
Abstract
The present studies examined the biology of the multiple sclerosis drug dimethyl-fumarate (DMF) or its in vivo breakdown product and active metabolite mono-methyl-fumarate (MMF), alone or in combination with proteasome inhibitors, in primary human glioblastoma (GBM) cells. MMF enhanced velcade and carfilzomib toxicity in multiple primary GBM isolates. Similar data were obtained in breast and colon cancer cells. MMF reduced the invasiveness of GBM cells, and enhanced the toxicity of ionizing radiation and temozolomide. MMF killed freshly isolated activated microglia which was associated with reduced IL-6, TGFβ and TNFα production. The combination of MMF and the multiple sclerosis drug Gilenya further reduced both GBM and activated microglia viability and cytokine production. Over-expression of c-FLIP-s or BCL-XL protected GBM cells from MMF and velcade toxicity. MMF and velcade increased plasma membrane localization of CD95, and knock down of CD95 or FADD blocked the drug interaction. The drug combination inactivated AKT, ERK1/2 and mTOR. Molecular inhibition of AKT/ERK/mTOR signaling enhanced drug combination toxicity whereas molecular activation of these pathways suppressed killing. MMF and velcade increased the levels of autophagosomes and autolysosomes and knock down of ATG5 or Beclin1 protected cells. Inhibition of the eIF2α/ATF4 arm or the IRE1α/XBP1 arm of the ER stress response enhanced drug combination lethality. This was associated with greater production of reactive oxygen species and quenching of ROS suppressed cell killing.
Abbreviations
| DMF | = | dimethyl-fumarate |
| MMF | = | monomethyl-fumarate |
| ERK | = | extracellular regulated kinase |
| MEK | = | mitogen activated extracellular regulated kinase |
| EGF | = | epidermal growth factor |
| PARP | = | poly ADP ribosyl polymerase |
| PI3K | = | phosphatidyl inositol 3 kinase |
| −/− | = | null / gene deleted |
| MAPK | = | mitogen activated protein kinase |
| PTEN | = | Phosphatase and tensin homolog |
| R | = | receptor |
| JNK | = | c-Jun NH2-terminal kinase |
| dn | = | dominant negative |
| P | = | phospho- |
| ca | = | constitutively active |
| WT | = | wild type |
Introduction
In the United States, glioblastoma multiforme (GBM) is diagnosed in ∼20,000 patients per annum. High-grade tumors such as anaplastic astrocytoma and GBM account for the majority of tumors.Citation1,2 Even under optimal circumstances, in which all of the tumor can be surgically removed and the patients are maximally treated with radiation and chemotherapy, the mean survival is only extended from ∼3 months to 1 y There is a major unmet need for new approaches to treat this lethal disease.
Dimethyl fumarate (DMF) is a methyl ester of fumaric acid and for many years has been used in Europe for the treatment of psoriasis.Citation3 More recently, DMF has been approved in The United States for the treatment of multiple sclerosis (Tecfidera).Citation4 The drug has immunomodulatory actions, e.g. T cell inactivation, that may be linked to increased expression of Nrf2 and HO-1 and an anti-oxidant response.Citation5,6 DMF, at the approved dose for multiple sclerosis therapy, is rapidly metabolized to mono-methyl fumarate (MMF), and has a C max in plasma of ∼15 μM, with an approximate steady state tissue and plasma concentration of 5 μM, though many studies using this compound have used the drug at much higher concentrations.Citation4,Citation7-10 In addition to its actions on immune cells, DMF also suppresses the inflammatory biology of microglia and astrocytes.Citation11-15 As activated microglia and reactive astrocytes play key roles in the biology and progression of GBM tumors in vivo, DMF represents one potential drug which could alter GBM growth and the growth of other tumor types in vivo.Citation16,17
Proteasome inhibitors e.g., velcade, carfilzomib inhibit the activity of the 20S proteasome.Citation18 Velcade is a reversible inhibitor; carfilzomib is an irreversible inhibitor and can kill tumor cells made resistant to velcade.Citation19-26 The ubiquitin-proteasome system regulates protein expression within cells and has a regulatory role in the apoptotic rheostat and the response to reactive oxygen species (ROS) and to DNA damage. The mechanisms by which proteasome inhibitors kill tumor cells are diverse and include endoplasmic reticulum stress; the generation of ROS; inhibition of NFκB; and modulation of signal transduction pathway activities. DMF has also been shown to inhibit NFκB.Citation17,27
The endoplasmic reticulum (ER) stress response prevents accumulation of unfolded proteins in the ER, and may lead to autophagy if unchecked. There are 3 primary UPR sensors: PERK, (PKR-like ER kinase), ATF6 (activating transcription factor 6) and IRE1.Citation28 As unfolded proteins accumulate, BiP (Grp78), the HSP70 ER resident chaperone, dissociates from PERK, ATF6 or IRE1.Citation29 BiP/Grp78 dissociation from PERK allows this protein to dimerize, autophosphorlate, and then phosphorylate eIF2α, the protein required for bringing the initiator methionyl-tRNA to the 40S ribosome.Citation30 Phosphorylated eIF2α thus leads to repression of global translation, helping to allow cells to recover from the accumulation of unfolded proteins. Reduced translation, however, can also lower expression of some pro-survival proteins such as MCL-1 leading to increased cell death.Citation31 Phosphorylation of eIF2α also leads to the increased transcription of activating transcription factor 4 (ATF4), which activates subsets of genes involved in metabolism, transport, redox reactions and ER stress-induced programmed cell death.Citation32 One of these proteins C/EBP homologous transcription factor (CHOP / GADD153) is implicated in both growth arrest and in apoptosis.Citation33 When Grp78/BiP dissociates from ATF6, this protein translocates to the Golgi complex, where it is cleaved by S1P and S2P.Citation34 This active form of ATF6 then translocates to the nucleus, where it binds to the ER stress response element (ERSE) to promote the transcription of ER-resident chaperones, e.g. Grp78/BiP, and other enzymes that assist in protein folding. After Grp78/BiP releases IRE1, this protein dimerizes, activating its cytosolic RNAse domain. This domain of IRE1 cleaves a sequence from the X-box DNA binding protein (XBP1).Citation35 The cleaved XBP1 translocates to the nucleus and binds the upstream DNA UPR element (UPRE) and thus is a potent activator of UPR genes. The UPR genes regulated by the UPRE are essential for protein folding, maturation and degradation. Although some ER stress responses lead to increased cell survival, under prolonged ER stress, the UPR can lead to reduced expression of pro-survival proteins and in addition, disruption of ER calcium homeostasis.Citation36
The present studies were designed to examine the biology of DMF in primary human GBM cells, and also when the drug is combined with therapeutic agents, particularly standard of care therapies as well as proteasome inhibitors. DMF modestly enhanced the lethality of ionizing radiation and temozolomide, and enhanced proteasome inhibitor toxicity. Cell killing was associated with increased autophagy and activation of the extrinsic apoptosis pathway. Endoplasmic reticulum stress was induced with the eIF2α and IRE1α arms of the stress response being protective against drug combination toxicity.
