
Abstract
Metastatic colon cancer has a 5-year survival of less than 10% despite the use of aggressive chemotherapeutic regimens. As signaling from epidermal growth factor receptor (EGFR) is often enhanced and epigenetic regulation is often altered in colon cancer, it is desirable to enhance the efficacy of EGFR-directed therapy by co-targeting an epigenetic pathway. We showed that the histone methyltransferase EZH2, which catalyzes methylation of histone H3 lysine 27 (H3K27), was upregulated in colon cancers in The Cancer Genome Atlas (TCGA) database. Since co-inhibition of both EGFR and EZH2 has not been studied in colon cancer, we examined the effects of co-inhibition of EGFR and EZH2 on 2 colon cancer cell lines, HT-29 and HCT-15. Co-inhibition of EZH2 and EGFR with the small molecules UNC1999 and gefitinib, led to a significant decrease in cell number and increased apoptosis compared to inhibition of either pathway alone, and similar results were noted after EZH2 shRNA knockdown. Moreover, co-inhibition of EZH2 and EGFR also significantly induced autophagy, indicating that autophagy may play a role in the observed synergy. Together, these findings suggest that inhibition of both EZH2 and EGFR serves as an effective method to increase the efficacy of EGFR inhibitors in suppressing colon cancer cells.
Abbreviations
| EGFR | = | epidermal growth factor receptor |
| H3K27 | = | histone H3 lysine 27 |
| HDAC | = | histone deacetylase |
| PRC2 | = | polycomb repressive complex 2 |
| TCGA | = | The Cancer Genome Atlas |
Introduction
Colon cancer is the third most common type of cancer in men and women and also represents the third most common cause of cancer related mortality in both sexes.Citation1 Despite aggressive chemotherapy regimens, metastatic colon cancer still has a poor 5-year survival of around 10%, illustrating the urgent need for more effective treatment regimens.Citation2 One class of targeted chemotherapeutic agents used for a subset of metastatic colorectal cancer is epidermal growth factor receptor (EGFR) inhibitors. EGFR inhibitors, including the antibodies cetuximab and panitumumab, are often added to traditional chemotherapy regimens such as FOLFIRI or FOLFOX in KRAS wild type colon cancers. Although no benefit is seen in KRAS mutant colon cancers, addition of an EGFR inhibitor has been shown to increase progression free survival and overall survival in patients with metastatic KRAS wild type tumors.Citation3 However, despite the survival benefits seen with the addition of EGFR inhibitors, metastatic colon cancer still has a poor prognosis, demonstrating the need for additional research in order to further improve therapy.
EGFR, also known as HER1, is a transmembrane protein that is a member of the ErbB family of receptors.Citation4 EGFR is overexpressed in multiple types of cancer and can be targeted by both antibodies directed at the extracellular domain, as well as small molecule inhibitors that target the intracellular tyrosine kinase domain.Citation4 EGFR inhibition leads to reduced signaling through both the PI3K/Akt and MAPK pathways, and ultimately has an anti-proliferative effect on cells.Citation4 In addition to decreasing proliferation, EGFR inhibition is also a potent inducer of autophagy, which is a basic cellular catabolic mechanism that is used to break down and recycle cellular components, and is often active during periods of cellular stress. EGFR inhibition induces autophagy through multiple mechanisms including mTor inhibition.Citation5 A recent report also shows that phosphorylated EGFR directly associates with Beclin-1 ultimately leading to its inactivation, and EGFR inhibition leads to liberation of Beclin-1 and promotes progression of autophagy.Citation6 Autophagy may be important in cellular resistance to EGFR inhibition in certain types of cancers, as recent studies in solid tumors such as breast and lung cancer, as well as glioblastoma, have demonstrated that inhibition of autophagy enhances the anti-proliferative effects of EGFR inhibitors.Citation7-11 On the other hand, it is also possible that other modulators of autophagy may serve to enhance the efficacy of EGFR inhibitors in certain types of cancer, however this has never been extensively studied in colon cancer cells.
