
Abstract
Extracellular vesicles (EVs) are nanovesicles secreted for intercellular communication with endosomal network regulating secretion of small EVs (or exosomes) that play roles in cancer progression. As an essential oncoprotein, Kirsten rat sarcoma virus (KRAS) is tightly regulated by its endosomal trafficking for membrane attachment. However, the crosstalk between KRAS and EVs has been scarcely discussed despite its endocytic association. An overview of the oncogenic role of KRAS focusing on its correlation with cancer-associated EVs should provide important clues for disease prognosis and inspire novel therapeutic approaches for treating KRAS mutant cancers. Therefore, this review summarizes the relevant studies that provide substantial evidence linking KRAS mutation to EVs and discusses the oncogenic implication from the aspects of biogenesis, cargo sorting, and release and uptake of the EVs.
The KRAS (Kirsten rat sarcoma virus) gene is one of the members of the Ras oncogene family that encodes for KRAS protein that belongs to the RAS protein subfamily. In this protein subfamily (KRAS, HRAS and NRAS), KRAS resembles the most frequently mutated protein in human cancers, especially in lung cancers, pancreatic cancers and colorectal cancers (CRCs) [Citation1]. Mutations in codons 12 and 13 of KRAS are most prevalently detected in cancers that lead to expression of amino acid substituted mutant KRAS protein variants such as KRASG12D, KRASG12V, KRASG12C and KRASG13D [Citation2]. Ras proteins in general are activated by surface receptors, and plasma membrane is the exclusive site for activation of these proteins. Wild-type KRAS protein cycles between the GDP-bound (inactive) and GTP-bound (active) state depending on ligand binding to the cell surface receptors [Citation3]. However, mutations of KRAS usually cause defective GTP hydrolysis resulting in permanent GTP-bound KRAS that is constitutively activated [Citation4]. KRAS is one of the upstream regulators of MAPK and phosphatidylinositol-3-kinase (PI3K) signaling pathways [Citation5]. Therefore, mutations of KRAS oncoproteins often induce hyperactivated MAPK and PI3K signaling that are involved in proliferation, survival and metastasis of cancers [Citation6]. Mutant KRAS was correlated with a more aggressive metastatic phenotype as well as poor disease prognosis in various cancers, such as CRC [Citation7]. Over the past decades, scientists poured considerable efforts into finding targeted therapeutics to combat mutant KRAS, and it was thought to be ‘undruggable’ [Citation8]. However, Shokat and his team set out to search for compounds that could serve as covalent inhibitors targeting cysteine 12 (C12) residue of KRASG12C protein [Citation9]. Based on Shokat’s team’s findings, Lanman and his group successfully identified a molecule that served as an effective covalent inhibitor of KRASG12C that later registered as AMG 510 or sotorasib for its potential clinical use [Citation10]. In May 2021, sotorasib was granted accelerated approval by the US FDA to treat patients diagnosed with local advanced and metastatic non-small-cell lung cancer (NSCLC) bearing KRASG12C mutation who had received at least one prior systemic therapy [Citation11]. Both phase I and II clinical trials of sotorasib (CodeBreaK 100) produced more than 30% confirmed response rate with 91.2% and 80.6% disease control rate, respectively [Citation12,Citation13]. Sotorasib permanently locks the KRAS in its inactive state by binding covalently to the C12 residue of the GDP-bound KRAS protein [Citation14]. Preclinical study confirmed that sotorasib selectively targets KRASG12C mutant cells in both in vivo and in vitro models. In the same study, sotorasib was also found to activate adaptive host immunity, suggesting its potential synergistic effect in immunotherapy [Citation15]. However, there are studies that have found the potential risk of this novel KRASG12C inhibitor. Cancer cells that exploit other signaling cascades such as PI3k/MAPK signaling [Citation16], or with activation of an epithelial-mesenchymal transition (EMT) program [Citation17], may not be sensitive to sotorasib treatment that specifically binds to KRASG12C. Malignancies with multiple KRAS mutations were also less responsive to sotorasib treatment [Citation18], on top of complications arising when different KRAS mutation variants were observed to give rise to distinct cancer phenotypes [Citation19] as well as sensitivity toward other chemotherapy [Citation20]. Constitutively activated KRAS could bypass signals from upstream effectors, such as EGFR, becoming the main cause of drug resistance in the treatments targeting the oncogenic receptors on cancer cells [Citation21]. Therefore, current research that is seeking effective treatments for KRAS mutant cancers focuses on combinational therapy targeting both KRAS and its downstream effectors such as MAPK [Citation22].
Endosomal trafficking is an essential cellular process regulating transportation of biomolecules, and studies revealed that the different trafficking, localization and membrane association of different RAS isoforms determine their signaling specificity [Citation23]. KRAS is the key mutated gene in pancreatic cancers, and recent research found that mutant KRAS promotes endosomal degradation in pancreatic ductal adenocarcinoma (PDAC) cell lines [Citation24]. Growing evidence indicates that endosomal/endocytic trafficking plays important roles in oncogenic signaling. For instance, endocytic degradation of EGFR is essential to recycle the receptors to the plasma membrane for prolonged signaling [Citation25]. Another endosomal trafficking-associated oncoprotein is c-Src, a tyrosine kinase activated under the plasma membrane and transduces oncogenic signals in the early stage of cancer development [Citation26]. c-Src has long been known to amplify EGFR activation oncogenic signals, and lately SRC was identified as one of the validated therapeutic targets in KRAS mutant cancers [Citation27,Citation28]. In most recent cancer research, extracellular vesicles (EVs), or more well known as exosomes, were hypothesized to facilitate the communication between cancers and neighboring cells as well as the tumor microenvironment (TME) in driving cancer progression [Citation29]. Both EGFR and SRC have been linked to biogenesis of EVs due to their associations with endosomal trafficking systems [Citation30–32]. Despite the fact that endocytic association was also reported in KRAS mutation, unlike EGFR and SRC, there is currently a lack of overview on the role of KRAS in regulation of EVs and their possible crosstalk in cancer biology.
EVs are small, cargo-bearing, membranous vesicles secreted by all types of cells with different sizes, membrane composition and vesicular content [Citation33]. All EVs can be categorized into three main groups: small EVs, microvesicles and apoptotic bodies. Classification of EVs is mostly based on their diameter, where small EVs have a diameter of 30 to 120 nm, microvesicles are usually larger with 50 to 1000 nm diameter and apoptotic bodies have a diameter that ranges from 50 to 2000 nm [Citation34]. Surface markers such as tetraspanins (CD9, CD63 and CD81), ALG-2-interacting protein X (ALIX) and tumor susceptibility gene 101 protein (TSG101) were often used to distinguish between small EVs and microvesicles, as these surface protein markers were usually expressed more prevalently in the small EV subpopulation [Citation35]. EVs were first discovered to discard unwanted molecules and cell components from cells, but recent studies provide a new perspective for the roles of EVs in mediating cell–cell communication and shaping the extracellular matrix (ECM) [Citation36]. Rapidly proliferating cancer cells in hypoxic condition tend to secrete higher amounts of EVs that carry vastly different cargo when compared with healthy cells under a normoxic condition [Citation37,Citation38]. Most of the cancer-derived EVs are small EVs and microvesicles. Cancer-derived EVs are inherently important for tumor initiation and progression, where recent research uncovered the essential role of these vesicles in promoting tumor proliferation, therapy resistance and metastatic phenotype [Citation39,Citation40].
It is becoming increasingly evident that EVs play a crucial role in pathophysiological development, and there are broad prospects for their clinical application in the diagnosis and therapy of various diseases, including different types of cancers [Citation36]. In this article, we review all of the recent studies involving both EVs and KRAS mutation in cancers and discuss the correlation between KRAS mutation and cancer-derived EVs systematically from the aspects of biogenesis, cargo sorting, and release and uptake of the EVs in different cancer types. Boolean search was performed on Google Scholar and PubMed using following search terms: (cancer* OR malignan* OR tumo*r* OR onco*) AND KRAS AND (EV* OR “extracellular vesicle*” OR “microvesicle*” OR “exosome*”). Peer-reviewed articles published from 2000 to 2021 were manually filtered by evaluating their title, abstract and discussion to determine their relevance to this review. Other than that, additional publications were also obtained from the reference list of selected articles. Collectively, it is hoped that a review of current studies of the oncogenic role of KRAS with a focus on its correlation with cancer-derived EVs might be promising in predicting disease prognosis and inspires novel approaches in the treatment of KRAS mutant cancers.
KRAS & EV biogenesis
Different subpopulations of EVs are produced via distinct biogenesis pathways in which microvesicles are formed through direct budding of plasma membranes, whereas the biogenesis of small EVs such as exosomes involves the endosomal network [Citation41]. In formation of small EVs, different cargo will be sorted through fusion of endocytic vesicles or intracellular vesicles with early endosomes. Late endosomes will then be derived from early endosomes after a series of maturation processes. There are two fates of late endosomes: lysosomal degradation or further sorting of its content into small vesicles to form multivesicular bodies (MVB) to be released extracellularly () [Citation42]. A recent study found exposure to EGF stimulated translocation of KRAS onto early endosomal membrane (A) [Citation43]. The hypervariable domain of KRAS protein undergoes multiple post-translational modifications, followed by interaction with the calcium/calmodulin system for correct translocation of KRAS from the endosomal membrane to the plasma membrane, which is essential for activation of KRAS signaling [Citation44]. Intriguingly, EGF-mediated KRAS translocation to the late endosome was observed to suppress lysosomal degradation activities (B) [Citation43]. Suppression of lysosomal degradation in the EGF-KRAS-endosomes axis would potentially facilitate formation of more MVBs, which subsequently increases the biogenesis of small EVs.