Results
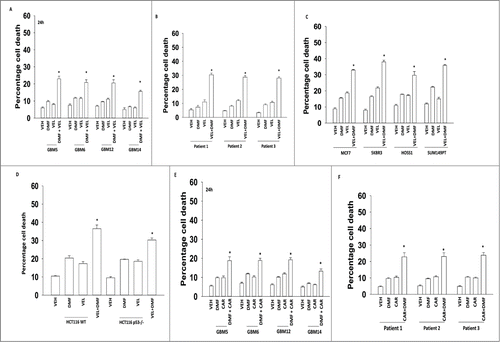
Initial studies examined the effect of DMF on velcade and carfilzomib toxicity. In semi-established GBM cells DMF and velcade interacted to cause increased levels of cell killing (). Similar data were obtained using freshly isolated GBM cell isolates (). Velcade and DMF interacted to kill breast, medulloblastoma and colon cancer cells; killing was marginally altered by loss of p53 function (). We next determined whether the irreversible proteasome inhibitor carfilzomib interacted with DMF; in semi-established GBM cells and in freshly isolated GBM cells carfilzomib interacted with DMF to cause tumor cell death ().
Figure 1. DMF enhances proteasome inhibitor toxicity in tumor cells. (A) GBM5/6/12/14 cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (B) Freshly isolated GBM cells (patients 1/2/3) were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (C) Breast cancer cells (MCF7, SKBR3; SUM149PT) or primary medulloblastoma cells (HOSS1) were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (D) HCT116 wild type and HCT116 p53 −/− colon cancer cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (E) GBM5/6/12/14 cells were treated with DMF (5 μM), carfilzomib (5 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (F) Freshly isolated GBM cells (patients 1/2/3) were treated with DMF (5 μM), carfilzomib (5 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone.
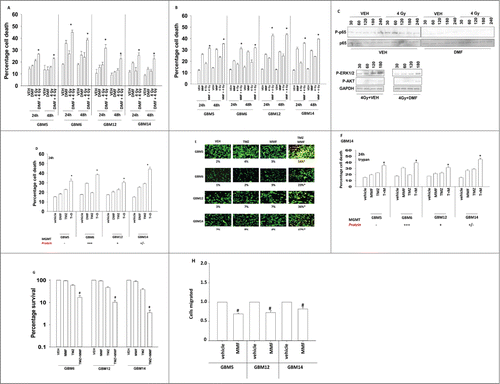
We next determined the impact of DMF and its breakdown product MMF on the toxic actions of standard of care therapies for GBM. DMF and MMF enhanced radiation toxicity in GBM cells (). These effects correlated with inhibition of radiation-induced NFκB p65; ERK1/2 and AKT phosphorylation (). DMF and MMF enhanced the toxicity of temozolomide in GBM cells (). MMF interacted with temozolomide to kill GBM cells as measured in colony formation assays after transient drug exposure (). DMF suppressed the invasiveness of GBM cells ().
Figure 2. DMF interacts with standard of care GBM therapeutics to enhance cell killing. (A) GBM5/6/ 12/14 cells were treated with DMF (5 μM) followed, as indicated, by irradiation (4 Gy). Cells were isolated 24 h and 48 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (B) GBM5/6/ 12/14 cells were treated with MMF (5 μM) followed, as indicated, by irradiation (4 Gy). Cells were isolated 24 h and 48 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (C) GBM6 cells were irradiated (4 Gy) in the presence of vehicle of DMF (5 μM). Cells were isolated at the indicated times and immunoblotting performed to determine the phosphorylation of p65 NFκB; ERK1/2 and AKT (n = 3). (D) GBM5/6/12/14 cells were treated with DMF (5 μM) and/or temozolomide (3 μM). Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than DMF alone. (E) GBM5/6/12/14 cells were treated with MMF (5 μM) and/or temozolomide (3 μM). Cells were isolated 24 h after treatment and viability determined by live / dead assay where green = alive; yellow = almost dead; red = dead (n = 3, +/− SEM) *P < 0.05 greater than MMF alone. (F) GBM5/6/12/14 cells were treated with MMF (5 μM) and/or temozolomide (3 μM). Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than MMF alone. (G) GBM6/12/14 cells that express MMGT were plated as single cells in 60 mm dishes (250–1,500 cell per dish). Twelve h after plating cells were treated with vehicle, MMF (5 μM), Temozolomide (TMZ, 3 μM) or the drugs in combination for 24 h. After 24 h the media was removed, the cells washed with drug free media and the media replaced using drug free media. Colonies were permitted to form for 10–14 days, after which time the colonies were fixed, stained and counted (n = 2 studies, in sextuplicate +/− SEM). # P < 0.05 less than TMZ alone treatment. (H) GBM5/12/14 cells were placed into chambers in 24 well plates and the migration of cells in the presence of vehicle or MMF (5 μM) was determined as in Methods (n = 3, +/− SEM) #P < 0.05 less than vehicle.
