
Abstract
Nanoparticle-based therapeutics are poised to play a critical role in treating disease. These complex multifunctional drug delivery vehicles provide for the passive and active targeted delivery of numerous small molecule, peptide and protein-derived pharmaceuticals. This article will first discuss some of the current state of the art nanoparticle classes (dendrimers, lipid-based, polymeric and inorganic), highlighting benefits/drawbacks associated with their implementation. We will then discuss an emerging class of nanoparticle therapeutics, bacterial outer membrane vesicles, that can provide many of the nanoparticle benefits while simplifying assembly. Through molecular biology techniques; outer membrane vesicle hijacking potentially allows for stringent control over nanoparticle production allowing for targeted protein packaged nanoparticles to be fully synthesized by bacteria.
Nanoparticles as therapeutics
There are many different types of nanoparticles that have been adapted for therapeutic use aiding in the delivery of small molecules, contrast agents, peptides, vaccines and proteins [Citation1–4]. Nanoparticles are attractive delivery vehicles since they can be produced from many different materials and can be formed in a wide range of sizes exhibiting diverse active and passive targeting capabilities [Citation5]. For these reasons nanoparticles are being utilized for a number of different therapeutic and medical diagnostic applications including cancer treatments, imaging tools, antibacterial agents and gene delivery vehicles, just to name a few [Citation6–8]. Properties that an ideal therapeutic nanoparticle should exhibit can be found in Box 1 [Citation9].
Nanoparticles can be used to deliver therapeutics in a variety of different ways either by encapsulation of a therapeutic agent within its core or through conjugation of a therapeutic agent to its exterior surface. Encapsulation of a therapeutic agent provides protection from proteolytic cleavage and immune recognition and generally improves the stability of compounds allowing for increased circulation times and larger amounts of active drug being delivered to its intended target [Citation10]. Outside facing targeting moieties provide for multivalent target–nanoparticle interactions to improve targeting specificity and avidity [Citation11]. High avidity display of enzymes on a nanostructure may also enhance enzymatic activity in addition to improving avidity of the nanoparticle for its target [Citation12]. The addition of surface-associated therapeutic agents allow for release of the active pharmaceutical agent while also not requiring opening of the nanoparticle, or diffusion from the nanoparticle core. However, the slow release capabilities associated with diffusion from within the nanoparticle core, or via hydrolyzable linkers, have also been utilized as a means of modulating pharmacokinetics (PK) and pharmacodynamics (PD) to reduce the need for frequent dosing of the therapeutic [Citation13,Citation14].
In many instances both interior loading and exterior targeting capabilities can be utilized simultaneously to create multifunctional nanoparticles that both selectively target a specific disease population and deliver a payload of encapsulated drug/protein to locally treat the disease [Citation15]. These tunable features allow for very precise control over the PK and PD of drug delivery as well as allow for the selective targeting of tissues and local multidrug delivery [Citation16]. Nanoparticles make these types of complex multifunctional therapeutics possible. This review seeks to shed light upon an emerging, unconventional, therapeutic delivery platform built around hijacking bacterial machinery and outer membrane vesicle (OMV) production to both express and package biologically synthesized therapeutic agents into nanoparticles for clinical use. Below we begin with a brief review of some current state of the art in nanoparticle therapeutic classes. We then utilize this as a basis to examine what OMVs have to offer from both an assembly and function standpoint.
Treating each type of disease comes with its own unique set of challenges making pharmacological target selection, drugs being delivered, imaging capabilities, pharmacokinetic and pharmacodynamic critical components that directly impact which nanoparticle platform is best suited for each disease application. Identified above are some therapeutic indications in which nanoparticle-based treatments may provide improved patient outcomes through reducing off-target toxicity by transporting pharmaceutical agents to specific biological compartments or cell types.
COPD: Chronic obstructive pulmonary disease.
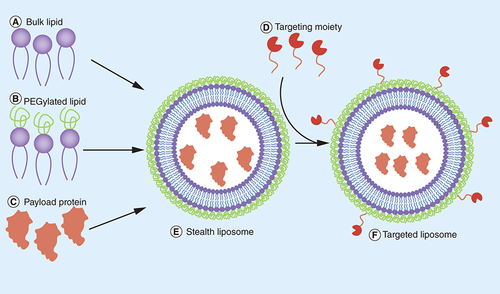
Step 1: each component of the liposome must be synthesized and purified separately: bulk lipid (A), PEG-conjugated lipid (B), recombinant protein being packaged (C) and the targeting ligand (D). Step 2: components (A) through (C) are mixed and liposomes are formed via extrusion, sonication, freeze thaw cycles or reverse-phase evaporation to make (E). Step 3: any unpackaged protein must then be removed in a purification step. Step 4: the resulting liposome is then incubated with the targeting ligand (D) and crosslinking reagents to conjugate to the liposome surface to make (F). Step 5: the liposome must then undergo another purification cycle to remove any remaining crosslinking reagents as well unconjugated targeting ligand.
PEG: Polyethylene glycol.
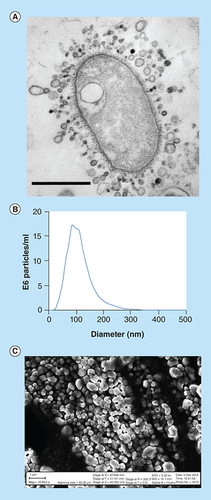
(A) Image of Gram-negative bacteria-shedding OMVs throughout its entire outer membrane, scale bar 1 µm. (B) Nanosight size distribution of bacterial OMVs indicating a size range of 30–200 nm with an average of approximately 80 nm. (C) SEM image of gold-coated lyophilized OMVs purified by ultracentrifugation. The size range of the OMVs pictured are 50–400 nm, slightly larger than the nanosight due to the freeze drying and gold coating process used for visualization (scale bar indicates 1 µm length). Panel A image is reproduced with permission from © American Society for Microbiology.
OMV: Outer membrane vesicle.
