Abstract
For small-molecule drugs the oral route of administration remains the most popular means of delivery for many indications. Optimizing oral bioavailability is, therefore, of critical importance in both drug discovery and development. However, while formulation development is routinely evaluated in clinical development, limited attention appears to be focused on improving exposure following oral delivery in early preclinical testing. The reasons for this appear to be limited compound availability and the requirement for a very rapid turnaround time. While some effort has been made to address solubility for intravenous formulation development, there is limited information available regarding formulation screening for oral delivery in drug discovery and preclinical development. In this brief article, we provide some details on our high-throughput, low compound requirement screen for oral formulation development. This screen has direct application in lead identification and development. The assay has been validated across multiple chemical series targeting different therapeutic areas, and is routinely applied with significant success in improving oral bioavailability.
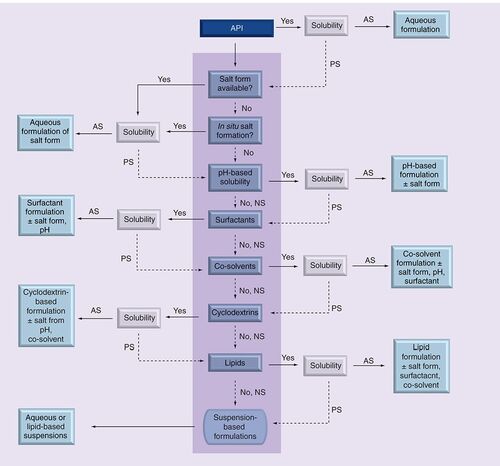
API: Active pharmaceutical ingredient; AS: Adequate solubility at target concentration; NS: Not soluble; PS: Partially soluble.
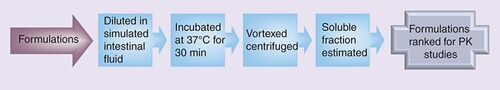
PK: Pharmacokinetic.
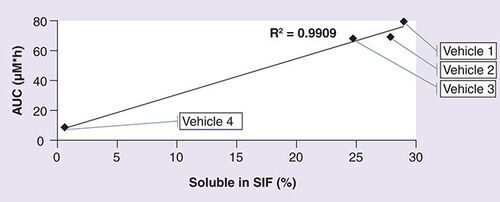
AUC: Total plasma concentration; SIF: Simulated intestinal fluid.
Cmax: Maximum plasma concentration; SIF: Simulated intestinal fluid.
The oral route of administration is one of the oldest and most popular routes for small drug molecules. Ease of administration, increased patient compliance and relatively lower development costs are responsible for the sustained interest in this route. There are, however, inherent limitations with this route of administration, such as solubility, permeability, GI metabolism, local irritation of the GI tract, stability of the drug in the GI environment, p-glycoprotein-mediated efflux effects and so forth. Over the years, a plethora of excipients and technologies has emerged to address some of these limitations, especially, solubility and permeability Citation[1–3]. However, most of these advances have been focused towards compounds in drug development and not in the discovery phase.
Poor aqueous solubility is a major factor limiting oral administration exposure. Advances in combinatorial chemical library synthesis and high-throughput in vitro screening of small molecules has resulted in a substantial increase in the number of compounds with poor aqueous solubility in drug-discovery pipelines. Limited compound availability, coupled with the requirement for a rapid turnaround time, has traditionally limited any significant efforts to improve oral bioavailability via formulation improvement in early drug discovery. Additionally, efforts to develop and screen oral formulations in drug discovery have been limited by the lack of appropriate and representative in vitro models. The importance of a focused formulation effort is often overlooked in the discovery phase of a program. Early formulations are usually prepared by biologists or pharmacologists to support their studies based on individual preferences with little or no help from pharmaceutical scientists Citation[4–6]. More recently, there has been increased awareness of the role of the pharmaceutical scientist in drug discovery Citation[7, 8].
Simple suspension formulations, or DMSO-based formulations, are widely employed in early stages of drug discovery Citation[5]. While suspension-based formulations, with cellulose-based excipients, have the advantage of revealing the intrinsic pharmacokinetic profile of the compound and providing early insights into potential issues with solid oral dosage forms, they do not consider the impact excipients can have on solubility. By enhancing the solubility of a compound, excipients can have a profound effect on bioavailability, pharmacokinetic and pharmacodynamic profiles of the compound Citation[9,10]. Solution formulations maximize the chances of improving exposure following oral administration and can enhance the onset of action due to the elimination of the dissolution phase. In addition, solution formulations enable low dose variability and enhance ease of administration. Suboptimal formulation approaches may result in the elimination of potentially promising drug candidates due to lower exposure and thereby adversely affect efficacy and toxicity assessment Citation[5].