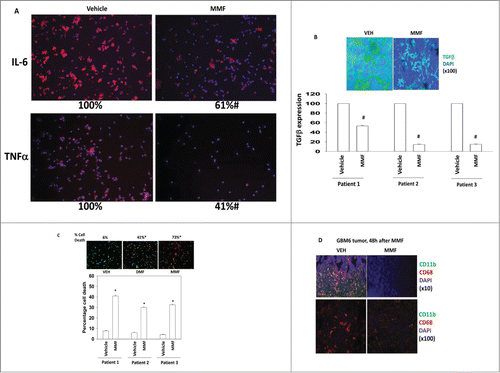
As noted in the Introduction, DMF / MMF has been reported to suppress the reactive phenotype of microglia and astrocytes. Using activated microglia freshly isolated from patient GBM tumors we found that MMF, within 12 h, suppressed the production of IL-6 and TNFα (). Similar data were obtained for TGFβ (). Identical data were obtained treating a mouse with DMF carrying a human GBM tumor growing in its brain (, upper panel). Treatment of activated microglia with DMF or with MMF caused significant amounts of microglia cell death within 24 h (). In orthotopic pre-formed GBM6 tumors in the brains of mice, a 48 h exposure to MMF resulted in a reduction in mouse microglia levels within the tumor ().
Figure 3. MMF kills freshly activated microglia from GBM tumors that is associated with reduced cytokine expression. Activated microglia were isolated from grade IV GBM tumors. (A) Cells were treated with vehicle or MMF (5 μM) and 24 h later cells examined using IHC for the expression of IL6 and TNFα (n = microglia from 3 tumors in triplicate +/− SEM) #P < 0.05 less than vehicle. (B) Cells were treated with vehicle or MMF (5 μM) and 24 h later cells examined using IHC for the expression of TGFβ (n = microglia from 3 tumors in triplicate +/− SEM) #P < 0.05 less than vehicle. (C) Upper images: activated microglia were treated with vehicle, DMF (5 μM) or MMF (5 μM) for 24 h after which they were isolated and cell viability determined by live / dead assay (n = microglia from 3 tumors in triplicate +/− SEM). Lower graph: activated microglia from 3 patients were treated with vehicle or MMF (5 μM) for 24 h after which they were isolated and cell viability determined by live / dead assay (n = 3, +/− SEM) *P < 0.05 greater than vehicle. (D) GBM6 cells (5 × 105) were infused into the right caudate putamen of athymic mice. Fourteen days after infusion animals were treated with either vehicle diluent (0.08% methocel) or with diluent containing MMF 30 mg/kg BID for 2 d PO. After 48 h, animals were sacrificed, their brains fixed and sectioned in a microtone (10 μm) and immuno-histochemistry performed against CD11b and CD68 in the tumors. Sections were counter-stained with DAPI. Images are at 10× and 100× magnification of the proliferating edge of the GBM6 tumor (n = 3).
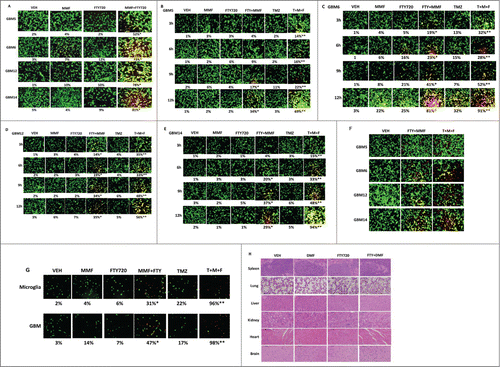
DMF is a recently approved medication for the treatment of multiple sclerosis; another recently approved drug for multiple sclerosis therapy is FTY720 (Fingolimod, Gilenya). Whereas DMF modulates cellular redox status to suppress immune cell activation, FTY720 acts to block sphingosine-1-phosphate signaling through inhibition of sphingosine kinases and down-regulation of sphingosine-1-phosphate receptors. MMF and FTY720 interacted to kill GBM cells within 24 h (). The lethality of the standard of care drug Temozolomide was also enhanced by [MMF + FTY720] treatment within 12 h (). Treatment of GBM cells for 12 h followed by drug free out-growth for 48 h still resulted in elevated levels of tumor cell killing (). Similar cell killing data to those with our PDX GBM isolates were obtained when we examined MMF / FTY720 / Temozolomide lethality in freshly isolated human GBM cells and in their associated purified activated microglia (). As judged using H&E staining of tissues isolated from drug treated animals, no normal tissue toxicity was observed using any of our drug combinations ( and data not shown).
Figure 4. MMF interacts with FTY720 (Fingolimod, Gilenya) to kill GBM cells. (A) GBM cells were treated with vehicle or MMF (5 μM), FTY720 (50 nM) or both drugs in combination and viability determined 24 h later using a live / dead assay (n = 3 +/− SEM) * P < 0.05 greater than MMF alone. (B–E) GBM5, GBM6, GBM12, GBM14 cells were treated with MMF (5 μM), FTY720 (50 nM), Temozolomide (TMZ, 3 μM) or in combination as indicated for the times (3 h–12 h) as indicated. Cell viability was assessed by live / dead assay. (n = 3 +/− SEM) *P < 0.05 greater than MMF; **P < 0.05 greater than MMF+FTY720 value. (F) GBM cells were treated with MMF+FTY720 or with MMF+FTY720+TMZ for 12 h. Cells were washed free of drug, and cell growth / repopulation permitted to occur for 48 h. Cell viability was assessed by live / dead assay. (G) GBM tumors, fresh from the operating room, were gently digested and dissociated, and microglia and primary GBM cells purified. Cells were plated and 12 h after plating were treated with MMF (5 μM), FTY720 (50 nM), Temozolomide (TMZ, 3 μM) or in combination as indicated for 12 h. Cell viability was assessed by live / dead assay. (n = 3 +/− SEM) *P < 0.05 greater than MMF; **P < 0.05 greater than MMF+FTY720 value. (H) BALB/c immune competent mice were treated for 14 d with: vehicle (cremophore); DMF (75 mg/kg); FTY720 (0.6 mg/kg) or the drugs in combination. After 14 d the mice were sacrificed and their organs fixed. Sections (10 μm) or each organ were taken and stained with H&E.