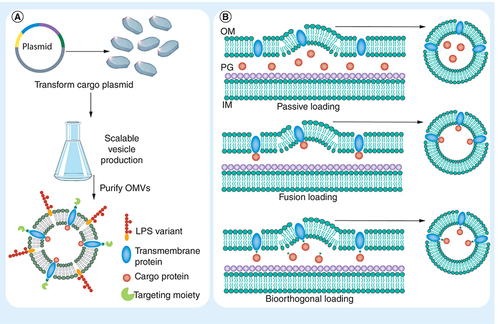
(A) Step 1: construction of a plasmid that encodes for the desired LPS variant, mutant transmembrane protein, packaged protein and targeting peptide/protein sequence is inserted into the bacteria. Step 2: bacteria containing the construct are selected and grown in a scalable culture flask or bioreactor. Step 3: the OMVs are then purified and are ready for use. (B) Methods for packaging protein cargo within OMVs: passive loading through overexpression of loaded protein, by creating a fusion of the protein being loaded with a known transmembrane or membrane-anchored protein, or via bioorthogonal loading of the protein which is overexpressed and then selectively conjugate to a mutant transmembrane or membrane-anchored protein.
IM: Inner membrane; LPS: Lipopolysaccharide; OM: Outer membrane; OMV: Outer membrane vesicle; PG: Peptidoglycan.
Nanoparticle classes
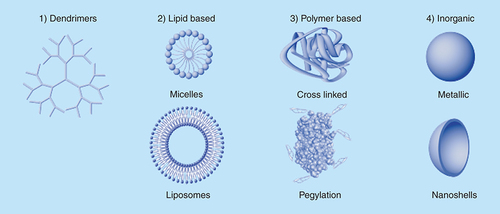
Nanoparticles can be divided into four primary classes: dendrimers, lipid-based nanoparticles, polymeric nanoparticles and inorganic nanoparticles (). These nanoparticle classes can then be divided further based on their material composition, size and function. Nearly all nanoparticle types can be fabricated across a wide size range from 10s to 100s of nanometers in diameter to meet the targeting and drug delivery needs of each unique therapeutic application. There also exist many other nanoparticle types, such as nanocrystals, nanoshells, fullerenes and multi-composite materials, which can be difficult to classify [Citation17]. Each type of nanoparticle has its own set of benefits and drawbacks for use, making the selection of a particular class of nanoparticle extremely application-specific. This list of nanoparticles and the following descriptions are not intended to be exhaustive but rather an informative selection of a range of nanoparticle types and uses to demonstrate their complexity and some of their current capabilities.
Dendrimers are highly branched, highly uniform nanoparticles that are classified based on their number of generations, indicated by the number of times a single arm branches, which is also a measure of its size [Citation18]. As an example, a fourth generation dendrimer made from a trivalent initiator, generation 0, will undergo four serial branching events making for a total of 48 terminal functional groups. The chemistries utilized to grow higher generation dendrimers are exceptionally efficient allowing for a high level of uniformity resulting in spherical and symmetrical dendrimers. The monodisperse nature of dendrimers allow for extremely tight control over functionalization and the resulting release and targeting characteristics make them especially well suited for therapeutic delivery applications [Citation19]. However, there can be issues regarding clearance of the nanoparticles once all of the therapeutic agent has been released due to the relatively chemically resistant and typically nonbiodegradable backbone of the dendrimer [Citation20].
Polymeric nanoparticles can be further split into three primary categories of crosslinked or biodegradable polymers and pegylated proteins [Citation21–23]. Both crosslinked and biodegradable polymeric nanoparticles can be synthesized through a variety of methods that include polymerization reactions, emulsification, nanoprecipitation or solvent evaporation [Citation24]. While the resulting nanoparticles can be fabricated of similar sizes and relative compositions as dendrimers, they lack the tight regulation over the number of generations associated with dendrimer synthesis making polymeric nanoparticles significantly more polydisperse. There also exist naturally occurring biodegradable polymers such as collagen and chitosan which have recently become more clinically prevalent [Citation25,Citation26]. While there are now a handful of US FDA-approved synthetic and naturally occurring polymers, the most widely known biodegradable polymers remain poly(lactic acid) and poly(glycolic acid) [Citation27]. Biodegradable polymers are unique since they degrade into their relatively inert subunits over a very predictable time period providing for controlled therapeutic release kinetics as well as easy biological clearance post delivery [Citation28]. Crosslinked polymers are made of nonbiodegradable subunits and result in similar limitations and clearance issues as dendrimers. Polymers are also utilized to modify proteins and nanoparticle surfaces to improve in vivo stability. The addition of a single or multiple polyethylene glycol chains, also known as pegylation, to therapeutic proteins has become common practice and is used to improve circulation times by reducing host recognition of the foreign proteins [Citation22]. This relatively simple technique can be utilized to tune half life effectively reducing the necessary dosing frequency needed to maintain therapeutic efficacy [Citation29].
The inorganic nanoparticle category can also be further split into a number of subgroups that include quantum dots, nonmagnetic and magnetic nanoparticles, iron oxides and various doped nanoparticles [Citation30–32]. Inorganic nanoparticles have primarily been used clinically as topical therapeutics and contrast agents allowing for visualization of anatomical structures under advanced imaging techniques such as MRI, ultrasound, CT and other radiological tools [Citation15,Citation33]. If administered without prior surface functionalization, nearly all inorganic nanoparticles display cellular toxicity limiting their unmodified use [Citation34]. Since their initial development, many approaches have been developed to coat inorganic nanoparticles endowing them with similar targeting and therapeutic drug loading characteristics as other nanoparticle classes while maintaining their imaging capabilities [Citation35]. Their inherent toxicity has also been utilized for therapeutic applications to kill targeted cell populations but due to their nonbiodegradable inorganic core there still remain issues regarding the biological clearance of the nanoparticles [Citation36,Citation37].
Lipid-based nanoparticles can be further divided into liposome and micellar nanoparticles. The primary difference between liposomes and micelles being that liposomes are comprised of a lipid bilayer that encapsulates an aqueous core whereas micelles do not possess a bilayer and have a hydrophobic core made up of the lipid tails. Liposomes, 30–500 nm in diameter, are therefore capable of delivering large hydrophilic molecules within their interior or conjugated to their surfaces. It is not surprising that liposomes are an attractive nanoparticle platform since they allow for the delivery capabilities of targeted therapeutics with either hydrophobic small molecules loaded within the lipid bilayer or hydrophilic small molecules and large proteins encapsulated within its aqueous core [Citation38]. The hydrophobic interior, and relatively small size of micelles (~15 nm), only allows for encapsulation of hydrophobic small molecules but is not amenable for the loading of proteins or hydrophilic small molecules within its lipid core. Many lipid-based nanoparticles also self assemble and their lipid subunits can be carefully selected to eliminate any nanoparticle clearances issues as the nanoparticles themselves can be made from the same endogenous lipids found within the body.