A recent report demonstrated that the epigenetic regulator EZH2 may have an important role in autophagy in hepatocellular carcinoma cells, where knockdown of EZH2 led to autophagy inhibition and ultimately sensitized these cells to other non-EGFR inhibitor chemotherapeutic agents.Citation12 EZH2 and the related EZH1 are catalytic subunits of the polycomb repressive complex 2 (PRC2) and serve as epigenetic regulators through their role as histone methyltransferases.Citation13 EZH2 and EZH1 are both responsible for H3K27-methylation, which leads to silencing of tumor suppressor genes, and EZH2 is known to be over-expressed in multiple malignancies including colon cancer.Citation14,15 Specifically targeting epigenetic mechanisms similar to EZH2 has recently been shown to be a promising and novel approach to enhancing the efficacy of current chemotherapeutic regimens.Citation16 An example of this new epigenetic approach includes the recently developed histone deacetylase (HDAC) inhibitors, such as Vorinostat, which have been shown to act synergistically when combined with other more conventional chemotherapeutic agents.Citation17 Use of the epigenetically-targeted HDAC inhibitors has also led to multiple phase I/II clinical trials that are currently underway evaluating the benefits of combining HDAC inhibitors with other chemotherapeutic agents in a variety of hematologic and solid malignancies.Citation18 Inhibition of EZH2 in colon cancer cell lines has been studied, and has shown some decrease in cell proliferation,Citation19-21 however there is no report evaluating the efficacy of co-inhibition of both EGFR and EZH2 in any cancer cell line.
Therefore, we set out to investigate the effects of co-inhibition of EZH2 and EGFR in colon cancer cell lines to determine if EZH2 inhibition could increase the anti-proliferative effects of EGFR inhibition.
Results
EZH2 is overexpressed in colon cancer, and can be inhibited by UNC1999.
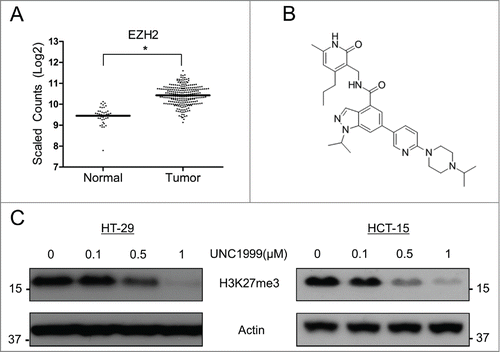
EZH2 is known to be over-expressed in multiple hematologic as well as solids tumors.Citation14,15 To determine its expression level in colon adenocarcinoma compared to non-cancerous colonic epithelial cells, The Cancer Genome Atlas (TCGA) database was analyzed. RNASeq data from this database demonstrated that EZH2 was 2.4-fold higher expressed in colon cancer tissue compared to non-cancerous colonic epithelium (). The relative expression of EZH1 was also analyzed and was found to be lower in colon cancer tissue compared to normal tissue, however this difference was much smaller than the difference observed with EZH2 expression (Fig. S1). Next, the small molecule UNC1999 (), which was recently shown to inhibit both EZH2 and EZH1 activity,Citation22 was utilized to inhibit EZH2/EZH1 in 2 colon cancer cell lines: KRAS wild type HT-29 cells, and KRAS mutant (G13D) HCT-15 cells. H3K27-trimethylation was used as a marker of EZH2/EZH1 inhibition, and in both HT-29 cells and HCT-15 cells UNC1999 demonstrated a dose-dependent inhibition of H3K27me3 (), indicating that this compound can effectively inhibit EZH2/EZH1 histone methyltransferase activity in these 2 colon cancer cell lines.
Figure 1. EZH2 is over-expressed in colon cancer and can be inhibited by UNC1999 in HT-29 and HCT-15 cells. (A) TCGA database analysis comparing EZH2 expression in non-cancerous colonic epithelial samples compared to colon cancer samples. Log2-transformed, normalized counts are shown for EZH2, dividing the plot to separate normal and tumor samples. *P = 3.97E-41. (B) Structure of UNC1999. (C) Total H3K27me3 levels in HT-29 and HCT-15 cells treated with varying concentrations of UNC1999 for 72 hours.
Gefitinib inhibits EGFR phosphorylation and induces autophagy in HT-29 and HCT-15 cells.