We next attempted to define the signaling and cell death pathways by which DMF and velcade interacted to kill GBM cells. The toxic interaction between DMF and velcade was suppressed by overexpression of BCL-XL or c-FLIP-s but not by expression of dominant negative caspase 9 (). DMF and velcade interacted to cause plasma membrane localization of the death receptor CD95, and knock down of CD95 or of the docking protein FADD suppressed the toxic interaction between the drugs (). Plasma membrane localization of CD95 was blocked by incubation of cells with the reactive oxygen species quenching agent N-acetyl cysteine and the inhibitor of ceramide synthase enzymes fumonisin B1. Knock down of CD95 or FADD protected cells from DMF and Velcade toxicity (). Quenching of reactive oxygen species protected cells from DMF and Velcade toxicity whereas surprisingly, inhibition of ceramide synthase enzymes increased cell killing (). The drug combination increased the numbers of autophagosomes and autolysosomes in cells, and knock down of the autophagy regulatory proteins Beclin1 or ATG5 suppress cell killing by DMF and velcade ().
Figure 5. The molecular mechanisms by which DMF/MMF and velcade interact to kill. (A) GBM cells (patient 2/3) were infected with recombinant adenoviruses to express: empty vector (CMV); the mitochondrial protective protein BCL-XL; the caspase 8 inhibitor c-FLIP-s; dominant negative caspase 9. Twenty 4 h after infection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) # P < 0.05 less than corresponding value in CMV infected cells. (B) GBM cells (patient 2) were treated with DMF (5 μM), velcade (10 nM) or the drug combination, or in parallel with N-acetyl cysteine (10 mM) or Fumonisin B1 (FB1, 25 μM). Cells were isolated 6 h after treatment and the cell surface levels of the death receptor CD95 determined (n = 3, +/− SEM) * P < 0.05 greater than DMF alone. (C) GBM cells (patient 3) were treated with DMF (5 μM), velcade (10 nM) or the drug combination, or in parallel with N-acetyl cysteine (10 mM) or Fumonisin B1 (FB1, 25 μM). Cells were isolated 6 h after treatment and the cell surface levels of the death receptor CD95 determined. (D) GBM cells (patient 2/3) were transfected with either a scrambled siRNA (siSCR) or siRNA molecules to knock down expression of CD95 or FADD. Thirty 6 h after transfection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) #P < 0.05 less than corresponding value in siSCR cells. (E) GBM cells (patients 2 and 3) were pre-treated with N-acetyl cysteine (10 mM) or fumonisin B1 (25 μM) and then treated with DMF (5 μM) and velcade (10 nM) in combination. Cells were isolated 6 h after treatment and the cell surface levels of the death receptor CD95 determined (n = 3, +/− SEM) #P < 0.05 less than corresponding value in VEH cells; *P < 0.05 greater than corresponding value in VEH cells. (F and G) GBM cells (patient 2/3) were transfected with a plasmid to express LC3-GFP-RFP. Twenty 4 h after transfection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were examined under a fluorescent microscope (×40) 12 h after drug treatment and the numbers of intense GFP+ and RFP+ punctae determined. (H) GBM cells (patient 2/3) were transfected with either a scrambled siRNA (siSCR) or siRNA molecules to knock down expression of Beclin1 or ATG5. Thirty 6 h after transfection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) #P < 0.05 less than corresponding value in siSCR cells.
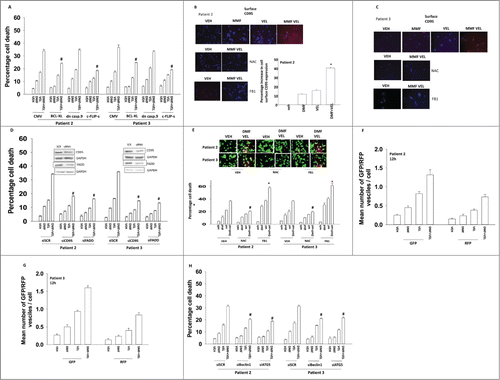
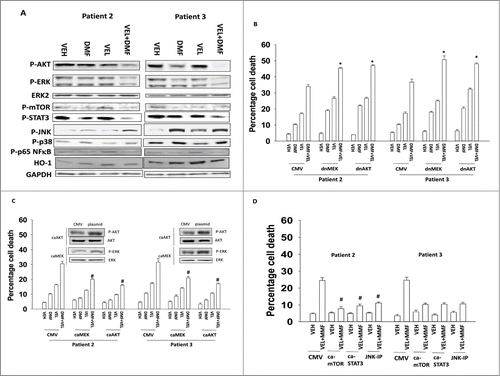
Treatment of GBM cells with DMF and velcade reduced the activities of the AKT and ERK1/2 pathways as well as the mTOR and STAT3 pathways and enhanced the activity of the JNK pathway (). Expression of dominant negative MEK1 or dominant negative AKT enhanced the toxicities of the individual drugs and the drug combination (). Expression of activated forms of MEK1 or AKT suppressed cell killing (). Expression of activated forms of mTOR or STAT3 suppressed cell killing, as did inhibition of the JNK pathway ().
Figure 6. Altered signaling by ERK, AKT, mTOR, NFκB and STAT3 are associated with drug combination toxicity. (A) GBM cells (patient 2/3) were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 12 h after exposure and subjected to SDS PAGE followed by immunoblotting to determine the expression and phosphorylation of the indicated proteins (n = 3). (B) GBM cells (patient 2/3) were infected with recombinant adenoviruses to express empty vector (CMV); dominant negative MEK; dominant negative AKT. Twenty 4 h after infection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than corresponding value in VEH cells. (C) GBM cells (patient 2/3) were infected with recombinant adenoviruses to express empty vector (CMV); constitutively active MEK; constitutively active AKT. Twenty 4 h after infection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) #P < 0.05 less than corresponding value in CMV cells. (D and E) GBM cells (patient 2/3) were transfected with empty vector plasmid (CMV) or plasmids to express constitutively active STAT3 or constitutively active mTOR. The JNK inhibitory peptide (JNK-IP) was used at 10 μM. Twenty 4 h after transfection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than corresponding value in CMV cells.