(A) Exposure to EGF promotes sorting of KRAS to early and late endosomes. (B) KRAS on the late endosomal membrane suppresses endosome-lysosome fusion and facilitates maturation of late endosome into MVBs to release small vesicles into the extracellular matrix. (C) KRAS promotes localization of Rab13, CD81 and EGFR at plasma membranes, resulting in release of Rab13+, CD81+, EGFR+ and CD63- small EVs through membrane budding [Citation33,Citation35].
EVs: Extracellular vesicles; KRAS: Kirsten rat sarcoma virus protein; MVBs: Multivesicular bodies.
![Figure 1. Kirsten rat sarcoma virus-associated endosomal networking for extracellular vesicles biogenesis. (A) Exposure to EGF promotes sorting of KRAS to early and late endosomes. (B) KRAS on the late endosomal membrane suppresses endosome-lysosome fusion and facilitates maturation of late endosome into MVBs to release small vesicles into the extracellular matrix. (C) KRAS promotes localization of Rab13, CD81 and EGFR at plasma membranes, resulting in release of Rab13+, CD81+, EGFR+ and CD63- small EVs through membrane budding [Citation33,Citation35].EVs: Extracellular vesicles; KRAS: Kirsten rat sarcoma virus protein; MVBs: Multivesicular bodies.](/cms/asset/11dab72d-4c6d-4237-81f9-4390b9cfa355/ifmc_a_12362049_f0001.jpg)
KRAS mutant cancer-related EVs
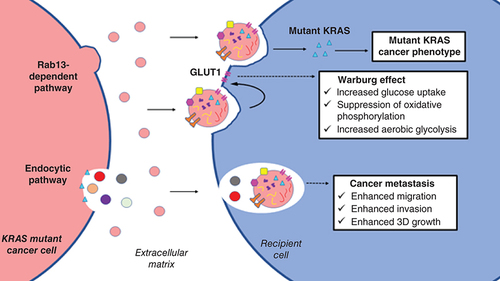
Tetraspanin proteins such as CD63 are key markers of small EVs (or more commonly known as exosomes) released from various types of normal cells. However, CRC cells bearing mutant KRAS secrete a distinct population of small EVs with the absence of CD63 [Citation45]. These EVs were observed being secreted through the KRAS-induced Rab13-dependent pathway (C). In the canonical pathway of small EV biogenesis, release of CD63+ exosomes rely on an endosomal network that leads to exocytic release of the MVB’s content [Citation41]. However, the study of Hinger and colleagues observed that the colocalization of Rab13 together with β1-integrin, CD81 and EGFR at the plasma membrane led to the formation of small EVs through budding, and knock-down experiments showed that the secretion of small EVs containing β1-integrin into the TME is Rab13 dependent. These Rab13+ small EVs showed different physical properties when compared with CD63+ exosomes [Citation45]. This unique population of small EVs is heterogeneous in size, where they were detected in both microvesicles and small EV fractions when differential centrifugation was used for EV isolation in the study [Citation45]. Within the small EV fraction, Rab13+ small EVs were detected with lower densities, a physical property closely associated with microvesicles or EVs with larger sizes implying that membrane composition of this unique population of small EVs may be more lipid rich when compared with that of CD63+ EVs with smaller diameter [Citation46]. Apart from the heterogeneity in size and density, missing of the established small EV marker CD63 can be considered as the common character of these Rab13+ small EVs. This observation proves that the small EVs released from KRAS mutant cancers are distinct from CD63+ exosomes [Citation45]. This finding evidently shows that oncogenic mutations promote secretion of distinct populations of EVs with different biological and physical properties.
Oncogenic properties of EV biogenesis in KRAS mutant cancers
KRAS increases production of EVs, and its mutation form triggers formation of a unique EV population. It was speculated that KRAS-mediated EV biogenesis may play a role in cancer progression. Petanidis and colleagues discovered that inhibition of EV biogenesis in KRAS mutant lung cancer cells suppressed the expression of chromatid remodeling associated genes such as SMARCE1, ARID1A and NCOR1 [Citation47]. These genes are important in controlling desired epigenetic regulation to maintain oncogenic phenotype in cancers [Citation48]. In nude mice bearing KRAS mutant lung cancer, inhibition of EV biogenesis was observed to significantly impact tumor growth and prolong survival of the tested animals. Further, an ablated pyruvate/pyruvate kinase M2 (PKM2) dependent shift in energy metabolism was observed in the same cancer model with co-inhibition of KRAS and EV biogenesis [Citation47]. PKM2 is an important rate-limiting enzyme in glycolysis that dictates the main energy metabolism pathways such as oxidative phosphorylation and lactate producing aerobic glycolysis [Citation49]. In cancer cells, PKM2 reprograms energy metabolism in favor of aerobic glycolysis for fast ATP production to support rapid cancer growth [Citation50]. However, depletion of miR-146 and restored miR-210 levels were observed in KRAS mutant lung cancer cells with inhibition of both KRAS and EV biogenesis [Citation47]. miR-146 promotes cancer by maintaining stemness of human breast cancer stem cells [Citation51], whereas miR-210 acts as tumor suppressor by inhibiting proliferation [Citation52] and promoting cell cycle arrest [Citation53]. Together, these findings suggest that EV biogenesis may contribute to mutant KRAS-related oncogenic properties through regulation of multiple facets of cancers including epigenetics, reprogramming of energy metabolism and miRNAs.
KRAS & cargo sorting of EVs
EVs contain specific sets of nucleic acids (DNA, RNA), proteins, lipids and metabolites depending on the donor cells, and the different composition of EV cargos moderates various responses in the recipient cells. The unique content of EVs highlights the specificity of sorting machinery in cells that coordinate the selective packaging of EV cargo [Citation54]. There are multiple roles for cancer-derived EVs, including removal of undesirable cell content, modulation of signaling and oncogenic transformation in recipient cells, and shaping of the TME [Citation55]. It was anticipated that various regulatory pathways may be involved in the cargo sorting of cancer-derived EVs, but the molecular machinery has remained obscure to date. In the superfamily of Ras proteins, families of Rab, Arf and Rho proteins are well known for their roles in intracellular trafficking [Citation56]. As part of this superfamily, the link between KRAS and endosomal trafficking was reported recently [Citation24]. In this section, we summarize and discuss the possible regulatory roles of KRAS in intracellular trafficking for sorting of various types of biomolecules in secretory vesicles. A general summary of KRAS-mediated differential sorting of proteins in EVs is tabulated in Supplementary Table 1.
KRAS regulates sorting of proteins in EVs
KRAS proteins
Mutant KRAS was found to promote its own sorting in EVs. A study discovered that EVs derived from mutant KRAS CRC cells were enriched with mutant KRAS proteins, but the proteins were barely detectable in EVs derived from CRC cells bearing wild-type KRAS. At the same time, enrichment of ubiquitinated proteins in the EVs containing mutant KRAS was also observed [Citation57]. EV sorting of other proteins such as phosphate and tensin homolog (PTEN) was reported to be monoubiquitination dependent [Citation58]. In addition, ubiquitination could enhance GTP loading of mutated KRAS and its binding to downstream effectors [Citation59]. Together, these observations imply that post-translational modification such as monoubiquitination may drive the differential sorting of mutant KRAS proteins in EVs to promote a cancer phenotype in recipient cells.
Argonaute 2
As a ribonuclease, argonaute 2 (Ago2) is an essential component of the RNA-induced silencing complex (RISC) regulating RNA-mediated gene silencing [Citation60]. Ago2 is widely considered as an oncoprotein, where it was found to assist in KRAS signaling by directly binding with KRAS and thus regulating KRAS-mediated gene expression to drive subsequent oncogenic signaling [Citation61]. Furthermore, Ago2 promotes tumorigenesis by facilitating expression of oncogenic miRNAs such as miR-19b [Citation62]. Acetylated Ago2 elevates miR-19b expression that is correlated with disease progression and poor prognosis in human lung cancer [Citation63]. Due to the RNA binding ability, Ago2 was hypothesized to regulate the sorting of miRNA in EVs derived from CRC with KRAS mutation. It was found that the packaging of Ago2 in EVs was promoted through MEK signaling in these cancer cells [Citation64]. Some studies show there are physiological and pathological effects on recipient cells with the uptake of miRNA bound to AGO2 through specific regulation [Citation65].
Amphiregulin
Amphiregulin (AREG) is one of the ligands of EGFR, promoting growth, survival, migration, invasion and inflammatory TME of cancer cells [Citation66]. In non-small-cell lung cancer (NSCLC), the EGFR signaling pathway is constitutively activated and the levels of AREG in plasma of NSCLC patients correlate with their poor prognosis. A recent study discovered the role of AREG carried by EVs in bone metastasis of NSCLC patients [Citation67]. The effect of AREG in promoting an invasive phenotype was also observed in breast [Citation68] and pancreatic [Citation69] cancers. Lately, a new mode of EGFR ligand signaling via EVs was revealed. In good agreement with the study of Baillo and colleagues [Citation68], EVs containing AREG significantly increased invasiveness of recipient breast cancer cells. The same response was observed in colon cancers bearing mutant KRAS [Citation70]. Notably, AREG was identified as a prognostic factor in CRC, where its elevated expression correlated with lower overall survival and recurrence-free survival [Citation71]. Both NSCLC and KRAS mutant CRC with constantly activated EGFR signaling release EVs contain significant high levels of AREG involved in promoting invasive phenotypes of cancers [Citation67,Citation70]. These findings collectively support the speculation that EGFR signaling via sending ligands such as AREG in EVs might contribute to cancer development via priming of the metastatic niche at distant sites.