With solution formulations, there is a risk of precipitation following administration potentially leading to low and variable exposure. In some of these cases, administration of a well- characterized suspension formulation may yield better results. Numerous in vitro methods have been described to address precipitation of injection formulations. For example, the visual appearance of precipitation following serial dilution in a buffer such as isotonic Sorensen’s phosphate buffer was compared with the occurrence of phlebitis following injection Citation[11–14]. A modification of the in vitro serial dilution method has been suggested for oral-solution formulation development using simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) Citation[6].
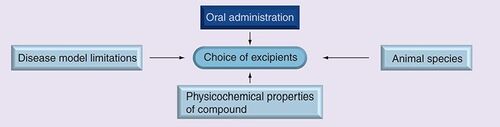
All excipients that are acceptable for oral administration in humans cannot be employed in discovery formulations. The choice of excipients for a particular compound depends on the animal species employed (e.g. mouse, rat or dog), the potential interactions the excipients could have on the disease model employed (e.g. corn oil should be avoided in hyperlipidemia models) and the physicochemical properties of the compound (e.g. salt forms are possible only in compounds with ionizable groups, simple aqueous formulations are difficult with increasing CLog P values) . US FDA-approved excipients for human use may not be well tolerated in certain animal species. For example, repeated oral administration of cyclodextrins has been shown to cause GI distress in rodents, leading to loss in body weight. The disease model employed could significantly dictate the exclusion of certain excipients from the formulation or limit the maximal amounts employed. The formulation should be designed to avoid contribution to any signs of toxicity that may be observed following administration and also avoid masking any pharmacological effects being investigated. For example, dextrose-based formulations are avoided in diabetes disease models, and ethanol is avoided when investigating behavioral effects of compounds. The disease model also dictates the duration of study, which is an important driver in limiting the concentrations of excipients employed. For long-term studies, milder formulations are preferred. For example, polyethylene glycol 400 (PEG 400) can be employed at 30% w/v concentration orally for a short-term study. However, in repeat-dose studies over a longer duration, it can lead to significant changes in GI motility, potentially confounding pharmacology and toxicological readings. Excipients in discovery formulations should be chosen with careful consideration for the impact the excipients could have on the efficacy and toxicity of the compound Citation[15,16].
Lipinski’s Rule of Five, predicts absorption or permeation based on the physicochemical parameters including molecular weight and CLogP values of the compound Citation[17]. The physicochemical properties of a compound also influence the choice of excipients employed. In general, compounds with low CLogP value (<3.5) can be solubilized in aqueous formulations with or without co-solvents or surfactants. With higher CLogP values, lipid- based formulations have a higher probability of solubilizing the compound. For compounds with ionizable groups, the calculated pKa value would determine the pH at which it has the greatest solubility.
Excipients and techniques commonly employed in the preparation of oral liquid formulations have been previously described and include Citation[6,18]:
pH adjustment and in situ salt formations
Surfactants
Co-solvents
Cyclodextrins
Lipids
Suspending agents
Our formulation strategy includes excipients from all of the above classes . Often, a combination of techniques stated above is utilized to achieve solubilization.
Materials & methods
A total of 26 structurally diverse compounds were obtained from Lexicon Pharmaceuticals. The compounds were selected from five projects representing different disease indications and contained six different chemotypes. Sterile water for injection (United States Pharmacopeia [USP]) was employed in the preparation of formulations, and all chemicals employed were of reagent or pharmacopeial grade. Solutol HS was obtained from BASF (NJ, USA), Labrasol was obtained from Gattefosse (NJ, USA), vitamin E TPGS was purchased from Eastman Chemical Company (TN, USA) and Captisol was purchased from CyDex Pharmaceuticals, Inc (KS, USA). Cremophor EL, carboxymethylcellulose sodium, corn oil, ethanol, 2-hydroxypropyl-β-cyclodextrin, methylcellulose, olive oil, PEG 400, propylene glycol and Tween 80 were purchased from Sigma (MO, USA). C57BL/6 albino mice were bred by Lexicon Pharmaceuticals.
