynamic behavior of GPCRs [Citation136]. Indeed, the protein-membrane interface can regulate the binding kinetics of lipophilic orthosteric ligands [Citation49] as well as the overall receptor activation and signaling [Citation137]. The effect that membrane lipids exert on the prototypic GPCR Rho has been extensively investigated [Citation138]. The polyunsaturated docosahexaenoic acid is abundant in the photoreceptor cells and influences the Rho functionality by specifically bind to localized sites on the protein surface [Citation139] that are more likely present when the Rho is in an inactive-like conformation [Citation140]. Cholesterol was probably the first membrane component to be computationally addressed by means of MD simulations. On Rho, its action mechanism is still unclear. It has been proposed either to regulate the membrane order and thickness [Citation139] or to specifically interact with Rho TM helices [Citation141]. For the β2-AR, seven different cholesterol-binding sites have been reported on the receptor surface, spread on both the intracellular and the extracellular leaflets [Citation142]. One spot in particular, which is located at the extracellular ends of TM1, TM2 and TM7 has been proposed to be involved in positive allosteric regulation of ligand binding. Moreover a distinct site, formed by TM1 and helix 8 (H8), putatively facilitates receptor dimerization, a phenomenon underlying receptor maturation and regulation [Citation143] that is likely to involve TM4 in several class A GPCRs [Citation144]. Cholesterol-binding sites were also located on the 5HT2A R (5-hydroxytryptamine receptor 2A) [Citation145] and the A2A AR [Citation146], where the lipid showed an increased simulated RT corresponding to three distinct spots without adopting a well-defined binding mode. The effects produced on the β2-AR activation mechanism by protein–lipid interactions was investigated by Neale et al. [Citation147] and, more recently, by Bruzzese et al. [Citation148]. Both these studies proposed that the phospholipids putatively regulate the activation or deactivation mechanism of the receptor, influencing the coupling to intracellular effectors. During MD simulations, negatively charged lipids were able to stabilize an active state of β2-AR, while neutral (zwitterionic) lipids generally favored the inactivation of the receptor, with an overall kinetics that was affected by the lipid headgroup charge distribution [Citation148]. A detailed study of the positive effects exerted by the negatively charged phosphatidylinositol-4,5-bisphosphate on Gs activation by the A2A AR, β1-AR and the neurotensin receptor 1 has been reported recently [Citation149].
Class B CPCRs
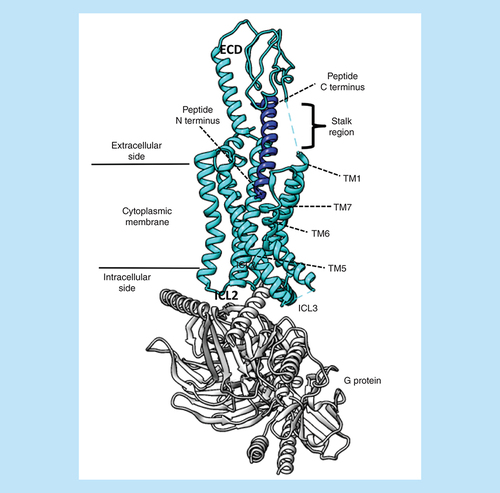
The small class B GPCR subfamily is composed of 15 receptors for endogenous peptides, structurally characterized by a large N-terminal extracellular domain (ECD – formed by up to 200 amino acids) connected to TM1 through a stalk region (). The pharmacological profile of class B GPCRs is strongly influenced by the capability, for some of them, to heterodimerize with receptor activity-modifying proteins (RAMPs) [Citation150], which are able to modulate the selectivity as well as the intracellular signaling profile. The peptide binding is proposed to take place through a two-step mechanism [Citation151]: the ligand C-terminus first binds the ECD, favoring the reorientation and the subsequent association of the peptide N terminus into the receptor TM bundle (). Since the early 2000s, several published structures of isolated ECDs (first), then isolated TMs, and more recently full-length receptors, have shed light on the different steps of this process [Citation64,Citation152], giving a new impulse to drug design efforts [Citation153,Citation154]. In this scenario, MD has been a helpful tool for rationalizing the wide amount of experimental data produced. The ECD of the apo GCGR (glucagon receptor) was observed in two preferred different orientations during simulations performed by Yang et al. [Citation155]. According to their findings, the endogenous peptide stabilizes the open form of the receptor, therefore acting through a conformational selection mechanism. Simulations of the CGRPR (calcitonin-gene-related peptide receptor) bound to adrenomedullin [Citation156] highlighted the possibility that conformational changes occurring at the ECLs upon agonist binding could be transmitted to the extracellular ends of TM5 and TM6, triggering, in turn, a reorganization of both hydrophobic and hydrophilic networks between TM3, TM5 and TM6. From this standpoint, RAMPs probably modulate the extracellular side of the TM bundle in a ligand-specific way. In the work from Bower et al. [Citation157], the first step of amylin binding to the ECD of the CTR (calcitonin receptor), both alone and in complex with RAMP1, was simulated by means of SuMD simulations. The authors proposed that the allosteric regulation exerted by the RAMP1 comprises modification in the electrostatics of the receptor and in the binding path of the peptide C-terminus.
The figure shows the GLP-1R in an active G-protein-bound state (Protein Data Bank code 6B3J). The N terminus of the agonist peptide binds in the TM bundle, while the C terminus interacts with the receptor ECD. The flexible stalk region and the ICL3 were not resolved in the cryo-electro microscopy map.
