Abstract
The technique of solid-state NMR, as applied to pharmaceutical analysis, has evolved from a sparingly used technique into an integrated, information-rich technique that uses a variety of sophisticated 1D and 2D experiments. In this article, several key developments in the field are reviewed, highlighting the increasing sophistication of the analyses being utilized and the detailed results obtained, in addition to future directions and developments.
The solid-state structure of a pharmaceutical product can influence its properties, manufacturability and stability Citation[1]. Since these characteristics can impact product performance and patient safety, regulatory agencies require control over solid-state properties in many situations. In the quality-by-design environment that is emphasized in modern pharmaceutical development, increased understanding of products gained by solid-state analysis is very useful when developing an active pharmaceutical ingredient (API) or a formulation process for solid-dose products, including oral, inhaled and injected products Citation[1,2]. Solid-state NMR (SSNMR) spectroscopy of pharmaceutical materials has evolved from a sparingly used technique into an important component of pharmaceutical solid-state analysis activities over the past decade, largely owing to its unique capabilities. Furthermore, continued innovation and application are occurring in the field that are likely to continue this trend. In this work, a summary of the path to the current status of applied pharmaceutical SSNMR is provided. The latest developments in the field are summarized, focusing on the utility of the technique during the development of a drug from the candidate stage through to a marketed, approved product. The dominant use of SSNMR in the pharmaceutical industry occurs during development, particularly during process development for API and drug products for human clinical trials. However, several examples are also given of the utility of SSNMR during the discovery stage, prior to candidate selection. Finally, future directions and developments in the field of pharmaceutical SSNMR are considered.
Solid-state NMR analysis of the 13C nucleus at natural abundance, using the crosspolarization with magic-angle spinning (CP-MAS) method, has been traditionally used to provide ‘fingerprint’ characterization of solid crystalline drugs and is widely used for this purpose today Citation[3–5]. In this role, SSNMR serves primarily as an orthogonal technique to the more routine and widely available techniques of vibrational spectroscopy (e.g., mid-infrared and Raman spectroscopy) and x-ray powder diffraction (XRPD). This is a very useful role, particularly since 13C SSNMR often offers the highest resolution of any technique between solid forms of interest, such as polymorphs, hydrates and solvates. However, the limited use of SSNMR as an orthogonal technique has restricted its overall acceptance, due to cost–benefit concerns, as an SSNMR instrument currently has a high installation and operational cost and lower throughput. Typically, a mid-field 400-MHz SSNMR system costs five- to ten-times more than an infrared or Raman spectrometer and two- to three-times more than an XRPD instrument. It is the additional capabilities of SSNMR that ultimately help justify its cost, including its ability to analyze complex solid-phase mixtures such as drug products and the detailed information content accessible from a wide range of nuclei using a variety of specialized 1D and 2D experiments. In these roles, it is superior to all other solid-state analysis techniques currently available that operate on powdered samples. Most of these advanced capabilities can be implemented on lower-cost 400- and 500- MHz systems, although higher fields are required in some cases, as discussed below. The flexibility of SSNMR allows a range of experiments to be sequentially performed on the same sample to extract out a variety of information beyond the conventional 1D spectrum. It also offers high spectral information content and benefits from extensive interpretability of the data using the vast, well-established empirical knowledge of NMR chemical shifts from solution-state NMR, as well as state-of-the-art theoretical calculations of NMR chemical shielding and quadrupolar interactions Citation[6–9]. This latter feature allows most SSNMR spectra to be ‘assigned’ so that each resonance is linked to an individual atom in the chemical structure of the drug molecule, as with solution-state NMR spectra of small- to mid-size molecules. Even in cases where resonances are overlapped in both large and small molecules, useful group assignments of SSNMR spectra can be made.
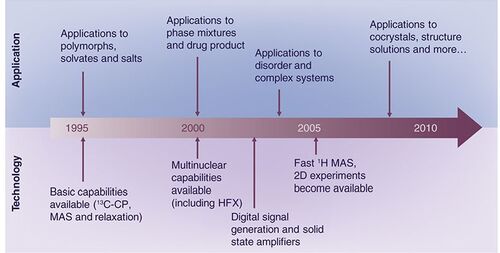
The evolution of pharmaceutical SSNMR over the past 15 years, from its use as a fingerprint technique to its current status, is sketched in a timeline shown in . necessarily focuses on several key technologies that led to significantly increased pharmaceutical applications and does not attempt to include every advance made in the field over this time period. Over the time span plotted in , there has also been a concurrent trend towards higher static magnetic fields. Generally, 1H frequencies of instruments used for pharmaceutical SSNMR were in the 300–400 MHz range in the mid-1990s, but as of 2010 are more typically in the range of 500 to 700 MHz (and even beyond).