Clinical implementation
These intricate multifaceted nanoparticles facilitate the creation of complex drug delivery systems that demonstrate clinical efficacy well beyond traditional therapeutics allowing for unique disease treatments that were previously impossible to attain. Due to the exceptional diversity across different classes of nanoparticles and varied applications, the FDA has struggled to create a standard set of guidelines for nanoparticle therapeutic approvals. This has historically made attaining FDA approval for these treatments exceedingly difficult as each nanoparticle therapeutic essentially requires its own unique approval pathway [Citation39]. Despite this fact, there were approximately 23 nanoparticle-based treatments that had gained FDA approval as of 2012 [Citation7]. Nearly half of the approved therapeutics are lipid-based micelles or liposomes as they have been the most widely studied and offer exceptional PK and PD with limited biological clearance issues [Citation40,Citation41]. The remaining approved nanoparticles are dominated by polymeric-based therapeutics, primarily comprised of pegylated proteins, but representatives from nearly all of the nanoparticle classes, including gold nanoparticles and nanocrystals, are also present.
The most widely known FDA-approved nanoparticle therapeutic is Doxil, which is a doxorubicin-loaded stealth liposome used to treat ovarian cancer and multiple myeloma, with annual sales of approximately US$500 million as of 2013 [Citation7]. By sequestering the doxorubicin within a liposome, the negative side effects associated with off target cell death from this chemotherapeutic agent can be reduced allowing for more selective targeting and a therefore more effective treatment of the disease [Citation11]. In 1995 it was the first nanoparticle-based chemotherapeutic formulation to be approved by the FDA for the treatment of cancer. Relatively few nanoparticle therapeutics are currently approved for clinical use compared with the number of traditional small molecule therapeutics that are FDA approved. With that said we should expect to see many more FDA-approved nanoparticle-based therapeutics as there is an ever increasing list of nanoparticle therapeutics actively seeking approval and countless more that exist in preclinical and early-stage development to address unmet clinical needs ().
General nanoparticle hurdles
With the added capabilities of targeted multifunctional nanoparticles, there also come many issues. Things to consider when deciding to develop a nanoparticle-based therapeutic include manufacturing complexity, cost, toxicity, intra-batch uniformity, inter-batch variation, synthetic yield, characterization/validation techniques, scalability and shelf life/stability [Citation9,Citation42]. These are just a few of the critical characteristics that each class of nanoparticle must address in order to develop less expensive and more robust protocols to make clinical implementation a more accessible reality. Each type of nanoparticle has its own set of complexities with certain classes of nanoparticles being more easily adaptable, and therefore more attractive than others, for therapeutic applications allowing them to remain clinically dominant [Citation43].
Therapeutic liposome production
Liposomes are of particular clinical importance as they can be made from naturally occurring lipids which eliminates many issues associated with immune activation, clearance or toxicity against the vehicle itself [Citation44]. They also allow for numerous loading strategies that include conjugating functional targeting moieties to their exterior surface, loading hydrophobic and insoluble drugs within their lipid bilayer or loading hydrophilic therapeutics and proteins within their aqueous core. Liposomes are essentially the only nanoparticle class that can carry proteins internally. All of these capabilities make liposomes an attractive therapeutic nanoparticle delivery platform yet they are not without fault.
To demonstrate this we will discuss the necessary workflow to produce a representative protein loaded, targeted, stealth liposome (). Liposomes are typically comprised of a combination of lipids mixed at desired mole ratios. For stealth targeted liposomes, one of the lipid components must be functionalized with polyethylene glycol and another lipid component must either be preconjugated to a targeting moiety or the targeting moiety can be conjugated post-liposome formation [Citation45,Citation46]. This targeting moiety can range from a small molecule or peptide to a full-length protein such as an antibody. Both the targeting moiety and the protein being loaded into the aqueous interior of the liposome must be produced separately. In the case of a peptide targeting ligand, solid-phase peptide synthesis is the most common synthetic technique that is utilized and requires separate production, purification and characterization. Recombinant protein expression for either a protein-based targeting moiety or the internally loaded protein requires its own unique set of molecular biology, purification and characterization techniques. Once all of the individual components have been made and validated separately the final liposome therapeutic can then be produced by either an extrusion process carried out at temperatures above the lipid phase transition temperature, sonication or by reverse-phase evaporation. Proteins are incapable of passively crossing the lipid bilayer due to their exceptionally large size and therefore must be incorporated into the lipid solution used to form the liposomes in order to be encapsulated within the aqueous core. The final combination therapeutic nanoparticle must itself be purified and undergo its own set of unique characterization techniques prior to clinical use.
Liposome hurdles
As outlined above there are major hurdles associated with using liposomes as they are exceptionally expensive to produce since they require complex production, purification and characterization techniques. Since many of the liposome and protein components must be produced separately and then packaged and purified there are many issues associated with inefficient and inconsistent loading of proteins and small molecules within each liposome and inconsistencies in the number of targeting moieties present on the surface of each liposome [Citation47]. There are also issues associated with the inactivation of targeting moieties and payload proteins due to exposure to the harsh conditions necessary to form the liposomes themselves. Enzymes can be particularly sensitive to inactivation at the high temperatures used for liposomal extrusion, multiple freeze thaw cycles or from exposure to organic solvents. Each additional cycle of fabrication, purification and characterization results in increased cost, reduced yields and increased complexity making these liposome-based therapeutics exceptionally difficult to produce. With that said, there has been a concerted effort to address each of these hurdles and extensive progress has been made to make this type of advanced nanoparticle therapeutic more clinically accessible [Citation48,Citation49]. Developing an orthogonal system to produce multifunctional lipid-based nanoparticles that eliminates numerous steps from the current liposome production process would be greatly beneficial. Methods of producing proteoliposomes through hijacking bacterial vesicle formation offers unique opportunities to address many of the production and packaging issues associated with liposome production to create complex, inexpensive nanoparticle therapeutics of the future.