Rab13
Hinger’s group has been working on isogenic CRC cell lines to profile KRAS-dependent cargo packing in EVs since 2013, and we discussed their recent finding on mutant KRAS promoting the biogenesis of a distinctive population of Rab13+ small EVs earlier [Citation45]. In the Ras superfamily, Rab GTPases are key regulators of cellular membrane trafficking. Members of this protein family are involved in all steps of membrane trafficking, including formation, transport and fusion of vesicles. One of the members, Rab13, has been identified as a potent driver of cancer progression, mainly or partly related to its regulatory roles in trafficking of several oncogenic proteins, such as integrins, surface delivery of GLUT4 glucose transporter and VEGFR [Citation72]. As a high-glucose affinity transporter, GLUT4 promotes glucose influx in cancer that results in metabolic reprogramming, which leads to increased proliferation, survival and metastasis [Citation73]. In animal models, Rab13 regulates RhoA trafficking to mediate VEGF-dependent directional angiogenesis, an oncogenic change for cancers to encounter stress from hypoxic environments [Citation74]. Additionally, Rab13 was identified to assist cancer migration and invasion through inhibition of cell–cell adhesion, initiating actin cytoskeletal remodeling [Citation75] and regulating integrin recycling [Citation76]. Hinger’s group successfully demonstrated that EVs derived by KRAS mutant cancers transfer functional cargo for uptake by recipient cells, but the mechanism of specific sorting of these EVs remains to be fully elucidated [Citation77]. The preceding studies reported that the function of Rab13 implicating this protein is probably the key Rab GTPase that regulates the trafficking of oncogenic cellular components (including itself) to be sorted in EVs of KRAS mutant cancers. After uptake by recipient cells, the functional EV cargo including Rab13 and its ‘crime partners’ may drive tumorigenesis in the cells.
Glucose transporter
Most cancers rely on high glucose utilization to support glycolysis instead of oxidative phosphorylation for ATP generation, which is known as the Warburg effect [Citation78]. Actively proliferating cancer cells require metabolites such as NADPH, which are produced more in cells generating ATP through anaerobic glycolysis [Citation79]. Therefore, overexpression of GLUT1 that promotes glucose uptake was observed in various cancers, including breast [Citation80] and ovarian [Citation81] cancers. Inhibition of GLUT1 suppressed the growth of human breast cancers [Citation82]. In general, this glucose transporter was found to promote proliferation [Citation83] and shield cancer against glucose deprivation-mediated stress [Citation84]. Notably, elevated GLUT1 expression was also observed in pancreatic cancer with KRAS mutation [Citation85]. Silencing of KRASG12D mutation in the mice model resulted in downregulation of GLUT1 expression and suppressed glycolysis [Citation86]. Moreover, in a study investigating colonic epithelial cells, increased levels of GLUT1 glucose transporter were detected both in CRC cells bearing mutant KRAS and in their EVs [Citation87]. GLUT1 in these EVs was observed as being full-length proteins rather than fragmented peptides, implying that these glucose transporters are functionally transferred from KRAS mutant cancers to trigger metabolic reprogramming in recipient cells.
KRAS regulates EV packaging of RNAs
One of the major cargos in EVs are RNAs in different forms, such as mRNA, lncRNA, miRNA and circRNA. In a recent study comparing RNA contents of EVs derived from CRC cells bearing different status of KRAS mutation, different mRNA and lncRNA enrichment was observed in EVs from cells containing heterozygous mutant and wild-type KRAS, respectively [Citation88]. Studies focused on lncRNA in EVs are rare. Therefore, the following sections focus on the discussion of mRNAs and miRNAs that were identified to be differentially sorted into EVs by mutated KRAS.
mRNA
When mutant KRAS was reported to promote sorting of Rab13 protein in EVs released by CRC with mutant KRAS, another study observed enrichment of Rab13 mRNA in these EVs [Citation88]. In addition, FAT1 and RTN4 mRNA were also found to be significantly upregulated in EVs derived from mutant KRAS CRC cells [Citation89]. FAT1 encodes for FAT atypical cadherin-1 (FAT1) protein modulating invasiveness in cancers such as colorectal [Citation90] and gastric cancer [Citation91]. RTN4, however, encodes for reticulon-4 (RTN4) oncoprotein that promotes cancer growth and resistance against the chemotherapeutic drug paclitaxel [Citation92]. Taken together, mutant KRAS promotes increased sorting of oncogene mRNA in EVs. These oncogenic mRNA-enriched EVs may be used as a means for intercellular signaling to promote tumorigenesis in recipient neighboring cells.
miRNAs
As one of the classes of noncoding RNAs, miRNA plays a major role in regulating gene expression mainly through post-transcriptional processing of mRNA [Citation93]. Recent studies discovered that cancer-derived EVs carry a unique population of miRNAs to be discarded from the donor cells [Citation94]. For instance, cancer cells were found to discard tumor-suppressing miR-23b [Citation95] and miR-202-3p [Citation96] through EV secretion. Conversely, cancers could pack miRNA in EVs for intercellular oncogenic signaling. For example, breast cancer secretes miR-125b containing EVs that induce differentiation to increase the population of cancer-associated fibroblasts [Citation97]. In CRC, increased sorting of miR-100-5p in EVs alongside its intracellular depletion was observed in cells with mutant KRAS [Citation98]. Several studies reported the role of miR-100-5p as a metastasis suppressor in numerous cancers, such as prostate [Citation99] and chordoma [Citation100] cancers. On the contrary, packaging of miR-10b in EVs was inhibited by mutant KRAS in CRC [Citation98]. miR-10b promotes cancer metastasis by activating an EMT program that leads to increased cancer mobility and invasiveness [Citation101]. Together, these studies infer that KRAS regulates the sorting of miRNA in EVs to remove undesired tumor-suppressing miRNAs, whereas oncogenic miRNAs that are favorable to cancer progression are retained in cancer cells. Intriguingly, increased 3′ trimming was observed in the miRNAs extracted from KRAS mutant cancer-derived EVs [Citation98]. This finding inspired interest in RNA trimming, which is worth further investigation for its potential as a form of post-transcriptional modification exploited by oncogenic genes like KRAS to regulate miRNA sorting in EVs.
KRAS & EV release/uptake
EV release by KRAS mutant cancer cells
Cells secrete EVs through either outward budding followed by fission of plasma membrane or membrane fusion of exosome-containing MVBs with plasma membrane resulting in exocytosis of small EVs [Citation102]. Many different factors are in play to regulate the type and concentration of EVs released by a cell. For instance, the endosomal sorting complex required for transport (ESCRT) may regulate EV release by mediating membrane curvature sorting, outward budding and fission [Citation103]. Cancer cells secrete EVs into the ECM, followed by diffusion of the vesicles into the bloodstream. Due to the small size, EVs can rapidly diffuse into various biofluids, and EVs have been detected in most biofluids, such as human urine, saliva, mucus and serum [Citation104]. In the mice bearing KRASG12D colon cancer, more small EVs were observed in their serum when compared with those with wild-type KRAS [Citation105]. In the study of Hinger and colleagues, EV secretion was found to be highly Rab13 dependent, in which knock-down of Rab13 significantly reduced EV release from KRAS mutant CRC cells, whereas the isogenic line bearing the wild-type protein was not affected [Citation45]. These studies support that there are unique mechanisms in KRAS mutant cancers regulating production of EVs that are carried by the circulatory system to be received by neighboring cells as well as by cells from distant sites.
EV uptake by KRAS mutant cancer cells
There are multiple ways in which a cell can uptake circulating EVs, such as direct membrane fusion, endocytosis, phagocytosis or micropinocytosis [Citation106]. Cancer-derived EVs were identified as transporters of signal molecules regulating tumorigenic biological processes such as PI3k-Akt or Wnt signaling pathways [Citation55]. Uptake of these circulating cancer-derived EVs by neighboring or distant recipient cells is thus important for cancer development locally as well as systemically. Primary human lung cancer cells carrying mutant KRAS were found to take up small EVs or exosomes more aggressively when compared with the cells bearing wild-type KRAS [Citation47]. This finding was further supported by another study that demonstrated mutant KRAS promotes uptake of circulating EVs through intensified micropinocytosis [Citation107]. In general, cancer cells with KRAS mutation tend to uptake more EVs regardless of their origin.
Therapeutic implications
Increased uptake of circulating EVs can be exploited in cancer treatment where the cell-derived vesicles could serve as a platform for delivery of targeted therapy to combat KRAS mutant malignancies. KRASG12D is the major form KRAS mutation found in PDAC where silencing of KRASG12D resulted in impeded tumor growth and recovered sensitivity to treatment with gemcitabine, a common chemotherapy drug that was used to treat PDAC [Citation108]. Kamerkar and his team engineered EVs known as iExosomes carrying siRNA to target the KRASG12D oncogenic allele in pancreatic cancers. Significant uptake of these engineered EVs was observed in pancreatic cells expressing KRASG12D when compared with the cell line carrying wild-type KRAS [Citation109]. When compared to cancer cells bearing wild type KRAS, more of these engineered EVs were uptake by cancer cells with mutated KRAS through enhanced micropinocytosis [Citation107,Citation109]. A phase I clinical trial utilizing iExosomes as a vehicle to deliver targeted therapy in KRASG12D mutant pancreatic cancer is under way (NCT03608631). Besides that, a study by McAndrews’ team successfully hampered tumor growth in a KRASG12D PDAC mice model via EV-mediated delivery of CRISPR/Cas9 [Citation110]. Together, these findings reinforce the great potential of EVs as effective vehicles to carry targeted therapy for KRAS mutant cancers.
Uptake of KRAS mutant cancer-derived EVs
Cancers secrete EVs into the ECM to shape the TME or promote cancer phenotypes in neighboring recipient cells through EV-mediated intercellular signaling [Citation29,Citation55,Citation102]. As mentioned in previous sections, cancers with mutant KRAS behaved differently in cargo sorting and secretion of EVs. In this section, we discuss the impact of EVs derived from KRAS mutant cancers on neighboring recipient cells. The main feature of this population of EVs is that they are able to promote numerous cancer phenotypes in recipient cells. It was discovered that EVs derived from human colon cancer cells carrying homozygous KRASG13D were able to transfer the mutated protein to recipient cells, whereas wild-type KRAS was not observed in EVs derived by non-KRAS mutant cancers. The mutant KRAS proteins carried by EVs are functional, as increased growth was observed in nontransformed rat intestinal epithelial cells with uptake of these EVs [Citation57].