Developments between 1995 & 2005
Between 1995 and 2005, the traditional ‘fingerprint’ role was expanded with applications to a variety of different drug systems that displayed polymorphism, hydration and solvation as well as different salt formation behavior Citation[10–18]. A feature of much of this work was spectral interpretation via careful interpretation of subtle spectral effects, such as hydrogen bonding and the use of more advanced 1D SSNMR experiments for spectral editing to enhance and confirm spectral assignments. This interpretation ultimately allows for useful structural conclusions. Several approaches were frequently employed in these studies, ranging from spectral editing to highlighting quaternary and methyl carbons to the investigation of solid-state molecular dynamics via 13C spectral lineshapes and 13C and 1H spin-lattice (T1) and other relaxation times. The techniques used in these studies were well established by the early 1990s. The assessment of phase purity, an important routine use of SSNMR, was also demonstrated in many of these examples. SSNMR compares favorably to the indexing of XRPD data for phase purity assessment, as phase impurities are usually immediately detected in an assigned 13C spectrum. Beyond 13C SSNMR, 15N CP-MAS analyses also appeared in many of these basic applications, owing to its sensitivity to hydrogen bonding both as a donor and an acceptor and to protonation effects in salts and zwitterions Citation[11,15,16,18].
Quantitative analysis methods were also demonstrated during this timeframe, illustrating the general applicability of SSNMR in these scenarios Citation[19,20]. SSNMR is particularly useful early in development, when there is little time or material for a standardized, calibrated quantitative method and a standardless quantitative output is desired Citation[19]. When development progresses and calibration standards can be prepared, XRPD and vibrational spectroscopy are more commonly used for quantitative analysis once a firm understanding of the solid form situation is gained, except in unusual cases (e.g., in drug products containing small amounts of drug, where 19F SSNMR might provide the only quantitative approach) Citation[19].
The development of triply tuned ‘HFX’ probes capable of simultaneously irradiation of 1H, 19F and an X nucleus (e.g., 13C) under high-power conditions also occurred between 1998 and 2005. While the development of HFX probes was certainly of interest to the wider SSNMR community, this technology had a disproportionately high impact on pharmaceuticals, given the frequent occurrence of fluorine in drugs and the emphasis given to fluorine in modern medicinal chemistry Citation[21,22]. Reliable, stable designs of HFX probes, first reported in 1998 Citation[23], achieved widespread commercial availability, with spinning rates up to 35 kHz, and had a substantial installed base by 2005. Pharmaceutical applications then became commonplace for both drug substance and drug product, the latter benefiting from the high sensitivity of 19F SSNMR Citation[19,24–26]. Although not apparent from the number of published reports, 19F SSNMR is heavily used during pharmaceutical development by many organizations.
Solid-state NMR has proven to be particularly successful in the characterization of amorphous forms, including samples that contain both amorphous and crystalline content produced by a multitude of methods, including desolvation and milling Citation[3,27,28]. This is to be expected, given the limitations of the other high-resolution technique (XRPD) in this area and the limitations of lower-resolution vibrational spectroscopic approaches. Various studies of amorphous forms have revealed interesting structural features of the amorphous state using 1D 13C SSNMR experiments Citation[3].
During the period of 1995–2005, the use of SSNMR to analyze drug products also increased markedly Citation[19,24–26,28–32]. The analysis of complex phase mixtures, such as many common solid oral-dosage forms, is an area where both the spectral resolution and experimental flexibility of SSNMR allow it to make a major contribution. This may seem surprising in light of the often quoted insensitivity of NMR. However, modern SSNMR systems have more than sufficient sensitivity to detect the drug in typical pharmaceutical formulations at 5–10% w/w levels using 13C detection Citation[26,29]. Both solid and semi-solid formulations can be readily analyzed Citation[19]. Developments in instrumentation, probes and higher static field strengths have contributed to the increasing use of SSNMR in drug product analysis. In particular, the advent of digital signal generation and detection and reliable high-power solid-state amplifiers has led to greater sensitivity, sharper lineshapes from high-power decoupling and higher quality spectral baselines for quantitative analysis Citation[33].