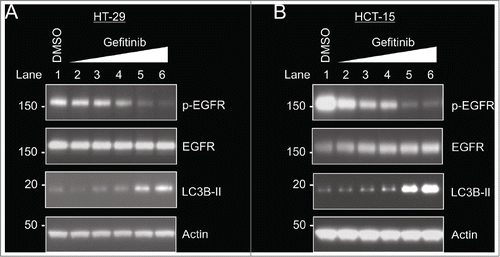
In order to confirm that the EGFR inhibitor gefitinib could adequately inhibit EGFR phosphorylation in HT-29 and HCT-15 cells, both cells lines were treated with increasing concentrations of gefitinib for 24 hours, leading to a dose-dependent decrease in EGFR phosphorylation (, Lanes 2–6). In both cell lines, gefitinib concentrations of at least 5 μM were needed to adequately inhibit EGFR phosphorylation. In addition, the ability of gefitinib to induce autophagy was also assessed through LC3B-II levels. Microtubule-associated protein 1 light chain 3 (LC3) has 3 different isoforms (A, B, C), and LC3B is proteolytically cleaved to form LC3B-I, which is then lipidated to LC3B-II and incorporated into the autophagosome.Citation23 Therefore, assessing levels of LC3B-II is a widely used method to monitor autophagy.Citation23 After treatment of both HT-29 and HCT-15 cells with gefitinib, increased levels of LC3B-II were noted in both cell lines in proportion to the level of EGFR inhibition, indicating that the EGFR inhibitor gefitinib induces autophagy in these 2 cell lines ().
Figure 2. Gefitinib inhibits EGFR phosphorylation and increases autophagy in HT-29 cells and HCT-15 cells. Cells were treated with DMSO (control) or varying concentrations of gefitinib (0.1 μM, 0.5 μM, 1 μM, 5 μM, 10 μM) for 24 hours. (A) HT-29 cells. (B) HCT-15 cells.
Co-inhibition of EZH2 and EGFR leads to increased toxicity in HT-29 cells and HCT-15 cells.
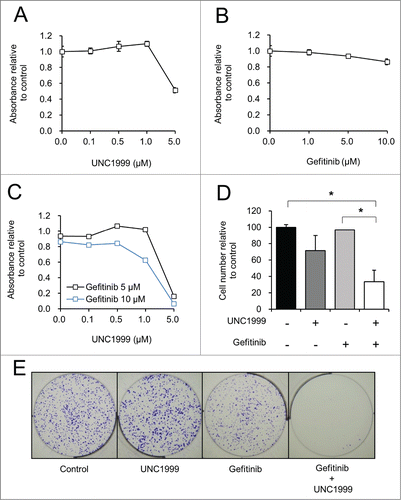
To determine if the EZH2 inhibitor affects the efficacy of the EGFR inhibitor gefitinib, the effect of co-inhibition of EZH2 and EGFR was studied on the proliferation of HT-29 and HCT-15 cells. EZH2 inhibition with UNC1999 had minimal effect on HT-29 cell proliferation up to 1 μM after 72 hours using the MTS assay, however higher doses did demonstrate some cellular toxicity (). Gefitinib alone also did not cause a significant decrease in HT-29 cell proliferation as assessed by the MTS assay, even up to a concentration of 10 μM (). The combination of UNC1999 and gefitinib at concentrations that effectively inhibit EGFR (5–10 μM) (), led to a synergistic decrease in proliferation via the MTS assay at 1 μM and 5 μM of UNC1999 (). This increased toxicity seen with the combination of UNC1999 plus gefitinib was also confirmed with direct cell counting, which demonstrated that treatment with UNC1999 plus gefitinib led to a significantly decreased cell number compared to control treated cells or gefitinib treated cells alone (). After long term treatment via a clonogenicity assay, there is also a clear synergy noted through EZH2 and EGFR inhibition, with nearly no viable colonies remaining after combination treatment with UNC1999 and gefitinib ().
Figure 3. Together UNC1999 and gefitinib significantly reduces the number of HT-29 cells compared to either compound alone. (A–C) HT-29 cells treated with varying concentrations of UNC1999, gefitinib, or the combination of UNC1999 and gefitinib. MTS assay was performed to assess cell proliferation after 72 hours. (D) Manual cell counting of live cells after treatment for 72 hours with 1 μM UNC1999, 5 μM gefitinib, or the combination of 1 μM UNC1999 and 5 μM gefitinib. *P < 0.05. (E) Clonogenicity assay with crystal violet staining after 10 days of treatment with DMSO (Control), 0.5 μM UNC1999, 5 μM gefitinib, or 0.5 μM UNC1999 and 5 μM gefitinib.