Earlier, GLUT1 glucose transporters sorted in EVs derived from KRAS mutant cancers were discussed. These EVs were found to alter the energy metabolism by switching from oxidative phosphorylation to aerobic glycolysis for ATP generation in non-KRAS mutant recipient cells [Citation87]. This in vitro study provided a fine piece of evidence to support that EVs secreted by KRAS mutant CRC cells are able to promote oncogenic transformation in recipient cells via metabolic reprogramming. Some other studies reported that KRAS mutant cells-derived EVs could induce metastasis in recipient cells. EVs isolated from DLD-1 colon cancer cells bearing heterozygous KRASG13D mutation were observed to increase invasiveness in human breast cancer cells [Citation70]. Moreover, EVs carrying mutated KRAS proteins also promoted migration and anchorage-independent growth in recipient colon cancer cells bearing wild-type KRAS and nontransformed rat intestinal epithelial cells [Citation57]. Therefore, oncogenic properties of EVs secreted by KRAS mutant cancers are evident regardless of the types of recipient cells. The reported impacts of these EVs are summarized in .
EVs: Extracellular vesicles; KRAS: Kirsten rat sarcoma virus.
KRAS mutant cancer-derived EVs & therapy resistance
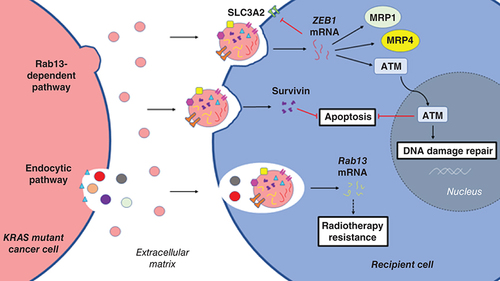
Resistance toward therapy such as chemoresistance or radiotherapy resistance remains as one of the most challenging hurdles in the battle against cancer. Due to the heterogeneity of malignant tumors, there is not a one-size-fits-all treatment for any cancers, as different populations within one tumor might have different sensitivity toward various chemotherapeutic agents [Citation111]. The population of cancer cells with primary resistance toward therapy may exploit EVs as vehicles to deliver signaling molecules that promote an acquired resistance phenotype in recipient cells. In our laboratory, EVs derived from SW480 colon cancer cells with KRASG12V mutation were found to carry acyl-CoA synthetase long-chain family 4 (ACSL4) proteins that promote cancer survival [Citation112]. Another study observed that exposure to EVs isolated from KRAS mutant cells promoted resistance against cisplatin and gemcitabine in normal human epithelial cells, which possibly regulated by ZEB1 mRNA received via EV uptake [Citation113]. ZEB1 encodes for zinc finger E-box binding homeobox 1 (ZEB1), which is a transcription factor that regulates multiple genes that contribute toward metastasis in cancer [Citation114]. Expression of transmembrane cell surface SLC3A2 was suppressed by ZEB1 that resulted in resistance against cisplatin-induced apoptosis in ovarian cancer cells [Citation115]. In a study investigating drug sensitivity of prostate cancer cells, correlation between ZEB1 level and resistance against apoptosis induction was observed [Citation116]. ZEB1 was also found to promote DNA damage repair by activating signaling of ataxia-telangiectasia mutated (ATM) in breast cancer cells detected with resistant phenotypes [Citation117]. More resistance-related proteins were discovered in EVs released by various cancers with KRAS mutation. Most (>90%) patients diagnosed with PDAC were found to carry KRAS mutation [Citation118], and the EVs extracted from their serum were observed with a high content of survivin [Citation119]. As part of the inhibitor of apoptosis family, survivin is a type of protein that regulates apoptosis and cell cycle progression. This protein is expressed in stem cells but absent in terminally differentiated cells [Citation120]. However, increased expression of survivin was observed in several cancers, including cervical [Citation121] and gastric [Citation122] cancers, correlating with poor prognosis of these cancers. Survivin promotes proliferation and inhibits apoptosis in cancers [Citation123]. More importantly, increased expression of survivin confers chemoresistance in multiple cancers, such as liver cancer [Citation124] and CRC [Citation125]. Survivin-enriched EVs released from KRAS mutant PDAC cells were found to promote insensitivity of recipient PDAC cells in treatment with paclitaxel and hydroxychloroquine [Citation119]. In good agreement with this finding, Kreger’s group observed that EV-mediated transfer of survivin between breast cancer cells increased their resistance against paclitaxel [Citation126]. Additionally, EVs isolated from various mutant KRAS expressing cell lines including human breast cancer cells MDA-MB-231 (KRASG13D), human lung adenocarcinoma cells A549 (KRASG12S) and H23 (KRASG12C) were enriched with survivin [Citation119]. Treatment resistance and poor prognosis are important features of cancers carrying KRAS mutations in which their resistance to radiotherapy remains poorly understood. Rab13 mRNA was detected in EVs derived by CRC cells expressing mutant KRAS [Citation88]. Concomitantly, upregulated Rab13 expression was observed in radiotherapy-resistant cancer cells [Citation127]. These studies provide clues implicating EV cargo unique to KRAS mutant cancers may contribute to radiation resistance. More profound experimental evidence was demonstrated for the role of survivin enriched in KRAS mutant cancer-derived EVs that confers chemoresistance in various types of cancers. Thus, inhibiting secretion of these EVs could potentially limit the development of tumor clusters with an acquired therapy-resistance phenotype. The potential roles of KRAS mutant cancer-derived EVs in promoting therapy resistance in recipient cells are summarized in .
ATM: Ataxia-telangiectasia mutated; EVs: Extracellular vesicles; KRAS: Kirsten rat sarcoma virus; MRP1: Multidrug resistance-associated protein 1; MRP4: Multidrug resistance-associated protein 4; SLC3A2: Solute carrier family 3 member 2.
KRAS mutant cancer-derived EVs & TME
The TME is important for tumor growth, survival, invasion and metastasis [Citation128]. In the process of shaping the TME, cancer cells interact with the ECM, fibroblasts, immune cells and the surrounding blood vessel network. Cancer cells secrete a variety of signaling molecules to prime the TME for transformation of immune cells such as macrophages and neutrophils to cancer-promoting types of cells. Tumor-associated macrophages (TAM) [Citation129] and tumor-associated neutrophils (TAN) [Citation130] are both important components of the TME that are favorable for tumor growth by secreting various types of cytokines. As discussed in previous sections, mutant KRAS regulates EV biogenesis, cargo sorting and secretion. Therefore, KRAS mutant cancer-derived EVs that carry unique cargo should play a role in conditioning of the TME through interactions with tumor-associated immune cells.
KRAS mutant cancer-derived EVs & TAM
After activation, macrophages go through maturation and form M1- or M2-like macrophages depending on their exposure to cytokines during the naive state [Citation131]. M1-like macrophages promote immune and inflammatory response, whereas M2-like macrophages are anti-inflammatory in nature and promote a post-inflammation recovery process [Citation132]. Thus, M2-like macrophages are generally considered as favorable for cancer development, as they secrete a wide range of cytokines that facilitate proliferation, growth and angiogenesis of tumor [Citation131]. A recent study revealed that the polarization of macrophages into pro-tumor M2-like TAM was mediated by KRASG12D mutation, and induction of autophagy-dependent ferroptosis in PDAC cells triggered the release of mutant KRASG12D protein into the TME via EV secretion [Citation133]. Ferroptosis is a recently described programmed cell death that requires iron-mediated accumulation of peroxidized lipids [Citation134]. The molecular mechanisms of ferroptosis are yet to be elucidated, but studies have shown that macrophages with ferroptosis induction released EVs that carry irons. These ferroptosis-dependent EVs were identified as the mutagenic factors in asbestos-associated cancers [Citation135]. Together, these findings signify the role of EVs as the carriers of mutagenic substances in the TME, and ferroptosis may be crucial in the production of oncogenic KRAS-associated EVs.
Macrophages that were recruited in the TME were found to ingest KRASG12D containing EVs through an advanced glycosylation end-product specific receptor (AGER)-dependent mechanism [Citation133]. AGER is a transmembrane surface receptor that mediates activation and differentiation of macrophages when exposed to chemokines or ligands [Citation136]. Ingested KRASG12D proteins facilitate phosphorylation and activation of STAT3 transcription factor through AGER, resulting in enhanced fatty acid oxidation in the macrophages that promoted its polarization to pro-tumor M2-like TAM. Investigation using a PDAC mice model showed that KRASG12D-mediated M2 TAM polarization is essential for in vivo macrophage-mediated pancreatic tumor growth [Citation133]. Other studies observed that high levels of M2-like TAM strongly correlate with shorter survival of PDAC patients [Citation137]. Growing evidence corroborates the role of M2-like TAM in promoting an aggressive phenotype in various malignancies, including PDAC [Citation137] and breast [Citation138] cancers. We here review a preliminary attempt to prove that mutant KRAS cancers may secrete EVs to transform pro-cancer TAM for conditioning the TME, and the possible mechanism is summarized in .