Apparent solubility assay
The apparent solubility assay was performed using a BD Gentest® solubility scanner utilizing scatter detection to determine compound precipitation in aqueous media. The compounds tested were run in serial dilution from DMSO stock solutions (10, 5, 2.5, 1.25, 0.625, 0.3125, 0.156, 0.078, 0.039 and 0.0195 mM) and diluted into phosphate buffered saline to a residual DMSO concentration of 1%. Amiodarone (low solubility, <3 µM) and verapamil (high solubility, 100 µM or greater) were employed as internal controls. The samples were run in duplicates in a 96-well format.
LogP
LogP was calculated using Scitegic Pipeline Pilot Version 7.0.1. This component uses the atom-based method published by Ghose and Crippen to calculate the octanol-water partition coefficient (LogP) Citation[19].
Formulation screen assay
Choice of excipients
We followed a stepwise procedure to test different classes of excipients. Based on the animal species employed, disease model limitations and physicochemical properties of the compound, certain excipients or classes of excipients were excluded from the scheme. For example, with hyperlipidemia targets, lipophilic excipients such as corn oil were avoided from the scheme. Similarly, the use of ethanol was avoided with certain immunology disease models, while osmotic agents such as PEG 400, which have the potential to cause GI disturbances possibly leading to loss in body weight, were avoided in obesity disease models.
In general, aqueous formulations were preferred over lipophilic preparations. In formulations containing organic co-solvents, the aqueous phase was 70% or greater. The final preparation was maintained between pH 4–8.
For compounds with poor aqueous solubility that contain ionizable groups, the solubility of a salt form when available was tested. If the salt form demonstrated inadequate solubility, pH adjustment was evaluated to study the impact on solubility. If a salt form was not available, in situ salt formation in the presence of a strong acid or base, depending on the functional groups present, was tested as a means to improve solubility. In the absence of any ionizable groups, this step was eliminated from the scheme.
The next class of excipients evaluated was surfactants, including Tween 80, Solutol HS and Cremophor EL. These were either tested alone, or in combination with pH and/or salt formation, depending on the effect of pH and/or salt formation on solubility. If surfactants failed to provide sufficient solubility, co-solvents such as PEG 400, ethanol or propylene glycol were tested either alone or in combination with surfactants, pH and/or salt formation. In the absence of adequate solubility in any of the above classes of excipients, cyclodextrins like Captisol and HP-β-CD were tested. These were either tested alone or in combination with co-solvents, pH and/or salt formation. Generally, combinations between cyclodextrins and surfactants were avoided. If cyclodextrin-based formulations did not provide adequate solubility, lipid formulations, either as single excipient preparations in corn oil or olive oil, or multi-excipient preparations containing solubilizers, surfactants or a combination of both were tested . Solution and suspension formulations obtained from the screen were tested in the in vitro formulation screen.
Formulation preparation
The selected excipients were utilized to prepare 0.3–0.5 ml of formulations. In multi-excipient formulations, the organic phase was added initially, followed by the aqueous phase. pH adjustments were performed after the addition of the aqueous phase. All formulations were prepared at room temperature. Vortexing and sonication was employed as required. Visual appearance of formulation was noted. Samples of the formulations were stored overnight at 4°C and visually examined for precipitation.
In vitro formulation screening
The prepared formulations were diluted with simulated intestinal fluid USP (Ricca Chemical Company, TX, USA) in a 1:9 ratio and incubated at 37°C for 0.5 h with shaking. The samples were vortexed for 3 min and then centrifuged at 14000 rpm for 10 min with a Beckman Coulter Microfuge 18 Centrifuge. Spectrophotometric analysis of the supernatant was performed on a Molecular Devices SpectraMax Plus384 utilizing a 96-well microplate. The calibration curves for the analyte were prepared in either acetonitrile or methanol over the concentration range of 0–250 µg/ml. Placebo formulations were prepared and processed in the same manner and these served as the blank for the experiment. Absorbance of the samples was measured between 200 and 400 nm and the wavelength that demonstrated linear absorption across the concentration range tested was chosen for further analysis. The percentage of compound remaining in solution was reported. The formulations were ranked based on the solubility data .
In vivo studies
All in vivo studies were conducted as per guidelines established by the Institutional Animal Care and Use Committee (IACUC) at Lexicon Pharmaceuticals. In general, four male C57BL/6 albino mice, 8–10 weeks of age were employed in each pharmacokinetic study. The mice were housed in micro-isolator cages with corn cob bedding in a temperature and light–dark cycle controlled environment. They were allowed access to standard chow diet and water ad libitum during the course of the study. Mice received a single dose of the selected formulation by oral gavage. All compounds were administered at a 10-mg/kg dose with a dose volume of 5 ml/kg, except compound 12, which was administered at a 30-mg/kg dose with a dose volume of 10 ml/kg. Blood samples were collected 0.25, 0.5, 1, 2, 4, 6, 24 h post-dose by either retro-orbital or sapheneous vein bleeds. Plasma was separated by centrifugation at 5000 rpm for 5 min and stored at -20°C until analysis by LC–MS/MS.