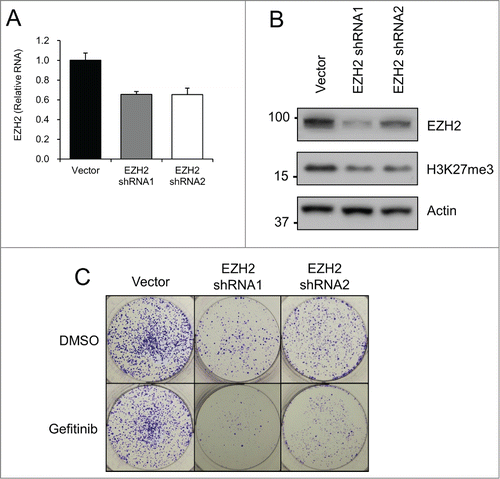
Given that small molecule inhibitors can have off-target effects, we utilized EZH2 knockdown as an alternative method of EZH2 inhibition, in order to confirm that the UNC1999 results were specific to EZH2 inhibition. EZH2 was knocked down in HT-29 cells utilizing 2 different lentiviral delivered shRNAs. After puromycin selection, RT-PCR showed consistent EZH2 RNA knockdown in HT-29 cells expressing EZH2 shRNA (). At the protein level EZH2 shRNA1 led to a more substantial knockdown compared to EZH2 shRNA2, however both EZH2 shRNAs were ultimately able to significantly decrease total H3K27me3 levels (). Although EZH2 knockdown alone did not have a significant effect on cell proliferation when combined with gefitinib over the short term (Fig. S2), long-term treatment via a clonogenicity assay showed a clear difference. Specifically, the addition of gefitinib to EZH2 shRNA expressing HT-29 cell lines led to a dramatic reduction in number of colonies formed, thus further demonstrating the anti-proliferative effects of EZH2 and EGFR co-inhibition ().
Figure 4. EZH2 shRNA decreases colony formation when combined with gefitinib. HT-29 cells were transfected with Vector or EZH2 shRNAs. (A) EZH2 RT-PCR after puromycin selection. EZH2 levels are relative to actin. (B) EZH2 western blotting after puromycin selection. (C) Clonogenicity assay with crystal violet staining after 10 days of treatment with DMSO (Control) or 5 μM gefitinib.
Similar results were also noted with the KRAS mutant colon cancer cell line HCT-15. Treatment of these cells with UNC1999 up to 5 μM demonstrated little toxicity (). The proliferation of HCT-15 cells was only minimally inhibited after treatment with 10 μM gefitinib, and was unaffected after treatment with lower concentrations of gefitinib (). The combination of UNC1999 and gefitinib, again at concentrations of gefitinib that effectively inhibit EGFR (5–10 μM) (), produced a more significant decrease in proliferation as assessed by the MTS assay () and by direct cell counting () compared to either compound alone. The enhanced toxicity of UNC1999 plus gefitinib was further magnified after long term treatment where clonogenicity assay demonstrated a decreased number and size of colonies after treatment with UNC1999 plus gefitinib compared to either compound alone ().
Figure 5. Together UNC1999 and gefitinib significantly reduces the number of HCT-15 cells compared to either compound alone. (A–C) HCT-15 cells treated with varying concentrations of UNC1999, gefitinib, or the combination of UNC1999 and gefitinib. MTS assay was performed to assess cell proliferation after 72 hours. (D) Manual cell counting of live cells after treatment for 72 hours with 1 μM UNC1999, 5 μM gefitinib, or the combination of 1 μM UNC1999 and 5 μM gefitinib. *P < 0.05. (E) Clonogenicity assay with crystal violet staining after 10 days of treatment with DMSO (Control), 0.5 μM UNC1999, 5 μM gefitinib, or 0.5 μM UNC1999 and 5 μM gefitinib.