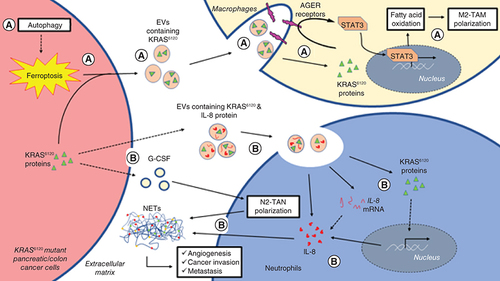
(A) Autophagy-dependent ferroptosis of KRASG12D mutant pancreatic cancer cells triggered the release of KRASG12D-containing EVs. (B) In recruited macrophages, ingested KRASG12D interacts with AGER receptors to promote M2-like TAM polarization. (C) KRASG12D colon cancer cells secrete EVs containing KRASG12D and IL-8 proteins as well as G-CSF into the TME. (D) Transfer of KRASG12D protein and IL-8 mRNA and protein elevates IL-8 expression in recruited neutrophils. (E) N2-like TAN polarization by G-CSF and upregulated IL-8 expression promote NET formation that facilitates angiogenesis, cancer migration, invasion and metastasis.
AGER: Advanced glycosylation end-product specific receptor; EVs: Extracellular vesicles; KRAS: Kirsten rat sarcoma virus; NET: Neutrophil extracellular trap; TAM: Tumor-associated macrophages; TAN: Tumor-associated neutrophils.
KRAS mutant cancer-derived EVs & TAN
Despite the advances in understanding of crosstalk between cancer and the immune system, the role of neutrophils in cancer development remains controversial. It was generally believed that neutrophils are anti-tumor in nature, where they could detect cancer cells followed by performing phagocytic removal, or cytotoxic elimination of the detected cells [Citation130]. However, a study discovered that neutrophils can be alternatively polarized into N2-like pro-tumor TAN when exposed to particular chemokines such as TGF-β in the TME [Citation139]. A different gene profile was detected in N2 TAN when compared with N1 anti-cancer neutrophils in a transcriptomics study [Citation140]. Those differentially expressed genes in N2 TAN were reported to play roles in cancer development by regulating secretion of growth-promoting chemokines [Citation141], immunosuppression [Citation142] and cancer invasion [Citation143].
Shang and colleagues discovered that colon cancer cells carrying KRASG12D mutation secrete elevated amounts of G-CSF [Citation105], which was found to promote neutrophil differentiation and polarization into N2 TAN [Citation144]. Enhanced neutrophil recruitment and N2 TAN polarization ultimately led to an increased neutrophil extracellular trap (NET) formation in a KRASG12D colon cancer mice model [Citation105]. NET is a net-like structure that is mainly composed of DNA-histone complexes and proteins that were released by activated neutrophils. Physiologically, NET was found to trap pathogens for prevention of infection. In cancers, findings suggest that NET facilitates angiogenesis [Citation145] and metastasis [Citation143]. In EVs secreted by KRASG12D mutant colon cancer cells, mutant KRAS protein and IL-8 mRNA in addition to other EV cargo were detected. Besides transferring mutant KRAS to promote cancer progression in recipient cells, these EVs stimulated IL-8 expression in recruited neutrophils, which ultimately led to increased NET formation [Citation105]. This was supported by other studies reporting that G-CSF [Citation146] and IL-8 [Citation147] are contributing to the formation of a cancer-associated NET structure. To date, study on the regulatory role of EVs in NET is scarce. A study demonstrated that EV-induced formation of NET regulates the prothrombotic phenotype in cancers [Citation146]. In this section, studies focused on mutant KRAS-associated EVs were reviewed, and the reported mechanism of its effects on N2 TAN recruitment and polarization as well as NET formation for cancer progression are summarized in .
KRAS mutant cancer-derived EVs & lymphocytes
T cells, B cells and natural killer cells are lymphocytes that are responsible for neutralizing invading foreign entities such as bacteria, viruses and toxins, as well as unhealthy host cells. Some lymphocytes, such as B cells, were reported to have a multifaced role in the cancer context. For instance, B cells may produce antibodies that targets tumor antigens [Citation148], and at the same time, tumor-infiltrating B cells were also found to produce cytokines such as IL-10 to promote tumor progression [Citation149] and suppress T-cell-mediated anti-tumor immunity [Citation150]. However, the role of T cells in anti-tumor immunity has been well documented [Citation151]. CD4+ T-helper cells promote cytotoxic T lymphocyte (CTL)-mediated anti-tumor immunity by facilitating CD8+ cytotoxic T-cell differentiation and proliferation [Citation152] or secrete anti-tumor cytokines such as INF-γ upon TH1 polarization [Citation153]. Notably, one study observed the conversion of CD4+ T cells into immune-suppressive Tregs when co-cultured with KRAS mutant human colon cancer cells [Citation154]. In another study, exosomes from NSCLC cells with mutated KRAS were observed to induce phenotypic change of CD4+ T-helper cells into immune-suppressive FOXP3+ Treg-like cells [Citation155]. Tumor-infiltrating Tregs were reported to promote malignancies by suppressing CTL-mediated anti-tumor immunity [Citation156]. Importantly, transfecting naive CD4+ T cells with mutant KRAS cDNA also triggered the phenotypic change, implying that mutant KRAS cancer-derived EVs might play a role in suppressing T-cell-mediated anti-tumor immunity by transferring the mutant KRAS gene to naive CD4+ T cells [Citation155].
Anti-cancer immune response is hugely dependent on CTL-mediated anti-tumor immunity [Citation157]. To date, there is a lack of study on B-cell-derived EVs in this context, but the effect of EVs secreted by KRAS mutant cancers on polarization of T cells into immune-suppressive Tregs were summarized in this section. This finding supports that EVs may play a role in facilitating the escape of cancers from host immunity and poses a challenge to cancer immunotherapy.
Conclusion
Important evidence for regulatory roles of mutant KRAS on biogenesis, cargo sorting, release and uptake of EVs was obtained from several studies focused on pancreatic and colon cancer models, which are the two cancer types with KRAS as the major form of mutations. Other relevant studies involving breast and lung cancers were also included in the discussion. Many of these studies employed isogenic cell lines bearing wild-type and mutated KRAS respectively to provide solid proof to support the oncogenic effect of KRAS mutation in EV biology. The most frequently mentioned one in this review would be Rab13. This GTPase regulates the production of the EV population (Rab13+, CD81+, CD63-) that is unique to KRAS mutation, and transferring of its mRNA via EVs may induce radiotherapy resistance in recipient cells. Many oncogenic molecules including mRNA, miRNA and proteins were found to be sorted specifically in the EVs secreted from KRAS mutant cancers including the mutated form of KRAS protein that could expand a mutant KRAS-associated cancer phenotype in neighboring or distant cells, and Ago2 as a component of the RNA-induced silencing complex that may facilitate the sorting of RNA species in KRAS-associated EVs. Intriguingly, mutant KRAS was found to selectively pack miRNA in EVs for removal of an undesired tumor suppressor (e.g., miR-100-5p; metastasis suppressor), whereas oncogenic miRNAs (e.g., miR-10b; EMT activator) are retained in the cells for cancer progression. Other functions of mutant KRAS-specific EV cargo include reprogramming energy metabolism (GLUT1), activating EGFR signaling (AREG) and inducing insensitivity to therapeutic agents (survivin) following uptake of the EV components in recipient cells.
Future perspective
The studies presented inspire several potential therapeutic applications of mutant KRAS-associated EVs. AREG carrying EVs could be the prognostic marker for poor response of patients to the treatments targeting EGFR, whereas increased survivin in EVs released from KRAS mutant cancers potentiates its use as a clinical indicator for chemoresistance. However, studies involving different cancer types demonstrated that KRAS mutation enhances uptake of EVs into cancer cells via micropinocytosis regardless of the origin of these vesicles, suggesting the great potential of EVs in the development of targeted therapy for KRAS mutant cancers. Cell–cell communication mediated by EVs includes that from the TME, such as macrophages and neutrophils. A few but significant findings evinced the effects of mutant KRAS-associated EVs in promoting transformation of the pro-tumor immune cells TAM and TAN in shaping the TME. More studies on the role of ferroptosis in the production of KRAS-associated EVs and the effects of these EVs in NET formation should warrant an in-depth understanding for mutant KRAS-mediated malignancies.
Extracellular vesicles (EVs) are small, cargo-bearing, membranous nanovesicles secreted by all types of cells for intercellular communication. EVs are different in size, membrane composition and vesicular content, which can be categorized into three main groups: small EVs, microvesicles and apoptotic bodies. Most of the cancer-derived EVs are small EVs and microvesicles, whereas the endosomal network is known to regulate secretion of small EVs (or more commonly known as exosomes).
The KRAS (Kirsten rat sarcoma virus) gene is one of the members of the Ras oncogene family that encodes for the KRAS protein that belongs to the RAS protein subfamily. Studies revealed that endosomal network-mediated trafficking, localization and membrane association of different RAS isoforms determine their signaling specificity. However, the crosstalk between KRAS and small EVs was scarcely discussed despite its endocytic association.
This review summarized the important evidence for regulatory roles of mutant KRAS on biogenesis, cargo sorting, release and uptake of small EVs obtained from several studies focused on pancreatic and colon cancer models, which are the two cancer types with KRAS as the major form of mutations. Related studies with a focus on breast and lung cancers were included in the discussion.
Tetraspanin protein CD63 is one of the key markers of small EVs released from various types of cells. However, colorectal cancer cells bearing mutant KRAS secrete a distinct population of small EVs with the absence of CD63. Rab13 (a GTPase) was found to regulate the production of the EV population (Rab13+, CD81+, CD63-) that is unique to KRAS mutation, and transferring of its mRNA via EVs may induce radiotherapy resistance in recipient cells.
Enrichment of mutant KRAS protein was observed in the EVs derived from mutant KRAS cells. In addition, many other oncogenic molecules including mRNA, miRNA and proteins were also found to be sorted specifically in the EVs secreted from KRAS mutant cancers. The EV cargo includes argonaute 2 (Ago2), which is an essential component of the RNA-induced silencing complex sorting of RNA species in EVs; amphiregulin (AREG), which is a ligand of EGFR regulating survival and invasion of cancer cells; Rab13, which is a GTPase regulating trafficking of oncogenic proteins; and GLUT1, which is a glucose transporter promoting glucose uptake for cancer cell growth. EV-mediated transferring of these molecules could possibly contribute to the expansion of a mutant KRAS-associated cancer phenotype in neighboring or distant cells.