ECD: Extracellular domain; ICL: Intracellular loop; TM: Transmembrane.
Considering the glucagon-like peptide-1 receptor (GLP-1R), simulation aimed at characterizing the role of the conserved polar residues located in the proximity of the TMs [Citation158] and at rationalizing the peptide-mediated signaling of the GLP-1R, proposing a key role for the polar network located in the TM core [Citation159]. The biased signaling of GLP-1R was later investigated in depth in the work by Wootten et al. [Citation160]. MD simulations of salmon calcitonin (sCT) and hCT (human calcitonin) pointed out the ability of the former to better interact and stabilize the receptor ECL2, explaining the higher affinity of salmon calcitonin [Citation161]. Weaver et al. [Citation162] combined mutagenesis data and MD simulation on homology models to address the molecular basis of selectivity between the parathyroid hormone and tuberoinfundibular peptide 39 peptides binding to parathyroid hormone 2 receptor (PTH2R); an active truncated peptide was used due to uncertainties in the binding mode of the peptide N-terminus.
Conclusion
Since the first GPCR crystal structures were published in the early 2000s, several cutting-edge methodologies have overcome the intrinsic difficulties that characterize membrane protein crystallography. Moreover, thanks to recent advances in cryo-EM, the protein structure can be determined in a more native environment. This is leading to an outbreak of GPCR structural knowledge and, in turn, to a broad application of MD-related methods to the study of this membrane protein superfamily. MD simulations can be considered as the computational method of election for the study of nonequilibrium phenomena, such as drug-receptor (un)binding events, the GPCR activation, and the allosteric structural communications. Moreover, MD simulations allow modeling the protein–solvent interfaces at atomic scale, enabling the study of how water and lipid molecules can influence the GPCRs.
Future perspective
Considering the continuous improvement in the informatics field, and the increasing computational capability, it is plausible that in the next few years MD will be increasingly used to translate the massive amount of static information into the dynamic language of biological systems. Thus, allowing a better description of several biological aspects that are still far from clear. In particular, the structural determinants for both biased signaling and allosterism may represent a more challenging topic in the GPCR field, and therefore the main targets of future computational efforts.
Recently, both experimental structural determinations and molecular dynamics (MD)-related methods are providing astonishing advances in the G-protein-coupled receptor (GPCR) field. MD allows the time-dependent (dynamic) description of biological systems at an atomistic scale.
The first step in the GPCRs signaling is the extracellular ligand recognition: MD is a valuable tool for investigating this process from the non equilibrium point of view.
MD simulations are shedding light on the GPCR activation mechanism, taking advantage of recent insights from x-ray crystallography and cryo-electron microscopy techniques.
Biased agonism, which can potentially lead to groundbreaking improvement in the design of new drugs, is still a poorly understood phenomena; MD simulations may contribute to an explanation of this aspect from a structural point of view.
To date, different allosteric mechanisms have been suggested by means of MD simulations.
MD investigations of class B GPCRs are gaining importance since the first experimental full-length active structures have been recently published.
Financial & competing interests disclosure
CAR is a Royal Society Industry Fellow and acknowledges support from the BBSRC (BB/M006883/1). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Fredriksson R Lagerström MC Lundin L-G Schiöth HB . The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints . Mol. Pharmacol.63 ( 6 ), 1256 – 1272 ( 2003 ).
- Kiselyov K Shin DM Muallem S . Signalling specificity in GPCR-dependent Ca2+ signalling . Cell Signal.15 ( 3 ), 243 – 253 ( 2003 ).
- Weis WI Kobilka BK . The molecular basis of G protein-coupled receptor activation . Annu. Rev. Biochem.87 , 897 – 919 ( 2018 ).
- Hilger D Masureel M Kobilka BK . Structure and dynamics of GPCR signaling complexes . Nat. Struct. Mol. Biol.25 ( 1 ), 4 – 12 ( 2018 ).
- Tesmer JJG . Hitchhiking on the heptahelical highway: structure and function of 7TM receptor complexes . Nat. Rev. Mol. Cell Biol.17 ( 7 ), 439 – 450 ( 2016 ).
- Katritch V Cherezov V Stevens RC . Structure–function of the G protein-coupled receptor superfamily . Annu. Rev. Pharmacol. Toxicol.53 , 531 – 556 ( 2013 ).
- Kobilka BK . G protein coupled receptor structure and activation . Biochim. Biophys. Acta1768 ( 4 ), 794 – 807 ( 2007 ).
- Hauser AS Attwood MM Rask-Andersen M Schiöth HB Gloriam DE . Trends in GPCR drug discovery: new agents, targets and indications . Nat. Rev. Drug Discov.16 ( 12 ), 829 – 842 ( 2017 ).
- Pándy-Szekeres G Munk C Tsonkov TM et al. GPCRdb in 2018: adding GPCR structure models and ligands . Nucleic Acids Res.46 ( D1 ), D440 – D446 ( 2018 ).
- Martínez-Rosell G Giorgino T Harvey MJ de Fabritiis G . Drug discovery and molecular dynamics: methods, applications and perspective beyond the second timescale . Curr. Top. Med. Chem.17 ( 23 ), 2617 – 2625 ( 2017 ).
- McCammon JA Gelin BR Karplus M . Dynamics of folded proteins . Nature267 ( 5612 ), 585 – 590 ( 1977 ).