Sample analysis
Analysis of the plasma samples was performed by LC–MS Citation[20]. Briefly, the proteins in the plasma sample were precipitated using acetonitrile: water (80:20) mixture containing verapamil as an internal standard. The HPLC system consisted of a Thermoelecton MS pump with a CTC HTS autosampler. Agilent SB C8 was the column employed. The MS was performed with either Thermo Electron TSQ Quantum Ultra AM or an Applied Biosystems 3000 mass spectrometer. Calibration curves of the compounds were prepared in naive blood plasma over the range of 1.3–10000 ng/ml. The calibration samples were processed in a manner similar to the test samples.
Data analysis
The pharmacokinetic parameters for the tested compounds were calculated using WinNonlin Professional, version 5.0 (Pharsight Corporation, USA). The formulations were ranked based on total plasma concentration (AUC) and maximum plasma concentration (Cmax). The pharmacokinetic data were compared with the ranking generated from the in vitro formulation screen.
Results & discussion
The physicochemical properties, apparent solubility, results from the formulation screening assay, and the in vivo pharmacokinetic data for the 26 compounds of diverse chemotypes (54 formulations) are summarized . The molecular weights of the compounds tested ranged from 245 to 530. The LogP values ranged from 2.5 to 6.4 and the PSA values ranged from 12 to 140 Å2. Apparent solubility data were measured for most of the compounds and the values obtained ranged from 2 to 53 µg/ml.
In total, 25 out of the 54 formulations prepared were solutions, and the remaining 29 were administered as suspensions. The formulation screen assay and the in vivo pharmacokinetic studies for compound 12 were run at a 30 mg/kg dose. All other compounds were tested at 10 mg/kg dose.
In general, formulations that exhibited higher solubility for the same compound in the in vitro formulation screen, gave higher in vivo exposure (i.e., AUC and Cmax). Out of the 54 formulations tested, only five of the formulations with lower predicted solubility gave higher AUC and/or Cmax (determined as AUC [Vehicle 1]/ AUC [Vehicle 2] < 0.9, Cmax [Vehicle 1]/ Cmax [Vehicle 2] < 0.9).
For two compounds (1 and 2), the solutions formulations were ranked lower that the suspension formulations from the in vitro formulation screen and this was validated in vivo. For both these compounds, the solution formulation demonstrated a higher solubility in the excipients when compared with the suspension formulation. However, on incubating with simulated intestinal fluid, the suspension formulations gave a higher soluble fraction than the solution formulation. This demonstrates the usefulness of dilution in a biologically relevant media mimicking the conditions following oral administration.
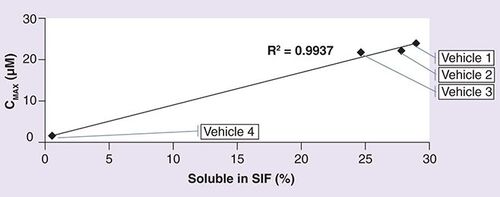
Compound 12 was a highly insoluble compound with an apparent solubility of 6 µg/ml. The target concentration was 3 mg/ml. The compound demonstrated enhanced solubility at acidic pH. It was soluble in PEG 400 at pH 4 (>30 mg/ml). Both 1:9 and 1:4 dilutions with water (resulting in a 10 and 20% PEG 400 concentrations) led to precipitation of the compound from solution. Compound 12 had a solubility of 2 mg/ml in 15% Captisol at pH 4. As part of the formulation screen, cyclodextrin alone (15% Captisol), combination of co-solvent with cyclodextrin (PEG 400: 15% Captisol [10:90] or PEG 400: 15% Captisol [20:80]) and a suspension formulation in 0.25% methylcellulose were tested. The results are summarized in . The soluble fraction obtained following incubation in simulated intestinal fluid is plotted against the AUC and Cmax obtained following oral administration of compound 12 in the different vehicles (, respectively.)
The results of this study have demonstrated the important effect formulations can have on exposure following oral administration. The specific objective of this study was to develop a high-throughput in vitro screening assay to enable compound exposure in discovery and preclinical development studies. It was critical that the assay had a rapid turnaround time and utilized minimal quantities of compound. The assay described utilizes 6 mg of compound and relies on the chemical structure and its calculated physicochemical properties. The total time required to run the assay is less than 3 h.