Developments since 2005
Since 2005, increased use of 2D SSNMR and other advanced methods has been observed in a number of published articles. The ability of 2D SSNMR methods to observe interactions between nuclei via the greater resolution and connectivity information in a 2D spectrum has allowed for more detailed structural analysis of pharmaceutical materials. Early work in this field was pioneered by Reutzel-Edens and co-workers using detailed interpretation of 1D SSNMR spectra in conjunction with crystallographic data Citation[31]. The advent of homonuclear 1H decoupling using the frequency-switched Lee–Goldburg method led to dipolar-based heteronuclear correlation (HETCOR) methods, which remove and reintroduce dipolar interactions to allow for detection of through-space 1H and 13C internuclear interactions within a few angstroms Citation[34]. An early example presented for a pharmaceutical hydrate illustrated the ability of this technique to selectively detect most hydrogen sites in the crystal structure Citation[35]. Use of 1H SSNMR has led to greater access to structural information using dipolar correlation, including applications of sensitive inverse-detected experiments Citation[36,37]. The increasing usage of advanced experiments in SSNMR studies is apparent in recent reviews of the field Citation[19,28]. This work has culminated in the ability to determine crystal structures using SSNMR Citation[38–43]. Methods to date have been based on spin-diffusion buildup rates that yield 1H-1H distances, chemical shift tensor measurements that are linked to structure via density functional theory simulations of chemical shielding, and other experiments that make use of through-space dipolar interactions to detect internuclear proximity. SSNMR approaches to crystal structure solution generally make use of relatively sophisticated software; a critical element of many studies has been the computational prediction of chemical shielding values from crystal structures for comparison and refinement against experimental chemical shift tensor values Citation[9,40–42]. Methods using molecular clusters replicate short-range effects in the crystal lattice, while other approaches, such as the gauge including projector augmented waves (GIPAW) method and the embedded-ion method (EIM), replicate longer range effects in crystals Citation[9,40–42]. Most SSNMR approaches are limited to molecules that are considerably smaller than typical drugs; a solution to this is to combine XRPD and SSNMR-based solution methods; an example crystal structure of a real pharmaceutical molecule solved using this approach with commercial software has been demonstrated Citation[41]. It should be noted that the determination of crystal structures using SSNMR and XRPD methods is a relatively narrow area of demand in practice, as many systems are amenable to the ‘gold standard’ of single-crystal x-ray diffraction (SCXRD), particularly with continual advances at the frontiers of this field, which have driven down crystal size and quality requirements Citation[43].
A broader area of potential focus for the more advanced 2D SSNMR approaches is the characterization of more complex systems, either to avoid the need for SCXRD entirely (by offering a faster means of answering a structural question) or to obtain structural data otherwise not available because of the complexity of the system. For example, the analysis of pharmaceutical cocrystals by SSNMR methods is greatly enhanced using a combination of techniques, including 1H fast MAS, 1H-1H dipolar correlation and 1H-13H and 1H-19F dipolar CP-HETCOR experiments Citation[44]. Cocrystals are currently an exciting area of research in both industry and academia as part of efforts to improve the solubility and physical properties of drugs Citation[45]. Similar applications have been demonstrated for complex disordered structures, including amorphous dispersions [Pham TN, Watson SA, Edwards AJ, Chavda M, Clawson JS, Strohmeier M, Vogt FG. Solid-state NMR analysis of amorphous solid dispersions. (Manuscript Submitted)]. Some of these techniques rely on spin diffusion, a well-known phenomena owing to its appearance in diverse fields from solution-state protein NMR to SSNMR of semi-crystalline polymers. In the context of pharmaceuticals, the detection of spin diffusion over a 5–10-Å range, easily accomplished with modern 2D experiments, allows for SSNMR to directly confirm the formation of a glass solution in a single sample without the need for difficult-to-prepare amorphous reference materials, answering one of the most important challenges in the characterization of amorphous dispersions [Pham TN, Watson SA, Edwards AJ, Chavda M, Clawson JS, Strohmeier M, Vogt FG. Solid-state NMR analysis of amorphous solid dispersions. (Manuscript Submitted)]. The advanced 2D SSNMR approaches also enable detailed structural analyses in concert with SCXRD for challenging systems such as composite crystals containing both salt and cocrystal environments Citation[46], disordered crystalline solid solutions Citation[47,48] and molecular conformation in amorphous dispersions Citation[28,30].