- Shaw DE . Millisecond-long molecular dynamics simulations of proteins on a special-purpose machine . Biophys. J.104 ( 2 ), 45a ( 2013 ).
- Deupi X Kobilka BK . Energy landscapes as a tool to integrate GPCR structure, dynamics, and function . Physiology (Bethesda)25 ( 5 ), 293 – 303 ( 2010 ).
- Doerr S De Fabritiis G . On-the-fly learning and sampling of ligand binding by high-throughput molecular simulations . J. Chem. Theory Comput.10 ( 5 ), 2064 – 2069 ( 2014 ).
- Bernardi RC Melo MCR Schulten K . Enhanced sampling techniques in molecular dynamics simulations of biological systems . Biochim. Biophys. Acta1850 ( 5 ), 872 – 877 ( 2015 ).
- De Vivo M Masetti M Bottegoni G Cavalli A . Role of molecular dynamics and related methods in drug discovery . J. Med. Chem.59 ( 9 ), 4035 – 4061 ( 2016 ).
- Deganutti G Moro S . Estimation of kinetic and thermodynamic ligand-binding parameters using computational strategies . Future Med. Chem.9 ( 5 ), 507 – 523 ( 2017 ).
- Tummino PJ Copeland RA . Residence time of receptor-ligand complexes and its effect on biological function . Biochemistry47 ( 20 ), 5481 – 5492 ( 2008 ).
- Copeland RA Pompliano DL Meek TD . Drug-target residence time and its implications for lead optimization . Nat. Rev. Drug Discov.5 ( 9 ), 730 – 739 ( 2006 ).
- Dahl G Akerud T . Pharmacokinetics and the drug-target residence time concept . Drug Discov. Today18 ( 15–16 ), 697 – 707 ( 2013 ).
- Copeland RA . The drug-target residence time model: a 10-year retrospective . Nat. Rev. Drug Discov.15 ( 2 ), 87 – 95 ( 2016 ).
- Casarosa P Bouyssou T Germeyer S Schnapp A Gantner F Pieper M . Preclinical evaluation of long-acting muscarinic antagonists: comparison of tiotropium and investigational drugs . J. Pharmacol. Exp. Ther.330 ( 2 ), 660 – 668 ( 2009 ).
- Tautermann CS Kiechle T Seeliger D et al. Molecular basis for the long duration of action and kinetic selectivity of tiotropium for the muscarinic M3 receptor . J. Med. Chem.56 ( 21 ), 8746 – 8756 ( 2013 ).
- Kapur S Seeman P . Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action . J. Psychiatr. Neurosci.25 ( 2 ), 161 – 166 ( 2000 ).
- Tarsy D Baldessarini RJ . Epidemiology of tardive dyskinesia: is risk declining with modern antipsychotics?Mov. Disord.21 ( 5 ), 589 – 598 ( 2006 ).
- Soethoudt M Hoorens MWH Doelman W Martella A van der Stelt M Heitman LH . Structure–kinetic relationship studies of cannabinoid CB2 receptor agonists reveal substituent-specific lipophilic effects on residence time . Biochem. Pharmacol.152 , 129 – 142 ( 2018 ).
- Schuetz DA de Witte WEA Wong YC et al. Kinetics for drug discovery: an industry-driven effort to target drug residence time . Drug Discov. Today22 ( 6 ), 896 – 911 ( 2017 ).
- Guo D Heitman LH IJzerman AP . Kinetic aspects of the interaction between ligand and G protein-coupled receptor: the case of the adenosine receptors . Chem. Rev.117 ( 1 ), 38 – 66 ( 2017 ).
- Swinney DC Haubrich BA Van Liefde I Vauquelin G . The role of binding kinetics in GPCR drug discovery . Curr. Top. Med. Chem.15 ( 24 ), 2504 – 2522 ( 2015 ).
- Strasser A Wittmann H-J Seifert R . Binding kinetics and pathways of ligands to GPCRs . Trends Pharmacol. Sci.38 ( 8 ), 717 – 732 ( 2017 ).
- Dror RO Pan AC Arlow DH et al. Pathway and mechanism of drug binding to G-protein-coupled receptors . Proc. Natl Acad. Sci. USA108 ( 32 ), 13118 – 13123 ( 2011 ).
- González A Perez-Acle T Pardo L Deupi X . Molecular basis of ligand dissociation in β-adrenergic receptors . PLoS ONE6 ( 9 ), e23815 ( 2011 ).
- Kruse AC Hu J Pan AC et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor . Nature482 ( 7386 ), 552 – 556 ( 2012 ).
- Radić Z Kirchhoff PD Quinn DM McCammon JA Taylor P . Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin . J. Biol. Chem.272 ( 37 ), 23265 – 23277 ( 1997 ).
- Carpenter B Lebon G . Human adenosine a2a receptor: molecular mechanism of ligand binding and activation . Front. Pharmacol.8 , 898 ( 2017 ).
- Guo D Pan AC Dror RO et al. Molecular basis of ligand dissociation from the adenosine A2A receptor . Mol. Pharmacol.89 ( 5 ), 485 – 491 ( 2016 ).
- Segala E Guo D Cheng RKY et al. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength . J. Med. Chem.59 ( 13 ), 6470 – 6479 ( 2016 ).
- Sabbadin D Ciancetta A Deganutti G Cuzzolin A Moro S . Exploring the recognition pathway at the human A2A adenosine receptor of the endogenous agonist adenosine using supervised molecular dynamics simulations . Medchemcomm6 ( 6 ), 1081 – 1085 ( 2015 ).