Bacterial vesicles
As a natural phenomenon, both Gram-positive and Gram-negative bacteria release OMVs at various stages of growth, under diverse environmental conditions, or in response to chemical signals () [Citation50]. These small approximately 30–200 nm vesicles are comprised of lipids, proteins and polysaccharides and carry various cargo () [Citation51,Citation52]. The reason that bacteria secrete these proteoliposomes is largely unknown but they have been implicated in cell–cell communication and gene transfer [Citation53–55], delivery of toxins and virulence factors [Citation56,Citation57], and host defense mechanisms [Citation58], and often are implicated in increased bacterial infection rates in pathogenic bacterial strains. The functional conditions and mechanisms for OMV production can be highly varied between bacterial species making discovery of conserved formation and packaging pathways difficult to determine. For the purpose of this discussion we will focus only on OMVs produced from Gram-negative bacteria as this class of bacteria includes the most common laboratory strains of Escherichia coli.
Though bacterial OMVs have been studied for decades, the variability in OMV composition between bacterial species has inhibited elucidation of a well-defined biosynthetic pathway. OMVs are released from the bacterial outer membrane (OM) and as such have a similar protein, lipopolysaccharide (LPS), and lipid composition as the parental cell membrane. Proteomic analysis of OMVs for E. coli has shown a high abundance of OM and periplasmic proteins as well as some inner membrane and cytoplasmic proteins within OMVs [Citation59,Citation60]. Proteomic comparison of OMVs to the cellular membrane and to the periplasmic space suggest that some packaging mechanism is employed for OMV loading since the protein composition does not proportionally correlate to protein abundance in either the OM or periplasmic space [Citation59,Citation61]. While the machinery involved in the packaging process has yet to be fully identified there are many known proteins that are present in high abundance in OMVs providing for numerous targets for synthetic modification to allow for controlled packaging of proteins of interest for therapeutic applications.
Bacterial vesicles as a therapeutic agent
The use of proteins expressed by bacteria as pharmaceutical agents began with the FDA approval of Humulin (1982), an E. coli-produced insulin used to treat Type I and Type II diabetes, opening the door for other bacterial-produced insulin formulations such as Humalog [Citation62]. While bacteria have been utilized to produce many peptides and proteins, bacterial vesicles themselves have largely not been utilized as therapeutic agents and their development as such has been extremely limited. OMVs are however, an attractive platform for nanoparticle synthesis of targeted protein encapsulated liposomes as the molecular machinery necessary to produce them has already naturally been fully developed by the bacteria. Additionally, numerous molecular biology tools for recombinant protein expression in bacteria are readily available providing avenues of producing and targeting proteins of interest to the periplasmic space and OM to drive OMV encapsulation. With slight modifications to these established protocols, the bacteria can be programmed to export and package recombinant proteins, either free or bound, to the inner or outer surfaces of the vesicle walls similar to liposomes. With the vast array of molecular tools for bacterial expression of recombinant proteins, the combinations of proteins, peptides and nucleic acids that can be packaged within OMVs are nearly limitless.
As previously stated, the pathway for OMV formation and packaging has yet to be elucidated. In an attempt to circumvent the lack of knowledge regarding the packaging mechanisms, researchers have tried to load OMVs simply by flooding the periplasmic space with the protein of interest [Citation63] or through in vitro loading of nucleic acids via electroporation [Citation64] (). While periplasmic loading with recombinant proteins has been shown to be successful by Kesty et al., in some cases this method is inefficient and cannot be applied across all recombinant proteins [Citation63]. As was shown in the Lee et al. proteomic studies, periplasmic proteins are not necessarily packaged into OMVs based solely on abundance [Citation59]. Therefore, while some examples of protein loading within OMVs have been reported, utilizing these methods offer no avenue of ensuring OMV loading and provide no method for separating ‘filled’ versus ‘empty’ OMVs. To utilize protein packaged OMVs as a therapeutic the packaging efficiency must be more strictly regulated.
Rather than focus on overproduction of a protein of interest, a potentially better approach is through direct anchoring of recombinant proteins to the OM itself. Two methods of OM anchoring that have seen some success, developed for nontherapeutic purposes, have been the inclusion of a lipid functional group attached to the target protein that drives membrane tethering or fusion of the target protein directly to a transmembrane OM protein [Citation65,Citation66]. Either method serves as an effective method of anchoring recombinant proteins to the bacterial OM (). While these methods may improve loading efficiency and subsequent abundance of recombinant proteins in the OMVs, a method of separating empty and filled OMVs will still be required. This will be particularly necessary for the development of OMV therapeutics to allow for precise quantitation of the loaded protein for dosage calculations. This issue has not been addressed to date but may be best addressed by linking an exterior-facing epitope tag in addition to the recombinant payload protein. Subsequent affinity purification utilizing the epitope tag would provide a rapid avenue for separation and concentration of functional protein-loaded OMVs over empty OMVs. Research utilizing OMVs as therapeutic agents is still in its infancy and issues such as these will have to be addressed as the field matures.
Among other things, OMVs are believed to be involved in both the delivery of toxins and for cellular communication between bacteria within a biofilm. To facilitate targeted delivery of both signal molecules and toxins, OMVs present small peptide sequences that serve as ligands that bind cellular receptors present on target cells. Following the models of nature, the exterior of OMVs can be modified with targeting ligands through either molecular techniques or through in vitro conjugation strategies. Molecularly, transmembrane and other surface-exposed proteins can be manipulated to encode specific peptide sequences known to direct bacterial vesicles to target cells or tissues. Kesty et al. showed that OMVs presenting the Ail peptide of Yersinia pestis on their surface could deliver green fluorescent protein to eukaryotic cells in vitro [Citation63]. Similarly, Gujrati et al. produced vesicles with an anti-HER2 affibody expressed on its surface and showed that OMV cargo could be selectively delivered to tumor cells [Citation64]. In addition to direct multivalent presentation of targeting moieties on the OMV surface, the abundance of OM proteins with exterior facing domains offer numerous avenues for engineering bioorthogonal tags and functional groups that can subsequently be targeted for in vitro conjugation or provide various OMV purification options [Citation67,Citation68].