Besides oncogenic mRNA (e.g., Rab13, FAT1 and RTN4), mutant KRAS was also found to selectively pack miRNA in EVs for removal of undesired tumor suppressor (e.g., miR-100-5p; metastasis suppressor), whereas oncogenic miRNAs (e.g., miR-10b; metastasis activator) are retained in the cells for cancer progression. Intriguingly, increased 3′ trimming was observed in the miRNAs enriched in KRAS mutant cancer-derived EVs. This may be a new form of post-transcriptional modification regulating miRNA sorting in cancer-derived EVs.
Mutant KRAS promotes uptake of circulating EVs through intensified micropinocytosis, and cancer cells with KRAS mutation tend to uptake more EVs regardless of their origin. Based on this property, EVs were engineered (iExosomes) to carry siRNA to target KRASG12D in pancreatic cancers. Significant uptake of iExsomes by mutant KRAS cells was observed, and the use of these engineered EVs in treating pancreatic cancers is in a phase I clinical trial (NCT03608631).
Cancer cells with primary resistance toward therapy may exploit EVs as vehicles to deliver signaling molecules that promote acquired resistance phenotype in recipient cells. Acyl-CoA synthase long-chain family 4 (ACSL4), zinc finger E-box binding homeobox (ZEB1) and survivin may play important roles in regulating drug responses of cancer cells with uptake of these protein/mRNA-enriched EVs secreted by mutant KRAS cancers. Many studies show that increased survivin in EVs released from KRAS mutant cancers potentiate its use as a clinical indicator for chemoresistance.
Cancer cells secrete a variety of signaling molecules to prime the tumor microenvironment (TME) for transformation of immune cells to cancer-promoting types of cells. Uptake of KRASG12D-enriched EVs enhances recruitment of macrophages in the TME through an advanced glycosylation end-product specific receptor (AGER)-dependent mechanism, whereas the polarization of macrophages into a pro-tumor M2-like tumor-associated macrophage was also found to be mediated by KRASG12D mutation. However, EVs secreted by KRASG12D mutant colon cancer cells stimulate IL-8 expression in tumor-associated neutrophils, which leads to increased formation of the neutrophil extracellular trap, which is a net-like structure found to facilitate angiogenesis and metastasis in cancers.
Exosomes from lung cancer cells with mutated KRAS were observed to transform CD4+ T-helper cells into immune-suppressive FOXP3+ Treg-like cells, implying that EVs may play a role in facilitating the escape of cancers from host immunity and pose a challenge to cancer immunotherapy.
An overview of the oncogenic role of KRAS focusing on its correlation with cancer-associated EVs should provide important clues for disease prognosis and inspire novel therapeutic approaches for treating KRAS mutant cancers.
Supplemental Text 1
Download MS Word (154 KB)Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at:www.tandfonline.com/doi/full/10.2217/epi-2016-0184
Financial & competing interests disclosure
This work was supported by Fundamental Research Grant Scheme grants (FRGS/1/2014/SKK01/MUSM/03/2) from the Ministry of Higher Education Malaysia. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Munoz-Maldonado C , ZimmerY , MedovaM. A comparative analysis of individual RAS mutations in cancer biology. Front. Oncol.9, 1088 (2019).
- Cefalì M , EpistolioS , PalmarocchiMC , FrattiniM , DeDosso S. Research progress on KRAS mutations in colorectal cancer. J. Cancer Metastasis Treat.7, 26 (2021).
- Pantsar T . The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J.18, 189–198 (2020).
- Waters AM , DerCJ. KRAS: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb. Perspect. Med.8(9), (2018).
- Nussinov R , TsaiCJ , JangH. Independent and core pathways in oncogenic KRAS signaling. Expert Rev. Proteomics13(8), 711–716 (2016).
- Khan AQ , KuttikrishnanS , SiveenKSet al. RAS-mediated oncogenic signaling pathways in human malignancies. Semin. Cancer Biol.54, 1–13 (2019).
- Peng J , LvJ , PengJ. KRAS mutation is predictive for poor prognosis in rectal cancer patients with neoadjuvant chemoradiotherapy: a systemic review and meta-analysis. Int. J. Colorectal Dis.36(8), 1781–1790 (2021).
- Chen F , AlphonseMP , LiuY , LiuQ. Targeting mutant KRAS for anticancer therapy. Curr. Top. Med. Chem.19(23), 2098–2113 (2019).
- Ostrem JM , PetersU , SosML , WellsJA , ShokatKM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature503(7477), 548–551 (2013).
- Lanman BA , AllenJR , AllenJGet al. Discovery of a covalent inhibitor of KRAS(G12C) (AMG 510) for the treatment of solid tumors. J. Med. Chem.63(1), 52–65 (2020).
- Nakajima EC , DreznerN , LiXet al. FDA approval summary: sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin. Cancer Res.28(8), 1482–1486 (2021).
- Hong DS , FakihMG , StricklerJHet al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med.383(13), 1207–1217 (2020).
- Skoulidis F , LiBT , DyGKet al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med.384(25), 2371–2381 (2021).
- Zhang SS , NagasakaM. Spotlight on sotorasib (AMG 510) for KRAS (G12C) positive non-small cell lung cancer. Lung Cancer (Auckl).12, 115–122 (2021).
- Canon J , RexK , SaikiAYet al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature575(7781), 217–223 (2019).
- Muzumdar MD , ChenPY , DoransKJet al. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun.8(1), 1090 (2017).
- Adachi Y , ItoK , HayashiYet al. Epithelial-to-mesenchymal transition is a cause of both intrinsic and acquired resistance to KRAS G12C inhibitor in KRAS G12C-mutant non-small cell lung cancer. Clin. Cancer Res.26(22), 5962–5973 (2020).
- Awad MM , LiuS , RybkinIIet al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med.384(25), 2382–2393 (2021).
- Zafra MP , ParsonsMJ , KimJet al. An in vivo Kras allelic series reveals distinct phenotypes of common oncogenic variants. Cancer Discov.10(11), 1654–1671 (2020).
- Jia Y , JiangT , LiXet al. Characterization of distinct types of KRAS mutation and its impact on first-line platinum-based chemotherapy in Chinese patients with advanced non-small cell lung cancer. Oncol. Lett.14(6), 6525–6532 (2017).
- Misale S , YaegerR , HoborSet al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature486(7404), 532–536 (2012).
- Molina-Arcas M , MooreC , RanaSet al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci. Transl. Med.11(510), eaaw7999 (2019).
- Prior IA , HancockJF. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol.23(2), 145–153 (2012).
- Teske C , SchweitzerC , PalamidessiAet al. Modulation of RAB5A early endosome trafficking in response to KRas mediated macropinocytic fluxes in pancreatic cancer cells. Biochem. Biophys. Res. Commun.493(1), 528–533 (2017).
- Tomas A , FutterCE , EdenER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol.24(1), 26–34 (2014).
- Sandilands E , FrameMC. Endosomal trafficking of Src tyrosine kinase. Trends Cell Biol.18(7), 322–329 (2008).
- Belli S , EspositoD , ServettoA , PesapaneA , FormisanoL , BiancoR. c-Src and EGFR inhibition in molecular cancer therapy: what else can we improve?Cancers (Basel)12(6), (2020).
- Saturno G , LopesF , Niculescu-DuvazIet al. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann. Oncol.32(2), 269–278 (2021).
- Wang SE . Extracellular vesicles and metastasis. Cold Spring Harb. Perspect. Med.10(7), a037275 (2020).
- Gurung S , PerocheauD , TouramanidouL , BaruteauJ. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun. Signal.19(1), 47 (2021).
- Hikita T , KuwaharaA , WatanabeR , MiyataM , OneyamaC. Src in endosomal membranes promotes exosome secretion and tumor progression. Sci. Rep.9(1), 3265 (2019).
- Jouida A , MccarthyC , FabreA , KeaneMP. Exosomes: a new perspective in EGFR-mutated lung cancer. Cancer Metastasis Rev.40(2), 589–601 (2021).
- Kalra H , DrummenGP , MathivananS. Focus on extracellular vesicles: introducing the next small big thing. Int. J. Mol. Sci.17(2), 170 (2016).
- Willms E , CabanasC , MagerI , WoodMJA , VaderP. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Front. Immunol.9, 738 (2018).
- Willms E , JohanssonHJ , MagerIet al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep.6, 22519 (2016).
- Hanayama R . Emerging roles of extracellular vesicles in physiology and disease. J. Biochem.169(2), 135–138 (2021).
- King HW , MichaelMZ , GleadleJM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer12, 421 (2012).
- Wang T , GilkesDM , TakanoNet al. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. U. S. A.111(31), E3234–3242 (2014).
- Becker A , ThakurBK , WeissJM , KimHS , PeinadoH , LydenD. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell30(6), 836–848 (2016).
- Maacha S , BhatAA , JimenezLet al. Extracellular vesicles-mediated intercellular communication: roles in the tumor microenvironment and anti-cancer drug resistance. Mol. Cancer18(1), 55 (2019).
- Akers JC , GondaD , KimR , CarterBS , ChenCC. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol.113(1), 1–11 (2013).
- Piper RC , KatzmannDJ. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol.23, 519–547 (2007).
- Lu A , TebarF , Alvarez-MoyaBet al. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol.184(6), 863–879 (2009).
- Tebar F , ChaveroA , AgellNet al. Pleiotropic roles of calmodulin in the regulation of KRas and Rac1 GTPases: functional diversity in health and disease. Int. J. Mol. Sci.21(10), (2020).
- Hinger SA , AbnerJJ , FranklinJL , JeppesenDK , CoffeyRJ , PattonJG. Rab13 regulates sEV secretion in mutant KRAS colorectal cancer cells. Sci. Rep.10(1), 15804 (2020).