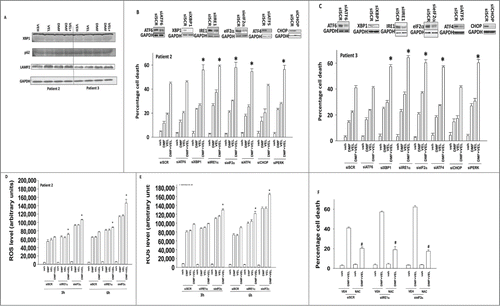
As the combination of DMF and velcade was increasing the levels of autophagosomes, and the fact that velcade increases the levels of unfolded proteins, we determined the relative roles of endoplasmic reticulum stress response and autophagy pathways in the survival/killing response to DMF and velcade treatment. DMF and velcade treatment increased the protein levels of p62 and LAMP2, suggestive of an autophagy flux stall (). We then knocked down the expression of individual proteins within the 3 arms of the ER stress response. Knockdown of ATF6 did not significantly alter the toxicity of DMF, velcade or DMF and velcade treatment (). In contrast to data examining the ATF6 pathway, knock down of the IRE1α/XBP1 or the eIF2α/ATF4 arms of the ER stress response increased the toxicity of DMF, velcade or DMF and velcade treatment. Knock down of CHOP did not alter the response of cells to DMF and velcade. Knock down of eIF2α or IRE1α enhanced the generation of reactive oxygen species following DMF and velcade treatment (). Quenching of ROS using N-acetyl cysteine protected cells from DMF and velcade toxicity ().
Figure 7. Endoplasmic reticulum stress pathways regulate the cellular response to DMF and velcade treatment. (A) GBM cells (patients 2 and 3) were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 12 h after exposure and subjected to SDS PAGE followed by immunoblotting to determine the expression and phosphorylation of the indicated proteins (n = 3). (B) GBM cells (patient 2) and (C) GBM cells (patient 3) were transfected with siRNA molecules to knock down the expression of: ATF6; XBP-1; IRE1α; eIF2α; ATF4; CHOP or with a scrambled siRNA (siSCR). Thirty 6 h after transfection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) *P < 0.05 greater than corresponding value in siSCR cells. (D and E) GBM cells (patient 2/3) were transfected with siRNA molecules to knock down the expression of: IRE1α; eIF2α or with a scrambled siRNA (siSCR). Thirty 6 h after transfection cells were treated with DMF (5 μM), velcade (10 nM) or the drug combination. The levels of reactive oxygen species in cells was determined 3 h after drug exposure using DCFH-DA. Data are plotted as the –Fold increase in ROS levels, with basal DCFH-DA fluorescence subtracted (n = 3, +/− SEM) *P < 0.05 greater than corresponding value in siSCR cells. (F) GBM cells (patient 2/3) were transfected with siRNA molecules to knock down the expression of: IRE1α; eIF2α or with a scrambled siRNA (siSCR). Thirty 6 h after transfection cells were pre-treated with N-acetyl cysteine (10 mM) and then treated with DMF (5 μM), velcade (10 nM) or the drug combination. Cells were isolated 24 h after treatment and viability determined by trypan blue exclusion assay (n = 3, +/− SEM) #P < 0.05 less than corresponding value in siSCR cells.
Discussion
The present studies were performed to determine whether the FDA approved drug for multiple sclerosis, DMF/MMF, could kill glioma cells and interact with other therapeutic agents to enhance GBM cell killing. In multiple primary human GBM cell isolates DMF/MMF interacted with proteasome inhibitors to cause GBM cell killing. DMF/MMF modestly enhanced the toxicity of standard of care GBM therapeutic modalities and killed freshly isolated activated microglia with a parallel reduction in the expression of growth promoting and inflammatory cytokines. Collectively our findings argue that DMF/MMF could represent a novel therapeutic response modifier for GBM treatment.
We determined that DMF/MMF and velcade lethality was reduced by protecting the mitochondrion (BCL-XL over-expression) or inhibition of caspase 8 (c-FLIP-s overexpression), but not impacted by inhibition of caspase 9 function. Based on our findings expressing c-FLIP-s, we determined that DMF/MMF and velcade treatment activated the death receptor CD95 and knock down of CD95 or the docking protein FADD suppressed killing by the drug combination. Activation of CD95 required the generation of reactive oxygen species and was blocked by inhibition of ceramide synthase enzymes. Velcade has been shown to kill in part through the generation of reactive oxygen species and mitochondrial dysfunction, but not through activation of death receptors. Recently velcade toxicity has been linked in pancreatic tumor cells to ceramide generation, though the velcade concentrations used were supra-physiologic. Many studies in tumor cells with DMF/MMF have used supra-physiologic concentrations of the agent, and in these studies DMF/MMF has been proposed to promote a more differentiated state, reduce reactive oxygen species generation and inactivate NFκB. Using lower concentrations of both agents we observed a cooperative interaction to increase CD95 activity and generate reactive oxygen species. Further investigations outside of the present manuscript will be required to fully define how reactive oxygen species and de novo ceramide generation are regulated by this drug combination to kill.
In addition to increased death receptor signaling we also observed that the drug combination increased the levels of autophagosomes and autolysosomes. Inhibition of autophagosome and autolysosome production via knock down of ATG5 or Beclin1 suppressed cell killing. In prior studies e.g., with lapatinib and obatoclax we have shown the drug combination increases autophagosome levels, with autophagosomes co-localizing with mitochondria, suggestive of mitophagy. In the present studies we also observed co-localization of autophagosomes with mitochondria (data not shown). Mitophagy can play both a survival role in response to modest levels of cell stress but can, in the face of higher amounts of stress; act as a cell executioner (observed in the present studies).Citation37 One possible explanation for our data is that as ubiquination of outer mitochondrial membrane proteins by Parkin plays a role in mitophagy, and velcade prevents the degradation of ubiquinated proteins, we would expect mitophagy levels to rise.