- Deganutti G Welihinda A Moro S . Comparison of the human A2A adenosine receptor recognition by adenosine and inosine: new insight from supervised molecular dynamics simulations . ChemMedChem.12 ( 16 ), 1319 – 1326 ( 2017 ).
- Sabbadin D Moro S . Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale . J. Chem. Inf. Model.54 ( 2 ), 372 – 376 ( 2014 ).
- Cuzzolin A Sturlese M Deganutti G et al. Deciphering the complexity of ligand–protein recognition pathways using supervised molecular dynamics (SuMD) simulations . J. Chem. Inf. Model.56 ( 4 ), 687 – 705 ( 2016 ).
- Christopoulos A Kenakin T . G protein-coupled receptor allosterism and complexing . Pharmacol. Rev.54 ( 2 ), 323 – 374 ( 2002 ).
- DeVree BT Mahoney JP Vélez-Ruiz GA et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs . Nature535 ( 7610 ), 182 – 186 ( 2016 ).
- Deganutti G Salmaso V Moro S . Could adenosine recognize its receptors with a stoichiometry other than 1:1?Mol. Inform.37 ( 8 ), e1800009 ( 2018 ).
- Igonet S Raingeval C Cecon E et al. Enabling STD-NMR fragment screening using stabilized native GPCR: a case study of adenosine receptor . Sci. Rep.8 ( 1 ), 8142 ( 2018 ).
- Deganutti G Zhukov A Deflorian F et al. Impact of protein–ligand solvation and desolvation on transition state thermodynamic properties of adenosine A2A ligand binding kinetics . In Silico Pharmacol.5 ( 1 ), 16 ( 2016 ).
- Yuan X Raniolo S Limongelli V Xu Y . The molecular mechanism underlying ligand binding to the membrane-embedded site of a G-protein-coupled receptor . J. Chem. Theory Comput.14 ( 5 ), 2761 – 2770 ( 2018 ).
- Hurst DP Grossfield A Lynch DL et al. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor . J. Biol. Chem.285 ( 23 ), 17954 – 17964 ( 2010 ).
- Stanley N Pardo L Fabritiis GD . The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor . Sci. Rep.6 , 22639 ( 2016 ).
- Bokoch MP Jo HI Valcourt JR et al. Entry from the lipid bilayer: a novel pathway for inhibition of a peptide G-protein coupled receptor by a lipophilic small molecule . Biophys. J.108 ( 2 ), 350a ( 2015 ).
- Bokoch MP Jo H Valcourt JR et al. Entry from the lipid bilayer: a possible pathway for inhibition of a peptide G protein-coupled receptor by a lipophilic small molecule . Biochemistry57 ( 39 ), 5748 – 5758 ( 2018 ).
- Bruce NJ Ganotra GK Kokh DB Sadiq SK Wade RC . New approaches for computing ligand-receptor binding kinetics . Curr. Opin. Struct. Biol.49 , 1 – 10 ( 2018 ).
- Pande VS Beauchamp K Bowman GR . Everything you wanted to know about Markov state models but were afraid to ask . Methods52 ( 1 ), 99 – 105 ( 2010 ).
- Plattner N Doerr S De Fabritiis G Noé F . Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling . Nat. Chem.9 ( 10 ), 1005 – 1011 ( 2017 ).
- Paul F Wehmeyer C Abualrous ET et al. Protein-peptide association kinetics beyond the seconds timescale from atomistic simulations . Nat. Commun.8 ( 1 ), 1095 ( 2017 ).
- Bortolato A Deflorian F Weiss DR Mason JS . Decoding the role of water dynamics in ligand–protein unbinding: CRF1R as a test case . J. Chem. Inf. Model.55 ( 9 ), 1857 – 1866 ( 2015 ).
- Doré AS Bortolato A Hollenstein K Cheng RKY Read RJ Marshall FH . Decoding corticotropin-releasing factor receptor type 1 crystal structures . Curr. Mol. Pharmacol.10 ( 4 ), 334 – 344 ( 2017 ).
- Rosenbaum DM Rasmussen SGF Kobilka BK . The structure and function of G-protein-coupled receptors . Nature459 ( 7245 ), 356 – 363 ( 2009 ).
- Pierce KL Premont RT Lefkowitz RJ . Seven-transmembrane receptors . Nat. Rev. Mol. Cell Biol.3 ( 9 ), 639 – 650 ( 2002 ).
- De Lean A Stadel JM Lefkowitz RJ . A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled beta-adrenergic receptor . J. Biol. Chem.255 ( 15 ), 7108 – 7117 ( 1980 ).
- Kobilka BK Deupi X . Conformational complexity of G-protein-coupled receptors . Trends Pharmacol. Sci.28 ( 8 ), 397 – 406 ( 2007 ).
- Latorraca NR Venkatakrishnan AJ Dror RO . GPCR dynamics: structures in motion . Chem. Rev.117 ( 1 ), 139 – 155 ( 2017 ).
- Venkatakrishnan AJ Deupi X Lebon G Tate CG Schertler GF Babu MM . Molecular signatures of G-protein-coupled receptors . Nature494 ( 7436 ), 185 – 194 ( 2013 ).
- de Graaf C Song G Cao C et al. Extending the structural view of class B GPCRs . Trends Biochem. Sci.42 ( 12 ), 946 – 960 ( 2017 ).
- Venkatakrishnan AJ Deupi X Lebon G et al. Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region . Nature536 ( 7617 ), 484 – 487 ( 2016 ).