- Ettelaie C , CollierME , MaraveyasA , EttelaieR. Characterization of physical properties of tissue factor-containing microvesicles and a comparison of ultracentrifuge-based recovery procedures. J. Extracell. Vesicles3, 10.3402/jev.v3.23592 (2014).
- Petanidis S , DomvriK , PorpodisKet al. Inhibition of kras-derived exosomes downregulates immunosuppressive BACH2/GATA-3 expression via RIP-3 dependent necroptosis and miR-146/miR-210 modulation. Biomed. Pharmacother.122, 109461 (2020).
- Nair SS , KumarR. Chromatin remodeling in cancer: a gateway to regulate gene transcription. Mol. Oncol.6(6), 611–619 (2012).
- Zahra K , DeyT , Ashish , MishraSP , PandeyU. Pyruvate kinase M2 and cancer: the role of PKM2 in promoting tumorigenesis. Front. Oncol.10, 159 (2020).
- Sun Q , ChenX , MaJet al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA108(10), 4129–4134 (2011).
- Tordonato C , MarziMJ , GiangrecoGet al. miR-146 connects stem cell identity with metabolism and pharmacological resistance in breast cancer. J. Cell Biol.220(5), e202009053 (2021).
- Tsuchiya S , FujiwaraT , SatoFet al. MicroRNA-210 regulates cancer cell proliferation through targeting fibroblast growth factor receptor-like 1 (FGFRL1). J. Biol. Chem.286(1), 420–428 (2011).
- Giannakakis A , SandaltzopoulosR , GreshockJet al. miR-210 links hypoxia with cell cycle regulation and is deleted in human epithelial ovarian cancer. Cancer Biol. Ther.7(2), 255–264 (2008).
- Anand S , SamuelM , KumarS , MathivananS. Ticket to a bubble ride: cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom.1867(12), 140203 (2019).
- Chang WH , CerioneRA , AntonyakMA. Extracellular vesicles and their roles in cancer progression. Methods Mol. Biol.2174, 143–170 (2021).
- Segev N . Coordination of intracellular transport steps by GTPases. Semin. Cell Dev. Biol.22(1), 33–38 (2011).
- Demory Beckler M , HigginbothamJN , FranklinJLet al. Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Mol. Cell. Proteomics12(2), 343–355 (2013).
- Putz U , HowittJ , DoanAet al. The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci. Signal.5(243), ra70 (2012).
- Sasaki AT , CarracedoA , LocasaleJWet al. Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal.4(163), ra13 (2011).
- Wang HW , NolandC , SiridechadilokBet al. Structural insights into RNA processing by the human RISC-loading complex. Nat. Struct. Mol. Biol.16(11), 1148–1153 (2009).
- Tien JC , ChughS , GoodrumAEet al. AGO2 promotes tumor progression in KRAS-driven mouse models of non-small cell lung cancer. Proc. Natl. Acad. Sci. USA.118(20), e2026104118 (2021).
- Yuan J , SuZ , GuWet al. MiR-19b and miR-20a suppress apoptosis, promote proliferation and induce tumorigenicity of multiple myeloma cells by targeting PTEN. Cancer Biomark.24(3), 279–289 (2019).
- Zhang H , WangY , DouJet al. Acetylation of AGO2 promotes cancer progression by increasing oncogenic miR-19b biogenesis. Oncogene38(9), 1410–1431 (2019).
- McKenzie AJ , HoshinoD , HongNHet al. KRAS-MEK signaling controls Ago2 sorting into exosomes. Cell Rep.15(5), 978–987 (2016).
- Prud’homme GJ , GlinkaY , LichnerZ , YousefGM. Neuropilin-1 is a receptor for extracellular miRNA and AGO2/miRNA complexes and mediates the internalization of miRNAs that modulate cell function. Oncotarget7(42), 68057–68071 (2016).
- Berasain C , AvilaMA. Amphiregulin. Semin. Cell Dev. Biol.28, 31–41 (2014).
- Taverna S , PucciM , GiallombardoMet al. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep.7(1), 3170 (2017).
- Baillo A , GirouxC , EthierSP. Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J. Cell. Physiol.226(10), 2691–2701 (2011).
- Wang L , WangL , ZhangHet al. AREG mediates the epithelialmesenchymal transition in pancreatic cancer cells via the EGFR/ERK/NFkappaB signalling pathway. Oncol. Rep.43(5), 1558–1568 (2020).
- Higginbotham JN , DemoryBeckler M , GephartJDet al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol.21(9), 779–786 (2011).
- Ohchi T , AkagiY , KinugasaTet al. Amphiregulin is a prognostic factor in colorectal cancer. Anticancer Res.32(6), 2315–2321 (2012).
- Ioannou MS , McPhersonPS. Regulation of cancer cell behavior by the small GTPase Rab13. J. Biol. Chem.291(19), 9929–9937 (2016).
- Gu CJ , XieF , ZhangBet al. High glucose promotes epithelial-mesenchymal transition of uterus endometrial cancer cells by increasing ER/GLUT4-mediated VEGF secretion. Cell Physiol. Biochem.50(2), 706–720 (2018).
- Wu C , AgrawalS , VasanjiAet al. Rab13-dependent trafficking of RhoA is required for directional migration and angiogenesis. J. Biol. Chem.286(26), 23511–23520 (2011).
- Sakane A , AbdallahAA , NakanoKet al. Rab13 small G protein and junctional Rab13-binding protein (JRAB) orchestrate actin cytoskeletal organization during epithelial junctional development. J. Biol. Chem.287(51), 42455–42468 (2012).
- Sahgal P , AlankoJ , IchaJet al. GGA2 and RAB13 promote activity-dependent beta1-integrin recycling. J. Cell Sci.132(11), jcs233387 (2019).
- Gou D , RamchandranR , PengXet al. miR-210 has an antiapoptotic effect in pulmonary artery smooth muscle cells during hypoxia. Am. J. Physiol. Lung Cell. Mol. Physiol.303(8), L682–691 (2012).
- Deberardinis RJ , LumJJ , HatzivassiliouG , ThompsonCB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab.7(1), 11–20 (2008).
- Tran Q , LeeH , KimCet al. Revisiting the Warburg effect: diet-based strategies for cancer prevention. Biomed. Res. Int.2020, 8105735 (2020).
- Brown RS , WahlRL. Overexpression of Glut-1 glucose transporter in human breast cancer. An immunohistochemical study. Cancer72(10), 2979–2985 (1993).
- Semaan A , MunkarahAR , ArabiHet al. Expression of GLUT-1 in epithelial ovarian carcinoma: correlation with tumor cell proliferation, angiogenesis, survival and ability to predict optimal cytoreduction. Gynecol. Oncol.121(1), 181–186 (2011).
- Wu Q , Ba-AlawiW , DebloisGet al. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun.11(1), 4205 (2020).
- Chen X , WangL , WangH. LINC01638 lncRNA promotes cancer cell proliferation in hepatocellular carcinoma by increasing cancer cell glucose uptake. Oncol. Lett.18(4), 3811–3816 (2019).
- Gonzalez-Menendez P , HeviaD , Alonso-AriasRet al. GLUT1 protects prostate cancer cells from glucose deprivation-induced oxidative stress. Redox Biol.17, 112–127 (2018).
- Basturk O , SinghR , KaygusuzEet al. GLUT-1 expression in pancreatic neoplasia: implications in pathogenesis, diagnosis, and prognosis. Pancreas40(2), 187–192 (2011).
- Ying H , KimmelmanAC , LyssiotisCAet al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell149(3), 656–670 (2012).
- Zhang Q , JeppesenDK , HigginbothamJNet al. Mutant KRAS exosomes alter the metabolic state of recipient colonic epithelial cells. Cell. Mol. Gastroenterol. Hepatol.5(4), 627–629.e626 (2018).
- Hinger SA , ChaDJ , FranklinJLet al. Diverse long RNAs are differentially sorted into extracellular vesicles secreted by colorectal cancer cells. Cell Rep.25(3), 715–725.e714 (2018).
- Dou Y , ChaDJ , FranklinJLet al. Circular RNAs are down-regulated in KRAS mutant colon cancer cells and can be transferred to exosomes. Sci. Rep.6, 37982 (2016).
- Pileri P , CampagnoliS , GrandiAet al. FAT1: a potential target for monoclonal antibody therapy in colon cancer. Br. J. Cancer115(1), 40–51 (2016).
- Kang MH , JeongGS , SmootDTet al. Verteporfin inhibits gastric cancer cell growth by suppressing adhesion molecule FAT1. Oncotarget8(58), 98887–98897 (2017).
- Pathak GP , ShahR , KennedyBEet al. RTN4 knockdown dysregulates the AKT pathway, destabilizes the cytoskeleton, and enhances paclitaxel-induced cytotoxicity in cancers. Mol. Ther.26(8), 2019–2033 (2018).
- O’Brien J , HayderH , ZayedY , PengC. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne)9, 402 (2018).
- Mills J , CapeceM , CocucciE , TessariA , PalmieriD. Cancer-derived extracellular vesicle-associated microRNAs in intercellular communication: one cell’s trash is another cell’s treasure. Int. J. Mol. Sci.20(24), 6109 (2019).
- Ostenfeld MS , JeppesenDK , LaurbergJRet al. Cellular disposal of miR23b by RAB27-dependent exosome release is linked to acquisition of metastatic properties. Cancer Res.74(20), 5758–5771 (2014).
- Farahani M , RubbiC , LiuL , SlupskyJR , KalakondaN. CLL exosomes modulate the transcriptome and behaviour of recipient stromal cells and are selectively enriched in miR-202-3p. PLoS One10(10), e0141429 (2015).