The discussion of SSNMR capabilities to this point has been limited to nuclei with a spin of ½, such as 1H, 13C, 15N, 19F and 31P. Although these nuclei continue to sit at the core of most pharmaceutical SSNMR analyses, increased attention has been focused on studies of quadrupolar nuclei in pharmaceutical solids. Activity in this growing field has been driven by the availability of higher static magnetic fields, reducing second-order quadrupolar broadening effects, as well as improved hardware and pulse sequences. As higher fields have become available, previously inaccessible nuclei have been targeted; for example, detailed 35Cl SSNMR studies of hydrochloride pharmaceutical salts have recently been reported Citation[49]. Quadrupolar nuclei that were previously thought to be inaccessible due to large quadrupolar broadening and low sensitivity can also be addressed. For example, a recent 65Cu SSNMR study of blue copper protein azurin showed the ability of this technique to probe the electronic state of copper sites in proteins Citation[50]. 23Na represents a more common, well-established quadrupolar nucleus of great interest in both pharmaceuticals and excipients. The ability to perform 1H-23Na HETCOR experiments offers insight into the hydrogen environment around this nucleus Citation[35]. Owing to their presence in water, quadrupolar 2H and 17O SSNMR studies have found use in the study of complex hydrate systems, both to probe mobility and to understand structure Citation[32,35,48,51]. The use of SSNMR to track moisture benefits from its unique ability to sensitively detect water isotopes and observe the physical state of water (e.g., surface vs bound), in addition to studies of crystalline hydrates. This allows studies of moisture ingress across semi-permeable membranes, such as dry micronized powder sealed in a device for inhalation or for studies of moisture uptake into an amorphous dispersion. 2H MAS experiments can also determine where water has come into contact with exchangeable groups via chemical exchange, even after the moisture is removed Citation[32,35,48]. Once a physical understanding of the water environment is gained using SSNMR, more efficient infrared, Raman and MS methods may be to enhance the speed of analysis for moisture tracking.
In the last 5 years, the active field of SSNMR of biomolecules has demonstrated a number of potential applications to drug-discovery activities Citation[52–56]. As noted previously, any industrial applications in this field would represent a shift into drug-discovery applications from the traditional drug-development role served by SSNMR in the pharmaceutical industry. This work also highlights the potential applications of increasingly high-end SSNMR instrumentation, normally procured for pharmaceutical development, to pharmaceutical drug discovery. Examples include the structural analysis of amyloid proteins Citation[52], membrane proteins Citation[53] and drug–receptor interactions between small molecules and proteins Citation[54,55]. Unlike the other applications of SSNMR described, these applications generally require extensive isotopic labeling of the molecules involved. Finally, recent experimental developments utilizing J-coupling and multidimensional SSNMR have opened up new avenues for even more detailed structural analysis of large biomolecules Citation[56].
Future perspective
Looking beyond 2010, several SSNMR applications for pharmaceutical development are expected to continue evolving. First, new experimental techniques are likely to open more territory for structural and dynamic studies. For example, recent pulse sequence developments have created the opportunity to work with the quadrupolar 14N nucleus Citation[57,58], which is much more abundant than the spin-half 15N isotope. The use of 1H SSNMR to probe formulations has been demonstrated and is expected to continue, providing sensitivity for low drug loads comparable to 19F SSNMR Citation[37] and allowing studies of the structure of nanoparticle delivery systems Citation[59]. Furthermore, as the advanced techniques described above become more embedded, they will likely find new applications to more complex systems. One example is the characterization of biopharmaceutical solids produced by lyophilization. Although these products are normally for intravenous use, it is still desirable to understand both mobility and structure of the solid material Citation[60], which can include a number of excipients in addition to a biomolecule (typically a protein, but potentially a peptide, oligonucleotide or antibody fragment). It is also important to understand the interactions between excipients and the biomolecule, as well as the solid-state form of the excipients, to ensure there is no potential impact on stability or reconstitution Citation[60]. Finally, increased applications of SSNMR in drug discovery may become more commonplace as methods are refined and more difficult targets are explored. The continued evolution of pharmaceutical SSNMR is thus likely to follow the trend of the past 15 years, with technology and applications advancing in concert to yield ever greater detail about the solid-state structures of these systems.