- Isberg V Mordalski S Munk C et al. GPCRdb: an information system for G protein-coupled receptors . Nucleic Acids Res.44 ( D1 ), D356 – D364 ( 2016 ).
- Dror RO Arlow DH Maragakis P et al. Activation mechanism of the β2-adrenergic receptor . Proc. Natl. Acad. Sci. USA108 ( 46 ), 18684 – 18689 ( 2011 ).
- Rasmussen SGF DeVree BT Zou Y et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex . Nature477 ( 7366 ), 549 – 555 ( 2011 ).
- Rasmussen SGF Choi H-J Fung JJ et al. Structure of a nanobody-stabilized active state of the β(2) adrenoceptor . Nature469 ( 7329 ), 175 – 180 ( 2011 ).
- Ghanouni P Gryczynski Z Steenhuis JJ et al. Functionally different agonists induce distinct conformations in the G protein coupling domain of the beta 2 adrenergic receptor . J. Biol. Chem.276 ( 27 ), 24433 – 24436 ( 2001 ).
- Huang W Manglik A Venkatakrishnan AJ et al. Structural insights into μ-opioid receptor activation . Nature524 ( 7565 ), 315 – 321 ( 2015 ).
- Sena DM Cong X Giorgetti A Kless A Carloni P . Structural heterogeneity of the μ-opioid receptor's conformational ensemble in the apo state . Sci. Rep.8 , 45761 ( 2017 ).
- Nygaard R Zou Y Dror RO et al. The dynamic process of β(2)-adrenergic receptor activation . Cell152 ( 3 ), 532 – 542 ( 2013 ).
- Kohlhoff KJ Shukla D Lawrenz M et al. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways . Nat. Chem.6 ( 1 ), 15 – 21 ( 2014 ).
- Li J Jonsson AL Beuming T Shelley JC Voth GA . Ligand-dependent activation and deactivation of the human adenosine A(2A) receptor . J. Am. Chem. Soc.135 ( 23 ), 8749 – 8759 ( 2013 ).
- Lee Y Choi S Hyeon C . Communication over the network of binary switches regulates the activation of A2A adenosine receptor . PLoS Comput. Biol.11 ( 2 ), e1004044 ( 2015 ).
- Miao Y Nichols SE Gasper PM Metzger VT McCammon JA . Activation and dynamic network of the M2 muscarinic receptor . Proc. Natl Acad. Sci. USA110 ( 27 ), 10982 – 10987 ( 2013 ).
- Jiménez-Rosés M Matsoukas M-T Caltabiano G Cordomí A . Ligand-triggered structural changes in the M2 muscarinic acetylcholine receptor . J. Chem. Inf. Model.58 ( 5 ), 1074 – 1082 ( 2018 ).
- Kruse AC Ring AM Manglik A et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor . Nature504 ( 7478 ), 101 – 106 ( 2013 ).
- Pupo AS Duarte DA Lima V Teixeira LB Parreiras-E-Silva LT Costa-Neto CM . Recent updates on GPCR biased agonism . Pharmacol. Res.112 , 49 – 57 ( 2016 ).
- Shukla AK Singh G Ghosh E . Emerging structural insights into biased GPCR signaling . Trends Biochem. Sci.39 ( 12 ), 594 – 602 ( 2014 ).
- Smith JS Lefkowitz RJ Rajagopal S . Biased signalling: from simple switches to allosteric microprocessors . Nat. Rev. Drug Discov.17 ( 4 ), 243 – 260 ( 2018 ).
- Liang Y-L Khoshouei M Glukhova A et al. Phase-plate cryo-EM structure of a biased agonist-bound human GLP-1 receptor-Gs complex . Nature555 ( 7694 ), 121 – 125 ( 2018 ).
- Violin JD Crombie AL Soergel DG Lark MW . Biased ligands at G-protein-coupled receptors: promise and progress . Trends Pharmacol. Sci.35 ( 7 ), 308 – 316 ( 2014 ).
- Klein Herenbrink C Sykes DA Donthamsetti P et al. The role of kinetic context in apparent biased agonism at GPCRs . Nat. Commun.7 , 10842 ( 2016 ).
- Grundmann M Kostenis E . Temporal bias: time-encoded dynamic GPCR signaling . Trends Pharmacol. Sci.38 ( 12 ), 1110 – 1124 ( 2017 ).
- Schneider S Provasi D Filizola M . How oliceridine (TRV-130) binds and stabilizes a μ-opioid receptor conformational state that selectively triggers G protein signaling pathways . Biochemistry55 ( 46 ), 6456 – 6466 ( 2016 ).
- Singla N Minkowitz HS Soergel DG et al. A randomized, Phase IIb study investigating oliceridine (TRV130), a novel μ-receptor G-protein pathway selective (μ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty . J. Pain Res.10 , 2413 – 2424 ( 2017 ).
- Miao Y McCammon JA . Mechanism of the G-protein mimetic nanobody binding to a muscarinic G-protein-coupled receptor . Proc. Natl Acad. Sci. USA115 ( 12 ), 3036 – 3041 ( 2018 ).
- Eichel K Jullié D Barsi-Rhyne B et al. Catalytic activation of β-arrestin by GPCRs . Nature557 ( 7705 ), 381 – 386 ( 2018 ).
- Latorraca NR Wang JK Bauer B et al. Molecular mechanism of GPCR-mediated arrestin activation . Nature557 ( 7705 ), 452 – 456 ( 2018 ).