Selection of the excipients is carried out with careful consideration to minimize any potential impact they could have on the disease model. The choice of excipients are limited to FDA-approved inactive ingredients and the formulation conditions are mild and controlled allowing easy translation to a first-in-human formulation if required.
The formulation screening assay has been developed and validated and is being used with considerable success in improving the oral bioavailability of a diversity of chemotypes in support of discovery and preclinical development.
Table 1. Examples of excipients employed in drug discovery.
Table 2. Results from in vitro formulation-screening assay and in vivo pharmacokinetic data.
Table 3. Results from in vitro formulation-screening assay and in vivo pharmacokinetic data for compound 12.
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Additional information
Funding
Bibliography
- Sastry SV , NyshadhamJR, FixJA. Recent technological advances in oral drug delivery – a review.Pharmaceut. Sci. Technol. Today3, 138–145 (2000).
- Gupta H , BhandariD, SharmaA. Recent trends in oral drug delivery: a review.Recent Pat. Drug Deliv. Formul.3, 162–173 (2009).
- Singh BN , KimKH. Drug delivery: oral route.Encyclopedia of Pharmaceutical Technology (Third Edition).1242–1265 (2006).
- Maas J , KammW, HauckG. An integrated early formulation strategy – from hit evaluation to preclinical candidate profiling.Eur. J. Pharm. Biopharm.66, 1–10 (2007).
- Neervannan N . Preclinical formulations for discovery and toxicology: physicochemical challenges.Expert Opin. Drug Metabol. Toxicol.2, 715–731 (2006).
- Li P , ZhaoL. Developing early formulations: practice and perspective.Int. J. Pharm.341, 1–19 (2007).
- Venkatesh S , LipperRA. Role of the development scientist in compound lead selection and optimization.J. Pharm. Sci.89, 145–154 (2000).
- Lee YC , ZocharskiPD, SamasB. An intravenous formulation decision tree for discovery compound formulation development.Int. J. Pharm.253, 111–119 (2003).
- Gonzalez RCB , HuwylerJ, WalterI, MountfieldR, BittnerB. Improved oral bioavailability of cyclosporin A in male Wistar rats: comparison of a Solutol HS 15 containing self-dispersing formulation and a microsuspension.Int. J. Pharm.245, 143–151 (2002).
- Klein S , WempeMF, ZoellerTet al. Improving glyburide solubility and dissolution by complexation with hydroxybutenyl-β-cyclodextrin. J. Pharm. Pharmacol.61, 23–30 (2009).
- Li P , VishnuvajjalaR, TabibiSE, YalkowskySH. Evaluation of in vitro precipitation methods. J. Pharm. Sci.87, 196–199 (1998).
- Irwin WJ , IqbalM. Bropirimine formulation: the dynamic testing of injections.Int. J. Pharm.83, 241–249 (1992).
- Ward GH , YalkowskySH. Studies in phlebitis. IV: injection rate and amiodarone-induced phlebitis.J. Parenter. Sci. Technol.47, 40–43 (1993).
- Ward GH , YalkowskySH. Studies in phlebitis. VI: dilution-induced precipitation of amiodarone HCl.J. Parenter. Sci. Technol.47, 161–165 (1993).
- Gad SC , CassidyCD, AubertN, SpainhourB, RobbeH. Nonclinical vehicle use in studies by multiple routes in multiple species.Int. J. Toxicol.25, 499–521 (2006).
- Pestel S , Martin H-J, Maier G-M, Guth B. Effect of commonly used vehicles on gastrointestinal, renal, and liver function in rats. J. Pharmacol. Toxicol. Methods54, 200–214 (2006).
- Lipinski CA , LombardoF, DominyBW, FeeneyPJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings.Adv. Drug Deliv. Rev.46, 3–26 (2001).
- Strickley RG . Solubilizing excipients in oral and injectable formulations.Pharm. Res.21, 201–230 (2004).
- Ghose AK , CrippenG M. Atomic physicochemical parameters for three-dimensional structure - directed quantitative structure–activity relationships I. Partition coefficients as a measure of hydrophobicity. J. Comput. Chem.7, 565–577 (1986)
- Sarawek S , LiL, Yu X-Q et al. Examination of the utility of the high throughput in vitro metabolic stability assay to estimate in vivo clearance in the mouse. Open Drug Metabol. J.3, 31–42 (2009).