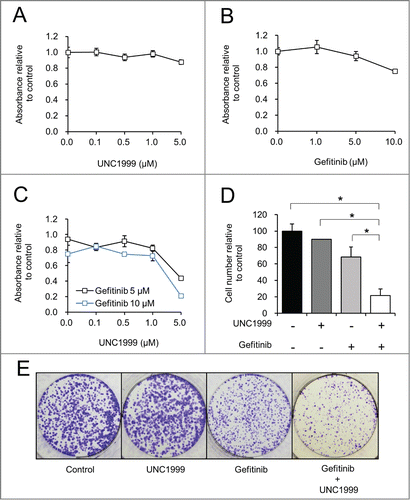
UNC1999 enhances gefitinib induced autophagy and apoptosis in colon cancer cell lines
Given the effects on proliferation with the combination of UNC1999 and gefitinib, we next aimed to study the effects of these compounds on H3K27 methylation and EGFR inhibition, as well as apoptosis and autophagy induction. To that end, in HT-29 cells UNC1999 effectively inhibited H3K27me3 alone and in the presence of gefitinib at both 24 hours () and 72 hours (). Similarly gefitinib at both 5 μM and 10 μM adequately inhibited EGFR phosphorylation when given as a single agent as well as when given in combination with UNC1999 at both 24 hours () and 72 hours (). Next, apoptosis induction was assessed through Parp cleavage. At 24 hours, cleaved Parp was only noted in cells treated with the combination of UNC1999 and 10 μM gefitinib (, Lane 6). After 72 hours of treatment, gefitinib 10 μM induced a small amount of Parp cleavage, consistent with some apoptosis induction (, Lane 4), however this cleavage was significantly increased with the addition of 1 μM UNC1999 (, Lane 6). Furthermore, while UNC1999 1 μM and gefitinib 5 μM alone did not induce any significant Parp cleavage, together these 2 inhibitors selectively induced Parp cleavage after 72 hours (, Lane 5). We also studied the effects of UNC1999 and gefitinib on autophagy using LC3B-II levels and p62. p62, also known as sequestrome 1 (SQSTM1), is degraded during autophagy and in combination with LC3B-II levels, these 2 proteins serve as useful markers for monitoring autophagy.Citation23,24 At both 24 hours and 72 hours, gefitinib caused a dose dependent increase in LC3B-II levels (, Lanes 3–4). UNC1999 alone caused only a small increase in LC3B-II levels, however addition of UNC1999 to either concentration of gefitinib at 24 hours or 72 hours led to a significant increase in LC3B-II levels consistent with increasing autophagy (, Lanes 5–6). The increase in autophagy was confirmed by the reduction in p62 levels with 10 μM gefitinib (, Lane 4) and a more substantial reduction with the combination of 10 μM gefitinib and UNC1999 (, Lane 6).
Figure 6. UNC1999 enhances gefitinib-induced autophagy and apoptosis in HT-29 cells. HT-29 cells were treated with DMSO, 1 μM UNC1999, gefitinib (5 μM or 10 μM), or a combination of UNC1999 + gefitinib, and then collected after (A) 24 hours, or (B) 72 hours.
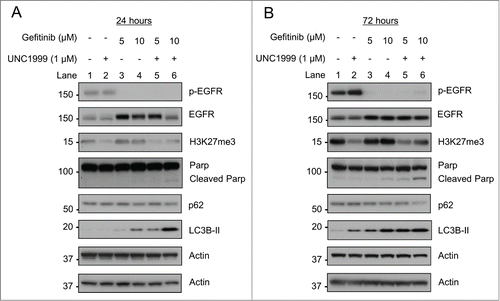
To confirm that the increased levels of LC3B-II were associated with an increase in autophagosome formation, HT-29 cells were transfected with a GFP-LC3B construct. While GFP-LC3B was diffusely distributed in the cytoplasm of DMSO treated cells, there were punctate foci that began to form after treatment with UNC1999 and gefitinib alone, and which were further enhanced after treatment with the combination of gefitinib and UNC1999, consistent with increasing autophagosome formation (). To verify that the effects on autophagy were not just non-specific effects of the small molecule UNC1999, EZH2 shRNA knockdown was again utilized in HT-29 cells. Similar to treatment with UNC1999, EZH2 shRNA knockdown also led to an increase in LC3B-II levels ().
Figure 7. EZH2 inhibition with UNC1999 or EZH2 shRNA knockdown increases autophagy. (A) HT-29 cells transfected with GFP-LC3B were treated for 48 hours with DMSO, 1 μM UNC1999, 10 μM gefitinib, or a combination of 1 μM UNC1999 + 10 μM gefitinib, then fixed and mounted with DAPI. 200× images. (B) HT-29 cells were transfected with Vector or EZH2 shRNAs. EZH2 western blotting was performed after puromycin selection.