Financial & competing interests disclosure
The author is an employee of GlaxoSmithKline plc. and owns stock in this corporation. The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
Bibliography
- Lee SL , RawAS, YuL. Significance of drug substance physiochemical properties in regulatory quality by design. In: Drugs and the Pharmaceutical Sciences 178 (Preformulation in Solid Dosage Form Development). Informa Healthcare, NY, USA, 571–583 (2008).
- Yu L . Pharmaceutical quality by design: product and process development, understanding and control.Pharm. Res.25(4), 781–791 (2008).
- Byrn SR , PfeifferRR, StowellJG. Solid-State Chemistry of Drugs. SSCI, West Lafayette, IN, USA (1999).
- Bernstein J . Polymorphism in Molecular Crystals. Oxford University Press, NY, USA (2002).
- Harris RK . NMR studies of organic polymorphs and solvates.Analyst131(3), 351–373 (2006).
- Kalinowski HO , BergerS, BraunS. Carbon-13 NMR Spectroscopy. Wiley, NY, USA (1988).
- Berger S , BraunS, KalinowskiHO. NMR Spectroscopy of the Non-Metallic Elements. Wiley, NY, USA (1997).
- Kaupp M , BühlM, MalkinVG. Calculation of NMR and EPR Parameters. Wiley-VCH, Weinheim, Germany (2004).
- Orendt AM , FacelliJC. Solid state effects on NMR chemical shifts.Ann. Rep. NMR Spectr.62, 115–178 (2007).
- Carss SA , HarrisRK, FlettonRA. Carbon-13 NMR investigations of three fluorinated steroids.Magn. Reson. Chem.33(7), 501–505 (1995).
- Reutzel-Edens SM , BushJK, MageePA, StephensonGA, ByrnSR. Anhydrates and hydrates of olanzapine: crystallization, solid-state characterization, and structural relationships.Cryst. Growth Des.3(6), 897–907 (2003).
- Apperley DC , FlettonRA, HarrisRK, LancasterRW, TavenerS, ThrelfallTL, Sulfathiazole polymorphism studied by magic-angle spinning NMR. J. Pharm. Sci.88(12), 1275–1280 (1999).
- Wenslow RM , BaumMW, BallRG, McCauleyJA, VarsolonaRJ. A spectroscopic and crystallographic study of polymorphism in an aza-steroid.J. Pharm. Sci.89(10), 1271–1285 (2000).
- Zell MT , PaddenBE, GrantDJW, SchroederSA, WachholderKL, PrakashI, MunsonEJ. Investigation of polymorphism in aspartame and neotame using solid-state NMR spectroscopy.Tetrahedron56(36), 6603–6616 (2000).
- Portieri A , HarrisRK, FlettonRA, LancasterRW, ThrelfallTL. Effects of polymorphic differences for sulfanilamide, as seen through 13C and 15N solid-state NMR, together with shielding calculations. Magn. Reson. Chem.42(3), 313–320 (2004).
- Vogt FG , CohenDE, BowmanJDet al. Structural analysis of polymorphism and solvation in tranilast. J. Pharm. Sci. 94(3), 651–665 (2005).
- Sheth AR , LubachJW, MunsonEJ, MullerFX, GrantDJW. Mechanochromism of piroxicam accompanied by intermolecular proton transfer probed by spectroscopic methods and solid-phase changes.J. Am. Chem. Soc.127(18), 6641–6651 (2005).
- Li ZJ , AbramovY, BordnerJ, LeonardJ, MedekA, TraskAV. Solid-state acid-base interactions in complexes of heterocyclic bases with dicarboxylic acids: crystallography, hydrogen bond analysis, and 15N NMR spectroscopy. J. Am. Chem. Soc.128(25), 8199–8210 (2006).
- Vogt FG , ClawsonJS, StrohmeierM, PhamTN, WatsonSA, EdwardsAJ. New approaches to the characterization of drug candidates by solid-state NMR. In: The Handbook of Pharmaceutical Formulations Wiley, NY, USA (2010) (In Press).
- Offerdahl TJ , SalsburyJS, DongZet al. Quantitation of crystalline and amorphous forms of anhydrous neotame using 13C CPMAS NMR spectroscopy. J. Pharm. Sci. 94(12), 2591–2605 (2005).