- Ballesteros JA Weinstein H . Integrated methods for the construction of three-dimensional models and computational probing of structure–function relations in G protein-coupled receptors . Methods Neurosci.25 , 366 – 428 ( 1995 ).
- Thal DM Glukhova A Sexton PM Christopoulos A . Structural insights into G-protein-coupled receptor allostery . Nature559 ( 7712 ), 45 – 53 ( 2018 ).
- van der Westhuizen ET Valant C Sexton PM Christopoulos A . Endogenous allosteric modulators of G protein-coupled receptors . J. Pharmacol. Exp. Ther.353 ( 2 ), 246 – 260 ( 2015 ).
- Wootten D Christopoulos A Sexton PM . Emerging paradigms in GPCR allostery: implications for drug discovery . Nat. Rev. Drug Discov.12 ( 8 ), 630 – 644 ( 2013 ).
- Katritch V Fenalti G Abola EE Roth BL Cherezov V Stevens RC . Allosteric sodium in class A GPCR signaling . Trends Biochem. Sci.39 ( 5 ), 233 – 244 ( 2014 ).
- Liu W Chun E Thompson AA et al. Structural basis for allosteric regulation of GPCRs by sodium ions . Science337 ( 6091 ), 232 – 236 ( 2012 ).
- Selent J Sanz F Pastor M De Fabritiis G . Induced effects of sodium ions on dopaminergic G-protein coupled receptors . PLoS Comput. Biol.6 ( 8 ), e1000884 ( 2010 ).
- Miao Y Caliman AD McCammon JA . Allosteric effects of sodium ion binding on activation of the m3 muscarinic g-protein-coupled receptor . Biophys. J.108 ( 7 ), 1796 – 1806 ( 2015 ).
- Yuan S Vogel H Filipek S . The role of water and sodium ions in the activation of the μ-opioid receptor . Angew. Chem. Int. Ed. Engl.52 ( 38 ), 10112 – 10115 ( 2013 ).
- Gutiérrez-de-Terán H Massink A Rodríguez D et al. The role of a sodium ion binding site in the allosteric modulation of the A(2A) adenosine G protein-coupled receptor . Structure21 ( 12 ), 2175 – 2185 ( 2013 ).
- Bartuzi D Kaczor AA Matosiuk D . Interplay between two allosteric sites and their influence on agonist binding in human μ opioid receptor . J. Chem. Inf. Model.56 ( 3 ), 563 – 570 ( 2016 ).
- Massink A Gutiérrez-de-Terán H Lenselink EB et al. Sodium ion binding pocket mutations and adenosine A2A receptor function . Mol. Pharmacol.87 ( 2 ), 305 – 313 ( 2015 ).
- Shang Y LeRouzic V Schneider S Bisignano P Pasternak GW Filizola M . Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions . Biochemistry53 ( 31 ), 5140 – 5149 ( 2014 ).
- Selvam B Shamsi Z Shukla D . Universality of the sodium ion binding mechanism in class A G-protein-coupled receptors . Angew. Chem. Int. Ed. Engl.57 ( 12 ), 3048 – 3053 ( 2018 ).
- Vohra S Taddese B Conner AC et al. Similarity between class A and class B G-protein-coupled receptors exemplified through calcitonin gene-related peptide receptor modelling and mutagenesis studies . J. R. Soc. Interface.10 ( 79 ), 20120846 ( 2013 ).
- Pasternak GW Snowman AM Snyder SH . Selective enhancement of [3H]opiate agonist binding by divalent cations . Mol. Pharmacol.11 ( 6 ), 735 – 744 ( 1975 ).
- Burgmer U Schulz U Tränkle C Mohr K . Interaction of Mg2+ with the allosteric site of muscarinic M2 receptors . Naunyn Schmiedebergs Arch. Pharmacol.357 ( 4 ), 363 – 370 ( 1998 ).
- Johansson B Parkinson FE Fredholm BB . Effects of mono- and divalent ions on the binding of the adenosine analogue CGS 21680 to adenosine A2 receptors in rat striatum . Biochem. Pharmacol.44 ( 12 ), 2365 – 2370 ( 1992 ).
- Mazzoni MR Martini C Lucacchini A . Regulation of agonist binding to A2A adenosine receptors: effects of guanine nucleotides (GDP[S] and GTP[S]) and Mg2+ ion . Biochim. Biophys. Acta1220 ( 1 ), 76 – 84 ( 1993 ).
- Ye L Neale C Sljoka A et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations . Nat. Commun.9 ( 1 ), 1372 ( 2018 ).
- Gentry PR Sexton PM Christopoulos A . Novel allosteric modulators of G protein-coupled receptors . J. Biol. Chem.290 ( 32 ), 19478 – 19488 ( 2015 ).
- Aurelio L Valant C Flynn BL Sexton PM Christopoulos A Scammells PJ . Allosteric modulators of the adenosine A1 receptor: synthesis and pharmacological evaluation of 4-substituted 2-amino-3-benzoylthiophenes . J. Med. Chem.52 ( 14 ), 4543 – 4547 ( 2009 ).
- Dror RO Green HF Valant C et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs . Nature503 ( 7475 ), 295 – 299 ( 2013 ).
- Shang Y Yeatman HR Provasi D et al. Proposed mode of binding and action of positive allosteric modulators at opioid receptors . ACS Chem. Biol.11 ( 5 ), 1220 – 1229 ( 2016 ).