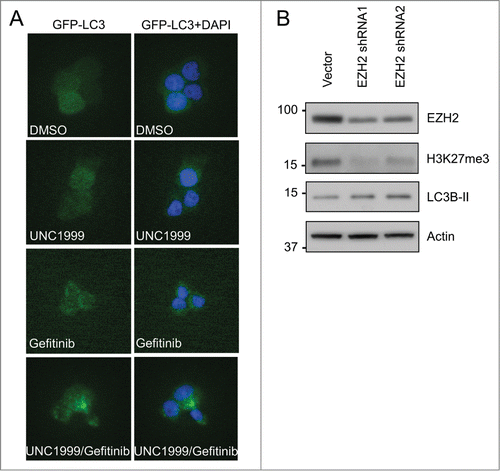
Similar results were also observed in HCT-15 cells where UNC1999 inhibited H3K27me3 both with and without gefitinib treatment (). Furthermore, gefitinib also inhibited EGFR at both 24 and 72 hours () independent of UNC1999 treatment. Less apoptosis induction was noted in HCT-15 cells compared to HT-29 cells with cleaved Parp only noted after 72 hours of treatment with both 10 μM gefitinib and 1 μM UNC1999 (, Lane 6). Despite decreased apoptosis, there was still a significant augmentation of gefitinib induced increases in LC3B-II levels when cells were co-treated with UNC1999 (, lanes 5–6). Again, the increased LC3B-II levels correlate with increased autophagy as demonstrated by p62 levels, which noticeably decreased after gefitinib 10 μM treatment, and further decreased with co-treatment with 10 μM gefitinib and 1 μM UNC1999 (, lanes 4 and 6).
Figure 8. UNC1999 enhances gefitinib-induced autophagy and apoptosis in HCT-15 cells. HCT-15 cells were treated with DMSO, 1 μM UNC1999, gefitinib (5 μM or 10 μM), or a combination of UNC1999 + gefitinib, and then collected after (A) 24 hours, or (B) 72 hours.
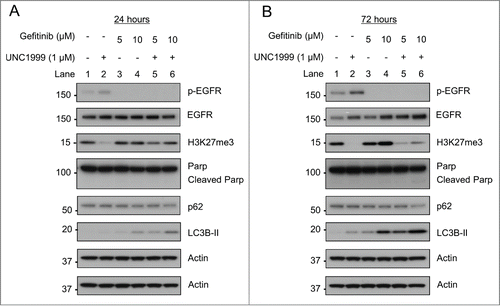
EZH2 inhibition with UNC1999 increases autophagic flux
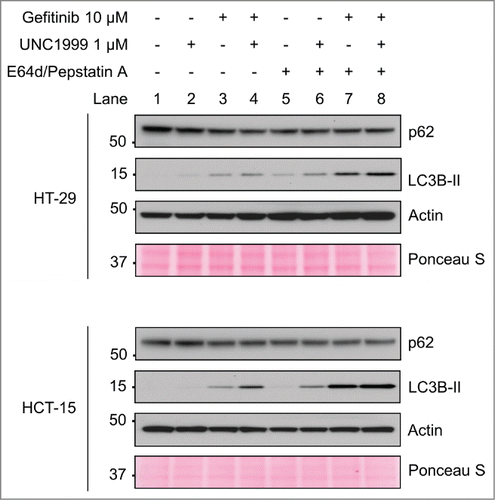
As reported previously in hepatocellular carcinoma cells, EZH2 knockdown was noted to inhibit autophagy by preventing the fusion of autophagosomes and lysosomes.Citation12 Given our differing results in these colon cancer cell lines, we therefore wanted to further confirm that EZH2 inhibition was inducing autophagy in colon cancer cells. To accomplish this an autophagy flux experiment was performed utilizing the protease inhibitors E64d and Pepstatin A, which are well known to inhibit lysosomal proteases, and prevent degradation of autophagolysome contents.Citation25 As noted previously, after 24 hours both UNC1999 and gefitinib induced an increase in LC3B-II levels with the combination of UNC1999 and gefitinib inducing more LC3B-II and a further decrease in p62 in both cell lines (, Lanes 2–4). When inhibiting lysosomal proteases with E64d and Pepstatin A, p62 levels remained constant as expected given that p62 is not broken down within the autophagolysosome (, Lanes 5–8). Furthermore, as expected, LC3B-II levels increased with gefitinib, indicating that gefitinib is inducing increased autophagic flux (, Lane 7). However, it was also noted that LC3B-II levels increased after UNC1999 treatment alone, providing additional evidence that UNC1999 is inducing autophagy rather than inhibiting it in this system (, Lane 6).