- Vu LT , PengB , ZhangDXet al. Tumor-secreted extracellular vesicles promote the activation of cancer-associated fibroblasts via the transfer of microRNA-125b. J. Extracell. Vesicles8(1), 1599680 (2019).
- Cha DJ , FranklinJL , DouYet al. KRAS-dependent sorting of miRNA to exosomes. Elife4, e07197 (2015).
- Ye Y , LiSL , WangJJ. miR-100-5p downregulates mTOR to suppress the proliferation, migration, and invasion of prostate cancer cells. Front. Oncol.10, 578948 (2020).
- Zhang H , YangK , RenTet al. miR-100-5p inhibits malignant behavior of chordoma cells by targeting IGF1R. Cancer Manag. Res.12, 4129–4137 (2020).
- Sheedy P , MedarovaZ. The fundamental role of miR-10b in metastatic cancer. Am. J. Cancer Res.8(9), 1674–1688 (2018).
- Yanez-Mo M , SiljanderPR , AndreuZet al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles4, 27066 (2015).
- Juan T , FurthauerM. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin. Cell Dev. Biol.74, 66–77 (2018).
- Jackson KK , PowellRR , BruceTF , MarcusRK. Rapid isolation of extracellular vesicles from diverse biofluid matrices via capillary-channeled polymer fiber solid-phase extraction micropipette tips. Analyst146(13), 4314–4325 (2021).
- Shang A , GuC , ZhouCet al. Exosomal KRAS mutation promotes the formation of tumor-associated neutrophil extracellular traps and causes deterioration of colorectal cancer by inducing IL-8 expression. Cell Commun. Signal.18(1), 52 (2020).
- Mulcahy LA , PinkRC , CarterDR. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles3, 10.3402/jev.v3.24641 (2014).
- Nakase I , KobayashiNB , Takatani-NakaseT , YoshidaT. Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci. Rep.5, 10300 (2015).
- Jiang W , LiH , LiuXet al. Precise and efficient silencing of mutant Kras(G12D) by CRISPR-CasRx controls pancreatic cancer progression. Theranostics10(25), 11507–11519 (2020).
- Kamerkar S , LebleuVS , SugimotoHet al. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature546(7659), 498–503 (2017).
- McAndrews KM , XiaoF , ChronopoulosA , LebleuVS , KugeratskiFG , KalluriR. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic Kras(G12D) in pancreatic cancer. Life Sci. Alliance4(9), e202000875 (2021).
- Dagogo-Jack I , ShawAT. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol.15(2), 81–94 (2018).
- Eng SK , ImtiazIR , GohBH , MingLC , LimYC , LeeWL. Does KRAS play a role in the regulation of colon cancer cells-derived exosomes?Biology (Basel)10(1), 58 (2021).
- Lobb RJ , Van AmerongenR , WiegmansA , HamS , LarsenJE , MollerA. Exosomes derived from mesenchymal non-small cell lung cancer cells promote chemoresistance. Int. J. Cancer141(3), 614–620 (2017).
- Drapela S , BouchalJ , JollyMK , CuligZ , SoucekK. ZEB1: a critical regulator of cell plasticity, DNA damage response, and therapy resistance. Front. Mol. Biosci.7, 36 (2020).
- Cui Y , QinL , TianDet al. ZEB1 promotes chemoresistance to cisplatin in ovarian cancer cells by suppressing SLC3A2. Chemotherapy63(5), 262–271 (2018).
- Orellana-Serradell O , HerreraD , CastellonEA , ContrerasHR. The transcription factor ZEB1 promotes chemoresistance in prostate cancer cell lines. Asian J. Androl.21(5), 460–467 (2019).
- Zhang X , ZhangZ , ZhangQet al. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis.9(2), 57 (2018).
- Lennerz JK , StenzingerA. Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist20(4), e8–e9 (2015).
- Chang WH , NguyenTT , HsuCHet al. KRAS-dependent cancer cells promote survival by producing exosomes enriched in Survivin. Cancer Lett.517, 66–77 (2021).
- Garg H , SuriP , GuptaJC , TalwarGP , DubeyS. Survivin: a unique target for tumor therapy. Cancer Cell Int.16, 49 (2016).
- Zhang Y , YanH , LiR , GuoY , ZhengR. High expression of Survivin predicts poor prognosis in cervical squamous cell carcinoma treated with paclitaxel and carboplatin. Medicine (Baltimore)98(20), e15607 (2019).
- Zhang J , ZhuZ , SunZ , SunX , WangZ , XuH. Survivin gene expression increases gastric cancer cell lymphatic metastasis by upregulating vascular endothelial growth factor-C expression levels. Mol. Med. Rep.9(2), 600–606 (2014).
- Dohi T , BeltramiE , WallNR , PlesciaJ , AltieriDC. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J. Clin. Invest.114(8), 1117–1127 (2004).
- Or YY , ChowAK , NgLet al. Survivin depletion inhibits tumor growth and enhances chemosensitivity in hepatocellular carcinoma. Mol. Med. Rep.10(4), 2025–2030 (2014).
- Rauch A , CarlstedtA , EmmerichCet al. Survivin antagonizes chemotherapy-induced cell death of colorectal cancer cells. Oncotarget9(45), 27835–27850 (2018).
- Kreger BT , JohansenER , CerioneRA , AntonyakMA. The enrichment of survivin in exosomes from breast cancer cells treated with paclitaxel promotes cell survival and chemoresistance. Cancers (Basel)8(12), 111 (2016).
- Kim HS , KimSC , KimSJet al. Identification of a radiosensitivity signature using integrative metaanalysis of published microarray data for NCI-60 cancer cells. BMC Genomics13, 348 (2012).
- Baghban R , RoshangarL , Jahanban-EsfahlanRet al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal.18(1), 59 (2020).
- Zhou J , TangZ , GaoS , LiC , FengY , ZhouX. Tumor-associated macrophages: recent insights and therapies. Front. Oncol.10, 188 (2020).
- Shaul ME , FridlenderZG. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol.16(10), 601–620 (2019).
- Van Dalen FJ , Van StevendaalM , FennemannFL , VerdoesM , IlinaO. Molecular repolarisation of tumour-associated macrophages. Molecules24(1), (2018).
- Jayasingam SD , CitartanM , ThangTH , MatZin AA , AngKC , Ch’ngES. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front. Oncol.9, 1512 (2019).
- Dai E , HanL , LiuJet al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy16(11), 2069–2083 (2020).
- Li J , CaoF , YinHLet al. Ferroptosis: past, present and future. Cell Death Dis.11(2), 88 (2020).
- Ito F , KatoK , YanatoriI , MuroharaT , ToyokuniS. Ferroptosis-dependent extracellular vesicles from macrophage contribute to asbestos-induced mesothelial carcinogenesis through loading ferritin. Redox Biol.47, 102174 (2021).
- Kierdorf K , FritzG. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol.94(1), 55–68 (2013).
- Hu H , HangJJ , HanT , ZhuoM , JiaoF , WangLW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol.37(7), 8657–8664 (2016).
- Klingen TA , ChenY , AasH , WikE , AkslenLA. Tumor-associated macrophages are strongly related to vascular invasion, non-luminal subtypes, and interval breast cancer. Hum. Pathol.69, 72–80 (2017).
- Sionov RV , FridlenderZG , GranotZ. The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron.8(3), 125–158 (2015).
- Shaul ME , LevyL , SunJet al. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: a transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology5(11), e1232221 (2016).
- Mishalian I , BayuhR , EruslanovEet al. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17 – a new mechanism of impaired antitumor immunity. Int. J. Cancer135(5), 1178–1186 (2014).
- Munder M , SchneiderH , LucknerCet al. Suppression of T-cell functions by human granulocyte arginase. Blood108(5), 1627–1634 (2006).
- Najmeh S , Cools-LartigueJ , RayesRFet al. Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int. J. Cancer140(10), 2321–2330 (2017).
- Takeshima T , PopLM , LaineA , IyengarP , VitettaES , HannanR. Key role for neutrophils in radiation-induced antitumor immune responses: potentiation with G-CSF. Proc. Natl. Acad. Sci. USA.113(40), 11300–11305 (2016).
- Jung HS , GuJ , KimJE , NamY , SongJW , KimHK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS One14(4), e0216055 (2019).
- Leal AC , MizuriniDM , GomesTet al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci. Rep.7(1), 6438 (2017).
- Alfaro C , TeijeiraA , OnateCet al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin. Cancer Res.22(15), 3924–3936 (2016).
- Gilbert AE , KaragiannisP , DodevTet al. Monitoring the systemic human memory B cell compartment of melanoma patients for anti-tumor IgG antibodies. PLoS One6(4), e19330 (2011).
- Ammirante M , LuoJL , GrivennikovS , NedospasovS , KarinM. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature464(7286), 302–305 (2010).
- Olkhanud PB , DamdinsurenB , BodogaiMet al. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res.71(10), 3505–3515 (2011).
- Tay RE , RichardsonEK , TohHC. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther.28(1-2), 5–17 (2021).
- Laidlaw BJ , CraftJE , KaechSM. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol.16(2), 102–111 (2016).
- Nonaka K , SaioM , UmemuraNet al. Th1 polarization in the tumor microenvironment upregulates the myeloid-derived suppressor-like function of macrophages. Cell. Immunol.369, 104437 (2021).
- Zdanov S , MandapathilM , AbuEid Ret al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol. Res.4(4), 354–365 (2016).
- Kalvala A , WalletP , YangLet al. Phenotypic switching of naive T cells to immune-suppressive Treg-like cells by mutant KRAS. J. Clin. Med.8(10), 1726 (2019).
- Nishikawa H , SakaguchiS. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol.27, 1–7 (2014).
- Raskov H , OrhanA , ChristensenJP , GogenurI. Cytotoxic CD8(+) T cells in cancer and cancer immunotherapy. Br. J. Cancer124(2), 359–367 (2021).