While only theoretical at this point, production of therapeutic OMVs would follow a simplified workflow compared with a similar synthetic liposome. Once a bacterial strain and expression system has been developed, the recombinant protein expression would be largely ‘plug-and-play’ for simple therapeutic proteins. Recombinant protein and OMV production can be strictly regulated and induced through any of several chemical (IPTG, arabinose, rhamnose, among others) or environmental (heat, cold, light, among others) conditions. The bacteria would synthesize and package the bulk membrane lipids, the externally facing targeting ligands and the encapsulated protein of interest eliminating multiple purification and synthetic steps associated with manufacturing a comparable liposome-derived therapeutic nanoparticle. Fully assembled exported OMV nanoparticles can then be purified directly from the culture medium, eliminating laborious cellular purification protocols and allowing for removal of a large number of contaminating substances in a single filtration step (). A comparison of the benefits and drawbacks associated with selection of each nanoparticle class, including OMVs, can be found in . Current OMV purification methods rely on ultracentrifugation which is a time intensive process, however, this purification technique can be circumvented through the development of alternate filtration and affinity purification protocols [Citation69]. OMVs, as with all recombinant therapeutics, would be subjected to stringent quality control methods to ensure removal of bacterial DNA and contaminating proteins prior to clinical use.
Bacterial vesicle hurdles
While bacterial vesicles as therapeutic agents are very attractive, they come with their own set of complexities when considering clinical implementation. One of the primary hurdles is the potential immunogenicity issue associated with the LPS and the presence of foreign proteins incorporated within the OMVs. The immunogenic issues associated with the LPS have already been investigated and there exist known LPS variants that have reduced or removed the immunogenicity of bacterial OMVs [Citation70]. It should be noted, however, that in some instances immune activation can be highly beneficial as it provides for endogenous clearance of targeted cells of interest. Modulating immune activation, and packaging specific proteins of interest, may also allow OMVs to be utilized as a unique vaccine development and delivery vehicle.
Hijacking bacterial vesicle production will still require the use of complex nanoparticle purification and characterization techniques [Citation71]. The purification process for the formed OMVs will not be that dissimilar to that of the final formed liposome therapeutic described above. Important things to consider for OMV use as a therapeutic is the removal of any genetic material or other nonencapsulated bacterial components present in the growth media. There already exist common methods for this as all recombinant therapeutic proteins must undergo a similar purification process. Other, not so easily surmountable, purification issues exist regarding the removal of bacterial proteins that are passively packaged within the vesicle or bound to the vesicle surface. For some applications these proteins may not be an issue from the perspective of immunogenicity but gaining FDA approval for such a therapeutic may be difficult due to potential consistency and purity issues. However, as the therapeutic OMV production process will utilize common laboratory strains that are maintained under highly controlled conditions, careful selection of an expression system, and through the development of highly mutated bacterial strains, much of the composition of the OMV proteins can be removed, modified or replaced with less immunogenic components.
Advanced capabilities of bacterial vesicles
Bacteria offer a well-developed platform for the production of therapeutics and novel reagents. The E. coli genome has been mapped in its entirety and there is already a vast suite of extrachromosomal DNA plasmids available for protein expression. Large-scale fermentation production systems and chemostat growth techniques are also well documented allowing for easily scalable bacterial growth and OMV recovery. By employing the cellular machinery of the bacterium, complex processes such as multiprotein packaging within OMVs, gene delivery and incorporation of targeting moieties, can readily be accomplished.
Simultaneous delivery of two or more proteins or enzymes to a target cell or tissue could prove beneficial in applications such as repairing biosynthetic pathways of anabolism/catabolism in target cells or removing bacterial or viral intracellular pathogens through synergistic avenues of attack. Creative molecular techniques could readily be employed to allow for the simultaneous packaging of two or more proteins in OMVs. This process would be similar to the work of Patterson et al. where protein–protein interactions were exploited to package three recombinant proteins in a P22 viral capsid [Citation72]. Similar techniques have been employed for sequestering proteins to the bacterial OM, utilizing paired alpha-helical fusions, split inteins or other known protein–protein interactions that would facilitate directing multiple components to the bacterial OMV. Fusion of a bacterially produced protein to the bacterial outer membrane through an engineered linker drives packaging of the protein of interest within OMVs and can also be designed to selectively release the encapsulated proteins via hydrolyzable linkers sensitive to various conditions such as fluctuations in pH or by incorporating known proteolytic cleavage peptide sequences recognized by endogenous enzymes.
While bacteria are capable of synthesizing and folding many complex proteins there are some small molecules and functional groups that cannot be produced by bacteria that are very useful for many therapeutic applications, such as chemotherapeutic agents. There are however, both post-production methods and molecular biology techniques that allow for incorporation of such functional groups into OMVs. A post-production method for internalizing a chemotherapeutic within the produced and purified OMV can be accomplished by utilizing transmembrane pH gradients to drive internalization; a similar method is used in the loading of doxorubicin into liposomes. Additionally, utilizing a molecular biology approach one could take advantage of the small peptide sequences present on the exterior surface of OMVs to incorporate bioorthogonal labeling targets, unnatural amino acids for ‘click’ chemistry and biotinylation sequences offering many avenues for functionalizing the exterior of OMVs with a plethora of peptides, proteins and chemical groups [Citation67,Citation68]. This conjugation technique also allows for development of advanced therapeutics that can include selectively hydrolyzable linkers for prodrug delivery. While functionalization of nanoparticle therapeutics is not novel, utilizing a single system for manufacture, loading and targeting is something that cannot readily be achieved by any other nanoparticle system. Bacterial OMVs can therefore exhibit nearly all of the desired properties of the ideal nanoparticle-based therapeutic while eliminating many labor- and cost-intensive synthetic and purification techniques typically associated with developing complex multifunctional therapeutic nanoparticles.
Conclusion
There are many potential benefits associated with bacterial production of OMVs for therapeutic use that include significantly simplified assembly without compromising overall functional capabilities, when compared with the other nanoparticle classes. Unlike other nanoparticle formulations, the bacteria themselves synthesize the lipids, produce the targeting ligands displayed on the outer surface of the vesicle, and express and package the recombinant proteins encapsulated within the vesicles to potentially greatly reduce the cost and complexity of production. For this reason a single round of purification of the final product can be carried out rather than producing and purifying each individual component and then purifying the final combined nanoparticle therapeutic, as is the case when utilizing traditional liposome-based nanoparticles. Similar to liposomes, bacterial OMVs can be utilized in both active and passive targeting applications and are amenable to multidrug delivery of nearly any combination of hydrophilic, hydrophobic or protein-based therapeutic agents. Due to their protein, lipid and polysaccharide composition there will likely be limited issues regarding OMV biocompatibility or biological clearance post delivery. Bacterial-based synthesis also has the added benefits of being easily scalable allowing for flexible production capabilities and reduced batch-to-batch variations due to large culture sizes.