- Böhm HJ , BannerD, BendelsSet al. Fluorine in medicinal chemistry. ChemBioChem 5(5), 637–643 (2004).
- Müller K , FaehC, DiederichF. Flourine in pharmaceuticals: looking beyond intuition.Science317(5846), 1881–1886 (2007).
- Stringer JA , DrobnyGP. Methods for the analysis and design of a solid-state nuclear magnetic resonance probe.J. Chem. Phys.69(9), 3384–3391 (1998).
- Wenslow RM . 19F solid-state NMR spectroscopic investigation of crystalline and amorphous forms of a selective muscarinic M3 receptor antagonist, in both bulk and pharmaceutical dosage form samples. Drug Dev. Ind. Pharm.28(5), 555–561 (2002).
- Farrer BT , PeresypkinA, WenslowRM. Quantitation of crystalline material within a liquid vehicle using 1H/19F CP/MAS NMR. J. Pharm. Sci.96(2), 264–267 (2007).
- Katrincic LM , SunYT, CarltonRA, DiederichAM, MuellerRL, VogtFG. Characterization, selection, and development of an orally-dosed drug polymorph from an enantiotropically-related system.Int. J. Pharm.366(1–2), 1–13 (2009).
- Lubach JW , XuD, SegmullerBE, MunsonEJ. Investigation of the effects of pharmaceutical processing upon solid-state NMR relaxation times and implications to solid-state formulation stability.J. Pharm. Sci.96(4), 777–787 (2007).
- Geppi M , MollicaG, BorsacchiS, VeraciniCA. Solid-state NMR studies of pharmaceutical systems.Appl. Spectros. Rev.43(3), 202–302 (2008).
- Harris RK , HodgkinsonP, LarssonT, MurugananthamA. Quantification of bambuterol hydrochloride in a formulated product using solid-state NMR.J. Pharm. Biomed. Anal.38(5), 858–864 (2005).
- Mollica G , GeppiM, PignatelloR, VeraciniCA. Molecular properties of flurbiprofen and its solid dispersions with Eudragit RL100 studied by high- and low-resolution solid-state nuclear magnetic resonance.Pharm. Res.23(9), 2129–2140 (2006).
- Reutzel-Edens SM , BushJK. Solid-state NMR spectroscopy of small molecules: from NMR crystallography to the characterization of solid oral dosage forms.Am. Pharm. Rev.5(2), 112–115 (2002).
- Vogt FG . A multi-disciplinary approach to the solid-state analysis of pharmaceuticals.Am. Pharm. Rev.11(5), 50–57 (2008).
- Vogt FG , Nuclear magnetic resonance. In: Ewing’s Analytical Instrumentation Handbook (Third Edition). Cazes J (Ed.). Marcel Dekker, NY, USA, 295–348 (2004).
- van Rossum BJ , FörsterH, de Groot HJM. High-field and high-speed CP-MAS 13C NMR heteronuclear dipolar-correlation spectroscopy of solids with frequency-switched Lee–Goldburg homonuclear decoupling. J. Magn. Reson.124, 516–519. (1997).
- Vogt FG , BrumJ, KatrincicLMet al. Physical, crystallographic, and spectroscopic characterization of a crystalline pharmaceutical hydrate: understanding the role of water. Cryst. Growth Des. 6(1), 2333–2354 (2006).
- Zhou DH , RienstraCM. Rapid analysis of organic compounds by proton-detected heteronuclear correlation NMR spectroscopy with 40 kHz magic-angle spinning.Angew. Chem. Int. Ed.47(38), 7328–7331 (2008).
- Griffin JM , MartinDR, BrownSP. Distinguishing anhydrous and hydrous forms of an active pharmaceutical ingredient in a tablet formulation using solid-state NMR spectroscopy.Angew. Chem. Int. Ed.46, 8036–8038 (2007).
- Elena B , EmsleyL. Powder crystallography by proton solid-state NMR.J. Am. Chem. Soc.127(25), 9140–9146 (2005).
- Elena B , PintacudaG, MifsudN, EmsleyL. Molecular structure determination in powders by NMR crystallography from proton spin diffusion.J. Am. Chem. Soc.128(29), 9555–9560 (2006).
- Pickard CJ , SalagerE, PintacudaG, ElenaB, EmsleyL. Resolving structures from powders by NMR crystallography using combined proton spin diffusion and plane wave DFT calculations.J. Am. Chem. Soc.129(29), 8932– 8933 (2007).