- Deganutti G Cuzzolin A Ciancetta A Moro S . Understanding allosteric interactions in G protein-coupled receptors using supervised molecular dynamics: a prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000 . Bioorg. Med. Chem.23 ( 14 ), 4065 – 4071 ( 2015 ).
- Saleh N Hucke O Montel F et al. Multiple binding sites contribute to the mechanism of mixed agonistic and positive allosteric modulators of the Cannabinoid CB1 receptor . Angew. Chem. Int. Ed. Engl.57 ( 10 ), 2580 – 2585 ( 2018 ).
- Vaidehi N Bhattacharya S . Allosteric communication pipelines in G-protein-coupled receptors . Curr. Opin. Pharmacol.30 , 76 – 83 ( 2016 ).
- Pandini A Fornili A Fraternali F Kleinjung J . Detection of allosteric signal transmission by information-theoretic analysis of protein dynamics . FASEB J.26 ( 2 ), 868 – 881 ( 2012 ).
- Rivalta I Sultan MM Lee N-S Manley GA Loria JP Batista VS . Allosteric pathways in imidazole glycerol phosphate synthase . Proc. Natl Acad. Sci. USA109 ( 22 ), E1428 – E1436 ( 2012 ).
- McClendon CL Friedland G Mobley DL Amirkhani H Jacobson MP . Quantifying correlations between allosteric sites in thermodynamic ensembles . J. Chem. Theory Comput.5 ( 9 ), 2486 – 2502 ( 2009 ).
- Bhattacharya S Vaidehi N . Differences in allosteric communication pipelines in the inactive and active states of a GPCR . Biophys. J.107 ( 2 ), 422 – 434 ( 2014 ).
- La Sala G Decherchi S De Vivo M Rocchia W . Allosteric communication networks in proteins revealed through pocket crosstalk analysis . ACS Cent. Sci.3 ( 9 ), 949 – 960 ( 2017 ).
- Venkatakrishnan AJ Ma A Fonseca R et al. Stable networks of water-mediated interactions are conserved in activation of diverse GPCRs . BioRxiv ( 2018 ). www.biorxiv.org/content/biorxiv/early/2018/06/20/351502.full.pdf
- Yuan S Palczewski K Peng Q Kolinski M Vogel H Filipek S . The mechanism of ligand-induced activation or inhibition of μ- and κ-opioid receptors . Angew. Chem. Int. Ed. Engl.54 ( 26 ), 7560 – 7563 ( 2015 ).
- Yuan S Filipek S Palczewski K Vogel H . Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway . Nat. Commun.5 , 4733 ( 2014 ).
- Lee Y Kim S Choi S Hyeon C . Ultraslow water-mediated transmembrane interactions regulate the activation of A2A adenosine receptor . Biophys. J.111 ( 6 ), 1180 – 1191 ( 2016 ).
- Abel R Young T Farid R Berne BJ Friesner RA . Role of the active-site solvent in the thermodynamics of factor Xa ligand binding . J. Am. Chem. Soc.130 ( 9 ), 2817 – 2831 ( 2008 ).
- Ramsey S Nguyen C Salomon-Ferrer R Walker RC Gilson MK Kurtzman T . Solvation thermodynamic mapping of molecular surfaces in AmberTools: GIST . J. Comput. Chem.37 ( 21 ), 2029 – 2037 ( 2016 ).
- López ED Arcon JP Gauto DF et al. WATCLUST: a tool for improving the design of drugs based on protein–water interactions . Bioinformatics31 ( 22 ), 3697 – 3699 ( 2015 ).
- Hu B Lill MA . WATsite: hydration site prediction program with PyMOL interface . J. Comput. Chem.35 ( 16 ), 1255 – 1260 ( 2014 ).
- Cuzzolin A Deganutti G Salmaso V Sturlese M Moro S . AquaMMapS: an alternative tool to monitor the role of water molecules during protein–ligand association . ChemMedChem.13 ( 6 ), 522 – 531 ( 2018 ).
- Zia SR Gaspari R Decherchi S Rocchia W . Probing hydration patterns in class-A GPCRs via biased MD: the A2A receptor . J. Chem. Theory Comput.12 ( 12 ), 6049 – 6061 ( 2016 ).
- Higgs C Beuming T Sherman W . Hydration site thermodynamics explain SARs for triazolylpurines analogues binding to the A2A receptor . ACS Med. Chem. Lett.1 ( 4 ), 160 – 164 ( 2010 ).
- Bortolato A Tehan BG Bodnarchuk MS Essex JW Mason JS . Water network perturbation in ligand binding: adenosine A(2A) antagonists as a case study . J. Chem. Inf. Model.53 ( 7 ), 1700 – 1713 ( 2013 ).
- Hedger G Sansom MSP . Lipid interaction sites on channels, transporters and receptors: recent insights from molecular dynamics simulations . Biochim. Biophys. Acta.1858 ( 10 ), 2390 – 2400 ( 2016 ).
- Escribá PV Wedegaertner PB Goñi FM Vögler O . Lipid–protein interactions in GPCR-associated signaling . Biochim. Biophys. Acta.1768 ( 4 ), 836 – 852 ( 2007 ).
- Mondal S Khelashvili G Weinstein H . Not just an oil slick: how the energetics of protein–membrane interactions impacts the function and organization of transmembrane proteins . Biophys. J.106 ( 11 ), 2305 – 2316 ( 2014 ).