This system also allows for enhanced enzyme and protein packaging control that can be achieved through known stoichiometry-based on mutant selection and the expression system that is utilized. Once the bacterial vesicle platform is established, incorporating various proteins and enzymes can be ‘plug-and-play’ while purification and production can remain relatively unchanged. This is often not the case when producing variations to nanoparticle formulations as each component change has a complex and often unforeseeable impact on the nanoparticle formation process. The use of bacterial OMVs as therapeutic agents is a long-term goal for the advanced treatment of disease as they offer many benefits over current nanoparticle formulations. These impressive bacteria possess all of the necessary components to produce exceptionally complex therapeutics at potentially greatly reduced production and purification cost.
Future perspective
Like all nanoparticle therapeutics, bacterial OMVs do not possess every characteristic of an ideal nanoparticle yet they hit many of the primary requirements making them an extremely promising drug delivery vehicle that is worth further development. Since OMV utilization is in its infancy there are many drug delivery parameters that have yet to be fully characterized such as PK, PD, circulation time, clearance, cellular uptake, immunogenicity, in vivo stability, shelf life and primary clinical targets for initial implementation. One of the only studies, to date, investigating in vivo OMV biodistribution and immune activation in nonimmunodeficient mice was carried out by injecting nontargeted, unmodified OMVs, purified from E. coli, and demonstrated accumulation of the nanoparticles in the liver and resulted in a tolerable systemic immune activation [Citation73]. We anticipate that many more of these studies will be carried out utilizing targeted OMVs that possess various mutations to modulate immune activation and drive accumulation of the nanoparticles to diseased sites similar to the study carried out by Gujrati et al. which used HER2-targeted OMVs in immunodeficient mice [Citation63]. Developing a catalog of the bacterial protein modifications necessary to create stealth, targeted OMVs and the mutations necessary to drive protein packaging will be the primary areas of study moving forward. While the cellular uptake rates, intracellular stability and degradation pathways for OMVs have not yet been thoroughly determined, we anticipate that these parameters will be very similar to current therapeutic liposomes and will depend heavily upon the targeted cell-surface receptor unique to each therapeutic application. Extensive preclinical research still remains necessary prior to bringing bacterial OMVs into the clinic yet there is promising evidence that through modifications to the OMV LPS and targeting ligands that the PK and PD of the OMV nanoparticles can be tightly regulated to allow for favorable drug release kinetics, controlled immune activation and enhanced local delivery of therapeutic agents.
Table 1. Nanoparticle delivery platforms
Active targeting: the use of targeting agents bound to the nanoparticle surface such as small molecules, peptides or proteins to direct interactions with specific cell/tissue populations.
Passive targeting: a nanoparticle that does not have targeting agents on its surface that relies on size-based localization of the nanoparticle to target a specific location within the body.
Avidity: a term used to describe affinity of multiple interactions between two bodies, in other words, multivalent affinity.
Pharmacokinetics: the study of the movement and distribution of pharmaceuticals throughout the body.
Pharmacodynamics: the study of the physiological effects that pharmaceuticals have on the body and their mechanisms of actions.
Outer membrane vesicle: a vesicle that is secreted by gram-negative and gram-positive bacteria that is shed by the bacteria to perform various functions.
Lipopolysaccharide: present on the bacterial surface, and subsequent outer membrane vesicles, the lipopolysaccharide is comprised of a lipid A and polysaccharide chain split into the inner core, outer core and O-antigen. The O-antigen is considered to be highly immunogenic.
Tightly controlled size distribution
Inexpensive and scalable production capabilities
Delivery of hydrophobic and hydrophilic small molecules
Delivery of large protein-based therapeutics
Simultaneous multidrug delivery capabilities
Active and passive targeting capabilities
Defined drug release kinetics
Defined pharmacokinetics and pharmacodynamics
Adjustable circulation time
Biologically compatible clearance
Long shelf life
Nanoparticles as therapeutics
Nanoparticles offer unique multifunctional targeting and drug delivery capabilities.
There are many different classes of nanoparticles (dendrimer, lipid-based, polymeric and inorganic) each with their own set of advantages and drawbacks for use.
Lipid-based nanoparticles currently are the dominant US FDA-approved nanoparticle type as they have been studied the longest and offer limited biological clearance issues.
Polymeric nanoparticles and various pegylated therapeutics have also demonstrated promise in clinical applications offering stealth and biodegradable capabilities.
No single class of nanoparticle can be applied across all therapeutic nanoparticle applications.
General nanoparticle hurdles
Things to consider when developing a nanotherapeutic include manufacturing complexity, cost, toxicity, intra-batch uniformity, inter-batch variation, synthetic yield, characterization/validation techniques, scalability and shelf life/stability.
FDA approval of complex nanoparticle formulations for therapeutic applications can be exceedingly difficult to attain.
Bacterial vesicles as a therapeutic agent
Bacterial outer membrane vesicle (OMV) production can be utilized to both synthesize and package proteoliposome-based therapeutics.
Common molecular biology techniques can be used to modify bacterial OMVs to target select cell types, deliver small-molecule pharmaceuticals and encapsulated proteins.
Hijacking OMV production eliminates many synthetic and purification steps necessary to make comparable liposome-based therapeutics with the potential to greatly reduce cost.
Bacterial vesicle hurdles
Methods for the removal of bacterial proteins and undesired genetic material from the host bacteria passively packaged in the OMVs will need to be developed.
OMV development as therapeutic nanoparticles is an emerging field and as such considerable preclinical validation still remains.
Financial & competing interests disclosure
This research was funded in part by the Office of Naval Research through core funding provided to the Naval Research Laboratory. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
- Lee N Choi SH Hyeon T . Nano-sized CT contrast agents. Adv. Mater.25 (19), 2641–2660 (2013).
- Rahman M Ahmad MZ Kazmi I et al. Emergence of nanomedicine as cancer targeted magic bullets: recent development and need to address the toxicity apprehension. Curr. Drug Discov. Technol.9 (4), 319–329 (2012).
- Shin MC Zhang J Min KA et al. Cell-penetrating peptides: achievements and challenges in application for cancer treatment. J. Biomed. Mater. Res. A.102 (2), 575–587 (2014).