Velcade and DMF/MMF treatment inactivated AKT, ERK, mTOR and STAT3. Expression of an activated form of MEK, and to a greater extent AKT suppressed cell killing. Expression of an activated form of mTOR or of STAT3 suppressed the increased levels of autophagosome and autolysosome production and blocked drug combination toxicity. The precise molecular mechanisms by which velcade and DMF modulate cell signaling are unclear, and will require additional study beyond the present manuscript.Citation38
It is now becoming widely appreciated that many anti-cancer drugs induce an endoplasmic reticulum stress / unfolded protein response. In the present studies we observed that inhibition of the ATF6 arm of the stress response did not appear to effect DMF/MMF and velcade toxicity. However, inhibition of either the IRE1α/XBP-1 arm or the PERK/eIF2α/ATF4 arm of the stress response enhanced the toxicities of the individual drugs and the drug combination. Knock down of eIF2α or IRE1α increased the levels of reactive oxygen species generated by drug treatment; quenching of reactive oxygen species prevented cell killing. The molecular mechanisms by which loss of the IRE1α/XBP-1 arm or the PERK/eIF2α/ATF4 arm enhances reactive oxygen species production at present remain unclear.
In conclusion, DMF/MMF can be combined with multiple standard of care and novel therapeutic agents to kill semi-established primary glioblastoma cell isolates. Killing involves inhibition of protective signaling pathways, the induction of autophagosome formation, death receptor activation and the actions of reactive oxygen species. Based on our encouraging data a Phase I dose-limiting toxicity trial is about to open at VCU/MCVH in newly diagnosed glioblastoma patients combining DMF with Temozolomide and radiotherapy.
Materials and Methods
Materials
Phospho-/total- antibodies were purchased from Cell Signaling Technologies (Danvers, MA) and Santa Cruz Biotech. (Santa Cruz, CA). All drugs were purchased from Selleckchem (Houston, TX). Commercially available validated short hairpin RNA molecules to knock down RNA / protein levels were from Qiagen (Valencia, CA). Antibody reagents, other kinase inhibitors, caspase inhibitors cell culture reagents, and non-commercial recombinant adenoviruses have been previously described.Citation39-42 Previously characterized semi-established GBM5 / GBM6 / GBM12 / GBM14 glioblastoma cells were supplied by Dr. C.D. James (University of California, San Francisco) and Dr. J.N. Sarkaria (Mayo Clinic, Rochester MN) and were not further characterized by ourselves. The primary human GBM isolates (patient 1; patient 2; patient 3) and primary human medulloblastoma isolates (HOSS1, VC312, CON1) were obtained / isolated from discarded tumor tissue after standard of care surgery. Patients had previously given informed consent under an IRB protocol to the use of tumor tissue. Tumor samples were made anonymous of all patient identifiers by the VCU TDAAC prior to hand-over to the Dent laboratory.
Methods
Cell culture and in vitro exposure of cells to drugs
All fully established cancer lines were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with 10% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. All primary human GBM cells were cultured at 37°C (5% (v/v CO2) in vitro using RPMI supplemented with 2% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. at 37°C (5% (v/v CO2). For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 × 103 per cm2 and 24 h after plating were treated with various drugs, as indicated. In vitro small molecule inhibitor treatments were from a 100 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study.
Cell treatments, SDS-PAGE and Western blot analysis
Cells were treated with various drug concentrations, as indicated in the figure legends. Samples were isolated at the indicated times and SDS PAGE and immunoblotting was performed as described in refs.Citation39-42 Blots were observed by using an Odyssey IR imaging system (LI-COR Biosciences, Lincoln, NE).
Recombinant adenoviral vectors; infection in vitro
We generated and purchased previously noted recombinant adenoviruses as per refs.Citation39-42 Cells were infected with these adenoviruses at an approximate m.o.i. as indicated in the figure legend (usually an moi of 50). Cells were incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.
Detection of cell death by Trypan Blue assay
Cells were harvested by trypsinization with Trypsin/EDTA for ∼10 min at 37°C. Harvested cells were combined with the culture media containing unattached cells and the mixture centrifuged (800 rpm, 5 min). Cell pellets were resuspended in PBS and mixed with trypan blue agent. Viability was determined microscopically using a hemocytometer. Five hundred cells from randomly chosen fields were counted and the number of dead cells was counted and expressed as a percentage of the total number of cells counted. Cell killing was confirmed using the Sceptor instrument (Millipore, Billerica MA) with a 60 μm tip, which measured tumor cell size/sub G1 DNA as an indication of tumor cell viability.
Assessment of autophagy
Cells were transfected with a plasmid to express a green fluorescent protein (GFP) and red fluorescent protein (RFP) tagged form of LC3 (ATG8). For analysis of cells transfected with the GFP-RFP-LC3 construct, the GFP/RFP -positive vesicularized cells were examined under the X40 objective of a Zeiss Axiovert fluorescent microscope.
Plasmid transfection
Plasmids
Cells were plated as described above and 24 h after plating, transfected. Plasmids (0.5 μg) expressing a specific mRNA or appropriate vector control plasmid DNA was diluted in 50 μl serum-free and antibiotic-free medium (1 portion for each sample). Concurrently, 2 μl Lipofectamine 2000 (Invitrogen), was diluted into 50 μl of serum-free and antibiotic-free medium. Diluted DNA was added to the diluted Lipofectamine 2000 for each sample and incubated at room temperature for 30 min. This mixture was added to each well/dish of cells containing 200 μl serum-free and antibiotic-free medium for a total volume of 300 μl and the cells were incubated for 4 h at 37°C. An equal volume of 2× medium was then added to each well. Cells were incubated for 48 h, then treated with drugs. To assess transfection efficiency of plasmids we used a plasmid to express GFP and defined the percentage of cells being infected as the percentage of GFP+ cells. For all cell lines the infection efficiency was >70%.
siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used (predominantly Qiagen, Valencia, CA; occasional alternate siRNA molecules were purchased from Ambion, Inc., Austin, Texas). Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added drop-wise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37°C for 2 h. One ml of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37°C for 24–48 h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with drugs (0–48 h). Trypan blue exclusion assays and SDS PAGE / immunoblotting analyses were then performed at the indicated time points.