- Grossfield A Feller SE Pitman MC . A role for direct interactions in the modulation of rhodopsin by omega-3 polyunsaturated lipids . Proc. Natl Acad. Sci. USA103 ( 13 ), 4888 – 4893 ( 2006 ).
- Salas-Estrada LA Leioatts N Romo TD Grossfield A . Lipids alter rhodopsin function via ligand-like and solvent-like interactions . Biophys. J.114 ( 2 ), 355 – 367 ( 2018 ).
- Khelashvili G Grossfield A Feller SE Pitman MC Weinstein H . Structural and dynamic effects of cholesterol at preferred sites of interaction with rhodopsin identified from microsecond length molecular dynamics simulations . Proteins76 ( 2 ), 403 – 417 ( 2009 ).
- Cang X Du Y Mao Y Wang Y Yang H Jiang H . Mapping the functional binding sites of cholesterol in β2-adrenergic receptor by long-time molecular dynamics simulations . J. Phys. Chem. B.117 ( 4 ), 1085 – 1094 ( 2013 ).
- Terrillon S Bouvier M . Roles of G-protein-coupled receptor dimerization . EMBO Rep.5 ( 1 ), 30 – 34 ( 2004 ).
- Milligan G . G protein-coupled receptor dimerisation: molecular basis and relevance to function . Biochim. Biophys. Acta1768 ( 4 ), 825 – 835 ( 2007 ).
- Shan J Khelashvili G Mondal S Mehler EL Weinstein H . Ligand-dependent conformations and dynamics of the serotonin 5-HT(2A) receptor determine its activation and membrane-driven oligomerization properties . PLoS Comput. Biol.8 ( 4 ), e1002473 ( 2012 ).
- Lee JY Lyman E . Predictions for cholesterol interaction sites on the A2A adenosine receptor . J. Am. Chem. Soc.134 ( 40 ), 16512 – 16515 ( 2012 ).
- Neale C Herce HD Pomès R García AE . Can specific protein-lipid interactions stabilize an active state of the beta 2 adrenergic receptor?Biophys. J.109 ( 8 ), 1652 – 1662 ( 2015 ).
- Bruzzese A Gil C Dalton JAR Giraldo J . Structural insights into positive and negative allosteric regulation of a G protein-coupled receptor through protein–lipid interactions . Sci. Rep.8 ( 1 ), 4456 ( 2018 ).
- Yen H-Y Hoi KK Liko I et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling . Nature559 ( 7714 ), 423 – 427 ( 2018 ).
- Liang Y-L Khoshouei M Deganutti G et al. Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor . Nature561 , 492 – 497 ( 2018 ).
- Hoare SRJ . Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors . Drug Discov. Today10 ( 6 ), 417 – 427 ( 2005 ).
- Hollenstein K de Graaf C Bortolato A Wang M-W Marshall FH Stevens RC . Insights into the structure of class B GPCRs . Trends Pharmacol. Sci.35 ( 1 ), 12 – 22 ( 2014 ).
- Bortolato A Doré AS Hollenstein K Tehan BG Mason JS Marshall FH . Structure of class B GPCRs: new horizons for drug discovery . Br. J. Pharmacol.171 ( 13 ), 3132 – 3145 ( 2014 ).
- Wootten D Miller LJ Koole C Christopoulos A Sexton PM . Allostery and biased agonism at class B G protein-coupled receptors . Chem. Rev.117 ( 1 ), 111 – 138 ( 2017 ).
- Yang L Yang D de Graaf C et al. Conformational states of the full-length glucagon receptor . Nat. Commun.6 , 7859 ( 2015 ).
- Woolley MJ Reynolds CA Simms J et al. Receptor activity-modifying protein dependent and independent activation mechanisms in the coupling of calcitonin gene-related peptide and adrenomedullin receptors to Gs . Biochem. Pharmacol.142 , 96 – 110 ( 2017 ).
- Bower RL Yule L Rees TA et al. Molecular signature for receptor engagement in the metabolic peptide hormone amylin . ACS Pharmacol. Transl. Sci.1 , 32 – 49 ( 2018 ).
- Wootten D Reynolds CA Smith KJ et al. Key interactions by conserved polar amino acids located at the transmembrane helical boundaries in Class B GPCRs modulate activation, effector specificity and biased signalling in the glucagon-like peptide-1 receptor . Biochem. Pharmacol.118 , 68 – 87 ( 2016 ).
- Wootten D Reynolds CA Koole C et al. A hydrogen-bonded polar network in the core of the glucagon-like peptide-1 receptor is a fulcrum for biased agonism: lessons from class B crystal structures . Mol. Pharmacol.89 ( 3 ), 335 – 347 ( 2016 ).
- Wootten D Reynolds CA Smith KJ et al. The extracellular surface of the GLP-1 receptor is a molecular trigger for biased agonism . Cell165 ( 7 ), 1632 – 1643 ( 2016 ).
- Dal Maso E Zhu Y Pham V et al. Extracellular loops 2 and 3 of the calcitonin receptor selectively modify agonist binding and efficacy . Biochem. Pharmacol.150 , 214 – 244 ( 2018 ).
- Weaver RE Mobarec JC Wigglesworth MJ Reynolds CA Donnelly D . High affinity binding of the peptide agonist TIP-39 to the parathyroid hormone 2 (PTH2) receptor requires the hydroxyl group of Tyr-318 on transmembrane helix 5 . Biochem. Pharmacol.127 , 71 – 81 ( 2017 ).