- van Riet E Ainai A Suzuki T Kersten G Hasegawa H . Combatting infectious diseases; nanotechnology as a platform for rational vaccine design. Adv. Drug Deliver. Rev.74, 28–34 (2014).
- Bertrand N Wu J Xu X Kamaly N Farokhzad OC . Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliver. Rev.66, 2–25 (2014).
- Guo S Huang L . Nanoparticles containing insoluble drug for cancer therapy. Biotechnol. Adv.32 (4), 778–788 (2014).
- Highsmith J . Nanoparticles In Biotechnology, Drug Development And Drug Delivery. BCC Research, MA, USA, 187 (2012).
- Lopez-Abarrategui C Figueroa-Espi V Reyes-Acosta O Reguera E Otero-Gonzalez AJ . Magnetic nanoparticles: new players in antimicrobial peptide therapeutics. Curr. Protein Pept. Sci.14 (7), 595–606 (2013).
- Hassan S Singh AV . Biophysicochemical perspective of nanoparticle compatibility: a critically ignored parameter in nanomedicine. J. Nanosci. Nanotechnol.14 (1), 402–414 (2014).
- Tu Q Zhang Y Liu R et al. Active drug targeting of disease by nanoparticles functionalized with ligand to folate receptor. Curr. Med. Chem.19 (19), 3152–3162 (2012).
- Steichen SD Caldorera-Moore M Peppas NA . A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci.48 (3), 416–427 (2013).
- Johnson BJ Algar WR Malanoski AP Ancona MG Medintz IL . Understanding enzymatic acceleration at nanoparticle interfaces: approaches and challenges. Nano Today9 (1), 102–131 (2014).
- Natarajan JV Nugraha C Ng XW Venkatraman S . Sustained-release from nanocarriers: a review. J. Control. Release193, 122–138 (2014).
- Spillmann CM Naciri J Algar WR Medintz IL Delehanty JB . Multifunctional liquid crystal nanoparticles for intracellular fluorescent imaging and drug delivery. ACS Nano8 (7), 6986–6997 (2014).
- Barar J Omidi Y . Surface modified multifunctional nanomedicines for simultaneous imaging and therapy of cancer. BioImpacts4 (1), 3–14 (2014).
- Duan X Li Y . Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small9 (9–10), 1521–1532 (2013).
- Dellinger A Zhou Z Connor J et al. Application of fullerenes in nanomedicine: an update. Nanomedicine (Lond.)8 (7), 1191–1208 (2013).
- Guo R Shi X . Dendrimers in cancer therapeutics and diagnosis. Curr. Drug Metab.13 (8), 1097–1109 (2012).
- Bharali DJ Khalil M Gurbuz M Simone TM Mousa SA . Nanoparticles and cancer therapy: a concise review with emphasis on dendrimers. Int. J. Nanomed.4 (1), 1–7 (2009).
- Madaan K Kumar S Poonia N Lather V Pandita D . Dendrimers in drug delivery and targeting: drug-dendrimer interactions and toxicity issues. J. Pharm. Bioallied Sci.6 (3), 139–150 (2014).
- Duncan R Vicent MJ . Polymer therapeutics-prospects for 21st century: the end of the beginning. Adv. Drug Deliver. Rev.65 (1), 60–70 (2013).
- Ginn C Khalili H Lever R Brocchini S . Pegylation and its impact on the design of new protein-based medicines. Future Med. Chem.6 (16), 1829–1846 (2014).
- Paraskar AS Soni S Chin KT et al. Harnessing structure-activity relationship to engineer a cisplatin nanoparticle for enhanced antitumor efficacy. Proc. Natl Acad. Sci. USA107 (28), 12435–12440 (2010).
- Gokmen MT Du Prez FE . Porous polymer particles–a comprehensive guide to synthesis, characterization, functionalization and applications. Prog. Polym. Sci.37 (3), 365–405 (2012).
- Nazemi K Azadpour P Moztarzadeh F Urbanska AM Mozafari M . Tissue-engineered chitosan/bioactive glass bone scaffolds integrated with plga nanoparticles: a therapeutic design for on-demand drug delivery. Mater. Lett.138, 16–20 (2015).
- Yang Y Wang S Wang Y Wang X Wang Q Chen M . Advances in self-assembled chitosan nanomaterials for drug delivery. Biotechnol. Adv.32 (7), 1301–1316 (2014).
- Gentile P Chiono V Carmagnola I Hatton PV . An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci.15 (3), 3640–3659 (2014).
- Mirakabad FST Nejati-Koshki K Akbarzadeh A et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac. J. Cancer Prev.15 (2), 517–535 (2014).
- Ikeda Y Nagasaki Y . Pegylation technology in nanomedicine. In : Polymers In Nanomedicine (Volume 247). KunugiSYamaokaT ( Eds). Springer Berlin Heidelberg, Berlin, Germany, 115–140 (2012).
- Acharya A . Luminescent magnetic quantum dots for in vitro/in vivo imaging and applications in therapeutics. J. Nanosci. Nanotechnol.13 (6), 3753–3768 (2013).
- Mok H Zhang M . Superparamagnetic iron oxide nanoparticle-based delivery systems for biotherapeutics. Expert Opin. Drug Del.10 (1), 73–87 (2013).
- Singh D McMillan JM Liu X-M et al. Formulation design facilitates magnetic nanoparticle delivery to diseased cells and tissues. Nanomedicine (Lond.)9 (3), 469–485 (2014).
- Zheng S-G Xu H-X Chen H-R . Nano/microparticles and ultrasound contrast agents. World J. Radiol.5 (12), 468–471 (2013).
- Cormode DP Skajaa GO Delshad A et al. A versatile and tunable coating strategy allows control of nanocrystal delivery to cell types in the liver. Bioconjugate Chem.22 (3), 353–361 (2011).
- Nam J Won N Bang J et al. Surface engineering of inorganic nanoparticles for imaging and therapy. Adv. Drug Deliver. Rev.65 (5), 622–648 (2013).
- Sengupta J Ghosh S Datta P Gomes A Gomes A . Physiologically important metal nanoparticles and their toxicity. J. Nanosci. Nanotechnol.14 (1), 990–1006 (2014).
- Simpson CA Salleng KJ Cliffel DE Feldheim DL . In vivo toxicity, biodistribution, and clearance of glutathione-coated gold nanoparticles. Nanomed. Nanotechnol.9 (2), 257–263 (2013).