Immunohistochemistry and live-dead assays
Immunohistochemistry and live-dead assays were performed in 96 well plates using a Hermes Wiscan instrument (IDEA Bio-medical, Rehovot, Israel). Cells were plated and treated with drugs for 12–24 h as indicated in the figure legend. After treatment plates were centrifuged to deposit floating cells onto the plate (800 rpm, 5 min). Cells were then either fixed in place (4% paraformaldehyde in PBS) or were subjected to live-dead assay using a commercially available kit and performed according to the manufacturer's method (Life Technologies, Grand Island, NY). Cell viability was measured using Wisoft software. Fixed cells were subjected to Ki67 / IL-6 / TNFα staining using standard procedures.
Isolation of activated microglia
Following acquisition of fresh GBM tumor tissue under an approved IRB protocol, we isolated activated microglia using CD11b microglia microbeads (MACS, Miltenyi Biotec., San Diego, CA). Tumor tissue was macerated and single cell suspensions obtained using a Miltenyi neural tissue dissociation kit.
Data analysis
Comparison of the effects of various treatments was performed using one way analysis of variance and a 2 tailed Student's t-test. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Differences with a P-value of <0.05 were considered statistically significant. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Support for the present study was funded from PHS grants from the National Institutes of Health [R01-CA141704, R01-CA150214, R01-DK52825]; the Department of Defense [W81XWH-10-1-0009]. PD is the holder of the Universal Inc. Chair in Signal Transduction Research.
References
- Field KM, Rosenthal MA, Yilmaz M, Tacey M, Drummond K. Comparison between poor and long-term survivors with glioblastoma: review of an Australian data set. Asia Pac J Clin Oncol 2013; 10:153-61 [Epub ahead of print]; PMID:23701649
- Juratli TA, Schackert G, Krex D. Current status of local therapy in malignant gliomas–A clinical review of three selected approaches. Pharmacol Ther 2013; 139:341-58; PMID:23694764; http://dx.doi.org/10.1016/j.pharmthera.2013.05.003
- Belge K, Brück J, Ghoreschi K. Advances in treating psoriasis. F1000Prime Rep 2014; 6:4. eCollection.
- Phillips JT, Fox RJ. BG-12 in multiple sclerosis. Semin Neurol 2013; 33:56-65; PMID:23709213; http://dx.doi.org/10.1055/s-0033-1343796
- Linker RA, Lee DH, Ryan S, Van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011; 134:678-692; PMID:21354971; http://dx.doi.org/10.1093/brain/awq386
- Schmidt MM, Dringen R. Fumaric acid diesters deprive cultured primary astrocytes rapidly of glutathione. Neurochem. Int., 2010; 57:460-467; http://dx.doi.org/10.1016/j.neuint.2010.01.006
- Li XN, Du ZW, Huang Q, Wu JQ. Growth-inhibitory and differentiation-inducing activity of dimethylformamide in cultured human malignant glioma cells. Neurosurgery 1997; 40:1250-8; PMID:9179899; http://dx.doi.org/10.1097/00006123-199706000-00027
- Ghods AJ, Glick R, Braun D, Feinstein D. Beneficial actions of the anti-inflammatory dimethyl fumarate in glioblastomas. Surg Neurol Int 2013; 4:160; PMID: 24404403; http://dx.doi.org/10.4103/2152-7806.123656
- Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012; 367:1098-107; PMID:22992073; http://dx.doi.org/10.1056/NEJMoa1114287
- Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, Polman CH, Schmierer K, Yousry TA, Yang M, et al. BG-12 Phase IIb Study Investigators. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008; 372:1463-72; PMID:18970976; http://dx.doi.org/10.1016/S0140-6736(08)61619-0
- Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflammation 2010; 19:7- 30.
- Lin SX, Lisi L, Dello Russo C, Polak PE, Sharp A, Weinberg G, Kalinin S, Feinstein DL. The anti-inflammatory effects of dimethyl fumarate in astrocytes involve glutathione and haem oxygenase-1. ASN Neuro 2011; 3. pii:e00055; http://dx.doi.org/10.1042/AN20100033
- Foresti R, Bains SK, Pitchumony TS, de Castro Brás LE, Drago F, Dubois-Randé JL, Bucolo C, Motterlini R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res 2013; 76:132-48; PMID:23942037; http://dx.doi.org/10.1016/j.phrs.2013.07.010
- Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther 2012; 341:274-84; PMID:22267202; http://dx.doi.org/10.1124/jpet.111.190132
- Moharregh-Khiabani D, Linker RA, Gold R, Stangel M. Fumaric acid and its esters: an emerging treatment for multiple sclerosis. Curr Neuropharmacol 2009; 7:60-4; PMID:19721818; http://dx.doi.org/10.2174/157015909787602788
- Van Nifterik KA, Van den Berg J, Slotman BJ, Lafleur MV, Sminia P, Stalpers LJ. Valproic acid sensitizes human glioma cells for temozolomide and γ-radiation. J Neurooncol 2012; 107:61-7; PMID:22037799; http://dx.doi.org/10.1007/s11060-011-0725-z
- Tsubaki M, Komai M, Itoh T, Imano M, Sakamoto K, Shimaoka H, Ogawa N, Mashimo K, Fujiwara D, Takeda T, et al. Inhibition of the tumour necrosis factor-alpha autocrine loop enhances the sensitivity of multiple myeloma cells to anticancer drugs. Eur J Cancer 2013; 49:3708-17; PMID:23932230; http://dx.doi.org/10.1016/j.ejca.2013.07.010
- Bedford L, Lowe J, Dick LR, Mayer RJ Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov 2011; 10:29-46; PMID:21151032; http://dx.doi.org/10.1038/nrd3321
- Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan AA, et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007; 110:3281-90; PMID: 17591945; http://dx.doi.org/10.1182/blood-2007-01-065888
- Zangari M, Aujay M, Zhan F, Hetherington KL, Berno T, Vij R, Jagannath S, Siegel D, Keith Stewart A, Wang L, et al. Alkaline phosphatase variation during carfilzomib treatment is associated with best response in multiple myeloma patients. Eur J Haematol 2011; 86:484-7; PMID:21477075; http://dx.doi.org/10.1111/j.1600-0609.2011.01602.x
- Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem 2003; 278:33714-23; PMID: 12821677; http://dx.doi.org/10.1074/jbc.M302559200
- Fribley A, Zeng Q, Wang CY. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol 2004; 24:9695-704; PMID:15509775; http://dx.doi.org/10.1128/MCB.24.22.9695-9704.2004
- Yu C, Rahmani M, Dent P, Grant S. The hierarchical relationship between MAPK signaling and ROS generation in human leukemia cells undergoing apoptosis in response to the proteasome inhibitor Bortezomib. Exp Cell Res 2004; 295:555-66; PMID:15093752; http://dx.doi.org/10.1016/j.yexcr.2004.02.001
- Dasmahapatra G, Lembersky D, Kramer L, Fisher RI, Friedberg J, Dent P, Grant S. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 2010; 115:4478-87; PMID: 20233973; http://dx.doi.org/10.1182/blood-2009-12-257261
- Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR, Sun SY. Proteasome inhibitor PS-341 (bortezomib) induces calpain-dependent IkappaB(alpha) degradation. J Biol Chem 2010; 285:16096-104; PMID:20335171; http://dx.doi.org/10.1074/jbc.M109.072694
- Peng H, Guerau-de-Arellano M, Mehta VB, Yang Y, Huss DJ, Papenfuss TL, Lovett-Racke AE, Racke MK. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF-κB) and extracellular signal-regulated kinase 1 and 2 (ERK12) and mitogen stress-activated kinase 1 (MSK1) signaling. J Biol Chem 2012; 287:28017-26; PMID:22733812; http://dx.doi.org/10.1074/jbc.M112.383380
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011; 334:1081-6; PMID:22116877; http://dx.doi.org/10.1126/science.1209038
- Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J Hepatol 2011; 54:795-809; PMID:21145844; http://dx.doi.org/10.1016/j.jhep2010.11.005
- Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal 2007; 9:2357-71; PMID:17760508; http://dx.doi.org/10.1089/ars.2007.1764
- Martín-Pérez R, Niwa M, López-Rivas A. ER stress sensitizes cells to TRAIL through down-regulation of FLIP and Mcl-1 and PERK-dependent up-regulation of TRAIL-R2. Apoptosis 2012; 17:349-63; http://dx.doi.org/10.1007/s10495-011-0673-2
- Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci 2010; 30:16938-48; PMID:21159964; http://dx.doi.org/10.1523/JNEUR-OSCI.1598-10.2010
- Qi Y, Xia P. Cellular inhibitor of apoptosis protein-1 (cIAP1) plays a critical role in β-cell survival under endoplasmic reticulum stress: promoting ubiquitination and degradation of CEBP homologous protein (CHOP). J Biol Chem 2012; 287:32236-45; PMID:22815481; http://dx.doi.org/10.1074/jbc.M11-2.362160
- Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6CHOP arm of ER-stress-response pathways. FASEB J 2010; 24:296-308; PMID:19723703; http://dx.doi.org/10.1096/fj.09-135087
- Wang FM, Chen YJ, Ouyang HJ. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem J 2011; 433:245-52; PMID:20955178; http://dx.doi.org/10.1042/BJ20-101293
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008; 7:1013-30; PMID:19043451; http://dx.doi.org/10.1038/nrd2755
- Sureshbabu A, Bhandari V. Targeting mitochondrial dysfunction in lung diseases: emphasis on mitophagy. Front Physiol 2013; 4:384; PMID:24421769; http://dx.doi.org/10.3389/fphys.2013.00384
- Yang F, Jove V, Chang S, Hedvat M, Liu L, Buettner R, Tian Y, Scuto A, Wen W, Yip ML, et al. Bortezomib induces apoptosis and growth suppression in human medulloblastoma cells, associated with inhibition of AKT and NF-ĸB signaling, and synergizes with an ERK inhibitor. Cancer Biol Ther 2012; 13:349-57; PMID:22313636; http://dx.doi.org/10.4161/cbt.19239
- Booth L, Cazanave SC, Hamed HA, Yacoub A, Ogretmen B, Chen CS, Grant S, Dent P. OSU-03012 suppresses GRP78BiP expression that causes PERK-dependent increases in tumor cell killing. Cancer Biol Ther 2012; 13:224-36; PMID:22354011; http://dx.doi.org/10.4161/cbt.13.4.18877
- Yacoub A, Park MA, Hanna D, Hong Y, Mitchell C, Pandya AP, Harada H, Powis G, Chen CS, Koumenis C, et al. OSU-03012 promotes caspase-independent but PERK-, cathepsin B-, BID-, and AIF-dependent killing of transformed cells. Mol Pharmacol 2006; 70:589-603; PMID:16622074; http://dx.doi.org/10.1124/mol.106.025007
- Booth L, Roberts JL, Cruickshanks N, Conley A, Durrant DE, Das A, Fisher PB, Kukreja RC, Grant S, Poklepovic A, et al. Phosphodiesterase 5 inhibitors enhance chemotherapy killing in gastrointestinal genitourinary cancer cells. Mol Pharmacol 2014; 85:408-19; PMID:24353313; http://dx.doi.org/10.1124/mol.113.090043
- Booth L, Roberts JL, Conley A, Cruickshanks N, Ridder T, Grant S, Poklepovic A, Dent P. HDAC inhibitors enhance the lethality of low dose salinomycin in parental and stem-like GBM cells. Cancer Biol Ther 2014; 15:305-16; PMID:24351423; http://dx.doi.org/10.4161/cbt.27309