- Vogt FG , KatrincicLM, LongSTet al. Enantiotropically-related polymorphs of {4-(4-chloro-3-fluorophenyl)-2-[4-(methyloxy)phenyl]-1,3-thiazol-5-yl} acetic acid. Crystal structures and multinuclear solid-state NMR. J. Pharm. Sci. 97(11), 4756–4782 (2008).
- Salager E , SteinRS, PickardCJ, ElenaB, EmsleyL. Powder NMR crystallography of thymol.Phys. Chem. Chem. Phys.11(15), 2610–2621 (2009).
- Riekel C , BurghammerM, SchertlerG. Protein crystallography microdiffraction.Cur. Opin. Struct. Biol.15(5), 556–562 (2005).
- Vogt FG , ClawsonJS, StrohmeierM, EdwardsAJ, PhamTN, WatsonSA. Solid-state NMR analysis of organic cocrystals and complexes.Cryst. Growth Des.9(2), 921–937 (2009).
- Miroshnyk I , MirzaS, SandlerN. Pharmaceutical co-crystals– an opportunity for drug product enhancement.Exp. Opin. Drug Delivery6(4), 333–341 (2009).
- Clawson JS , VogtFG, BrumJet al. Formation and characterization of crystals containing a pleuromutilin derivative, succinic acid and water. Cryst. Growth Des. 8(11), 4120–4131 (2008).
- Vogt FG , VenaJA, ChavdaM, ClawsonJS, StrohmeierM, BarnettME. Structural analysis of 5-fluorouracil and thymine solid solutions.J. Mol. Struct.932(1–3), 16–30 (2009).
- Vogt FG , CopleyRCB, MuellerRLet al. Isomorphism, disorder and hydration in the crystal structures of racemic and single-enantiomer carvedilol phosphate. Cryst. Growth Des. In press (2010).
- Hamaed H , PawlowskiJM, CooperBFT, FuRQ, EichhornSH, SchurkoRW. Application of solid-state 35Cl NMR to structural characterization of hydrochloride pharmaceuticals and their polymorphs. J. Am. Chem. Soc.130(33), 11056–11065 (2008).
- Lipton AS , HeckRW, de Jong WA et al. Low temperature 65Cu NMR spectroscopy of the Cu+ site in azurin, J. Am. Chem. Soc.131(39), 13992–13999 (2009).
- Vogt FG , Dell‘OrcoPC, DiederichAMet al. A study of variable hydration states in topotecan hydrochloride. J. Pharm. Biomed. Anal. 40(5), 1080–1088 (2006).
- Heise H . Solid-state NMR spectroscopy of amyloid proteins,ChemBioChem9(2), 179–189 (2008).
- Middleton DA . NMR methods for characterising ligand-receptor and drug-membrane interactions in pharmaceutical research.Ann. Rep. NMR Spectros.60, 39–75 (2006).
- Baldus M . Molecular interactions investigated by multi-dimensional solid-state NMR.Curr. Opin. Struct. Biol.16(5), 618–623 (2006).
- Williamson PTF . Solid-state NMR for the analysis of high-affinity ligand/receptor interactions.Concepts Magn. Reson.34A(3), 144–172 (2009).
- Chen L , KaiserJM, PolenovaT, YangJ, RienstraCM, MuellerLJ. Backbone assignments in solid-state proteins using J-based 3D heteronuclear correlation spectroscopy.J. Am. Chem. Soc.129(35), 10650–10651 (2007).
- Gan Z , AmoureuxJP, TreboscJ. Proton-detected 14N MAS NMR using homonuclear decoupled rotary resonance. Chem. Phys. Lett.435(1–3), 163–169 (2007).
- O‘Dell LA , SchurkoRW. Fast and simple acquisition of solid-state 14N NMR spectra with signal enhancement via population transfer. J. Am. Chem. Soc.131(19), 6658–6659 (2009).
- Io T , FukamiT, YamamotoKet al. Homogeneous nanoparticles to enhance the efficiency of a hydrophobic drug, antihyperlipidemic probucol, characterized by solid-state NMR. Mol. Pharm. 7(1), 299–305 (2010).
- Pikal MJ , RigsbeeD, RoyMLet al. Storage stability of freeze-dried human growth hormone (hGH) with structure and dynamics in the glassy solid. J. Pharm. Sci. 97, 5106–5121 (2008).