- Torchilin VP . Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov.4 (2), 145–160 (2005).
- Dawidczyk CM Kim C Park JH et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J. Control. Release187, 133–144 (2014).
- Ang CY Tan SY Zhao Y . Recent advances in biocompatible nanocarriers for delivery of chemotherapeutic cargoes towards cancer therapy. Org. Biomol. Chem.12 (27), 4776–4806 (2014).
- Anselmo AC Mitragotri S . An overview of clinical and commercial impact of drug delivery systems. J. Control. Release190, 15–28 (2014).
- Cooper DL Conder CM Harirforoosh S . Nanoparticles in drug delivery: mechanism of action, formulation and clinical application towards reduction in drug-associated nephrotoxicity. Expert Opin. Drug Del.11 (10), 1661–1680 (2014).
- Kraft JC Freeling JP Wang Z Ho RJY . Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci.103 (1), 29–52 (2014).
- Chen WC May JP Li S-D . Immune responses of therapeutic lipid nanoparticles. Nanotechnol. Rev.2 (2), 201–213 (2013).
- Allen TM . Long-circulating (sterically stabilized) liposomes for targeted drug-delivery. Trends Pharmacol. Sci.15 (7), 215–220 (1994).
- Marques-Gallego P de Kroon AIPM . Ligation strategies for targeting liposomal nanocarriers. Biomed Res. Int.2014, 1–12 (2014).
- Barenholz Y . Liposome application: problems and prospects. Curr. Opin. Colloid In.6 (1), 66–77 (2001).
- Kiziltepe T Ashley JD Stefanick JF et al. Rationally engineered nanoparticles target multiple myeloma cells, overcome cell-adhesion-mediated drug resistance, and show enhanced efficacy in vivo. Blood Cancer J.2 (4), e64 (2012).
- Ashley JD Stefanick JF Schroeder VA et al. Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo. J. Control. Release196, 113–121 (2014).
- Beveridge TJ . Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol.181 (16), 4725–4733 (1999).
- Kulkarni HM Jagannadham MV . Biogenesis and multifaceted roles of outer membrane vesicles from gram-negative bacteria. Microbiology160 (Pt 10), 2109–2121 (2014).
- Kulp A Kuehn MJ . Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol.64 (1), 163–184 (2010).
- Remis JP Wei D Gorur A et al. Bacterial social networks: structure and composition ofmyxococcus xanthusouter membrane vesicle chains. Environ. Microbiol.16 (2), 598–610 (2014).
- Yaron S Kolling GL Simon L Matthews KR . Vesicle-mediated transfer of virulence genes from escherichia coli O157:H7 to other enteric bacteria. Appl. Environ. Microb.66 (10), 4414–4420 (2000).
- Mashburn-Warren L Howe J Garidel P et al. Interaction of quorum signals with outer membrane lipids: insights into prokaryotic membrane vesicle formation. Mol. Microbiol.69 (2), 491–502 (2008).
- Kato S Kowashi Y Demuth DR . Outer membrane-like vesicles secreted by actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb. Pathogenesis32 (1), 1–13 (2002).
- Horstman AL . Enterotoxigenic escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J. Biol. Chem.275 (17), 12489–12496 (2000).
- Park AJ Surette MD Khursigara CM . Antimicrobial targets localize to the extracellular vesicle-associated proteome of pseudomonas aeruginosa grown in a biofilm. Front. Microbiol.5, 464 (2014).
- Lee E-Y Choi D-S Kim K-P Gho YS . Proteomics in gram-negative bacterial outer membrane vesicles. Mass Spectrom. Rev.27 (6), 535–555 (2008).
- Lee E-Y Bang JY Park GW et al. Global proteomic profiling of native outer membrane vesicles derived from escherichia coli. Proteomics7 (17), 3143–3153 (2007).
- Haurat MF Aduse-Opoku J Rangarajan M et al. Selective sorting of cargo proteins into bacterial membrane vesicles. J. Biol. Chem.286 (2), 1269–1276 (2011).
- Simpson D McCormack PL Keating GM Lyseng-Williamson KA . Insulin lispro–a review of its use in the management of diabetes mellitus. Drugs67 (3), 407–434 (2007).
- Kesty NC Kuehn MJ . Incorporation of heterologous outer membrane and periplasmic proteins into escherichia coli outer membrane vesicles. J. Biol. Chem.279 (3), 2069–2076 (2003).
- Gujrati V Kim S Kim S-H et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano8 (2), 1525–1537 (2014).
- Ghrayeb J Inouye M . Nine amino acid residues at the NH2-terminal of lipoprotein are sufficient for its modification, processing, and localization in the outer-membrane of Escherichia coli. J. Biol. Chem.259 (1), 463–467 (1984).
- Francisco JA Campbell R Iverson BL Georgiou G . Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl Acad. Sci. USA90 (22), 10444–10448 (1993).
- Sapsford KE Algar WR Berti L et al. Functionalizing nanoparticles with biological molecules: developing chemistries that facilitate nanotechnology. Chem. Rev.113 (3), 1904–2074 (2013).
- Algar WR Prasuhn DE Stewart MH et al. The controlled display of biomolecules on nanoparticles: a challenge suited to bioorthogonal chemistry. Bioconjug. Chem.22 (5), 825–858 (2011).
- Alves NJ Cusick W Stefanick JF Ashley JD Handlogten MW Bilgicer B . Functionalized liposome purification via liposome extruder purification (LEP). Analyst138 (17), 4746–4751 (2013).
- Steeghs L Kuipers B Hamstra HJ Kersten G van Alphen L van der Ley P . Immunogenicity of outer membrane proteins in a lipopolysaccharide-deficient mutant of neisseria meningitidis: influence of adjuvants on the immune response. Infect. Immun.67 (10), 4988–4993 (1999).
- Sapsford KE Tyner KM Dair BJ Deschamps JR Medintz IL . Analyzing nanomaterial bioconjugates: a review of current and emerging purification and characterization techniques. Anal. Chem.83 (12), 4453–4488 (2011).
- Patterson DP Prevelige PE Douglas T . Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage P22. ACS Nano6 (6), 5000–5009 (2014).
- Jang SC Kim SR Yoon YJ et al. In vivo kinetic biodistribution of nano-sized outer membrane vesicles derived from bacteria. Small11 (4), 456–461 (2015).