Figure 9. UNC1999 increases autophagy flux in HT-29 and HCT-15 cells. HT-29 and HCT-15 cells were treated for 24 hours with DMSO, 1 μM UNC1999, 10 μM gefitinib, or a combination of UNC1999 + gefitinib, both with and without lysosomal protease inhibitors E64d/Pepstatin A (10 μg/mL).
Discussion
Metastatic colon cancer continues to cause significant morbidity and mortality, and therefore finding better treatment regimens for this condition is imperative. EGFR inhibitors are one class of targeted agents that have proven useful for treatment of KRAS wild type metastatic colon cancer, however they have not been shown to be beneficial in KRAS mutant tumors.Citation3 One mechanism to potentially enhance the efficacy of EGFR inhibitors is to co-target an epigenetic mechanism, as demonstrated with HDAC inhibitor enhancement of EGFR inhibitor efficacy in multiple hematologic and solid malignancies.Citation17,18 Therefore in an attempt to find a novel epigenetic mechanism through which to improve the efficacy of EGFR inhibitors in colon cancer, we examined co-inhibition of both EGFR and EZH2. Our results demonstrate that EZH2 inhibition with UNC1999 or EZH2 shRNA knockdown can be combined with the EGFR inhibitor gefitinib to decrease proliferation and induce apoptosis in 2 different colon cancer cell lines, and the observed effects may be partially related to increased autophagy. These results are significant for the following reasons: First, to our knowledge this is the first report to show that co-inhibition of EGFR and EZH2 can reduce cell number and increase apoptosis in colon cancer cells. Secondly, this is the first use of the novel EZH2/EZH1 inhibitor UNC1999 in any solid tumor cell line. Thirdly, in this report we demonstrate for the first time that EZH2 inhibition promotes autophagy in colon cancer cell lines.
EZH2 and EZH1, which are both histone methyltransferases that are components of the PRC2 complex, are important epigenetic regulators that serve their function through methylation of H3K27.Citation13 EZH2 has been more extensively studied than EZH1, and EZH2 has been shown to be overexpressed in multiple different hematologic and solid malignancies.Citation14,15 We analyzed the colon cancer and non-cancerous colonic epithelial specimens within the TCGA database, and we showed that EZH2 was in fact significantly overexpressed in colon cancer tissue whereas there was only a small difference in EZH1 levels between cancerous and non-cancerous tissue. This suggests that EZH2 may be an important epigenetic mechanism in colon cancer pathogenesis.
We then demonstrated that UNC1999 effectively inhibits EZH2 H3K27-methylation in HT-29 and HCT-15 colon cancer cell lines, and co-inhibition of EGFR and EZH2 significantly decreases the cell number of both of these colon cancer cell lines. Not only does co-inhibition decrease cell number, but it also leads to increased apoptosis and a dramatic increase in autophagy. EGFR inhibitors are well known to induce autophagy through effects on mTorCitation5 and more recently through liberation of sequestered Beclin-1.Citation6 We confirmed that the EGFR inhibitor gefitinib induced autophagy in a dose dependent manner in both HT-29 and HCT-15 cells. However, the effect of EZH2 inhibition on autophagy is far less clear. Although a prior report in hepatocellular carcinoma cells has shown that EZH2 knockdown inhibited autophagy,Citation12 in colon cancer cell lines EZH2 inhibition with either the small molecule UNC1999 or EZH2 shRNA appears to increase autophagy. This was further studied through an autophagy flux experiment where lysosomal proteases were inhibited with E64d and Pepstatin A, which further confirmed these results. It is possible that autophagy can be differentially regulated by EZH2 inhibition in different tumor tissues. Furthermore, this also demonstrates that the mechanism through which EZH2 regulates autophagy needs to be studied in greater detail in a variety of cancers that overexpress this epigenetic regulator.
Autophagy is often active during cell stress and facilitates catabolic processes such as removal of damaged organelles or recycling of other intracellular components. There is evidence in the literature that autophagy has an important role in cancer pathogenesis, and that it can serve as a protective factor that promotes tumor survival under stressful conditions such as EGFR inhibition.Citation26 However, excessive autophagy can have the opposite effect on cells, and rather than promoting cell survival it can serve tumor suppressive functions through inducing cell death or cell senescence.Citation27 Therefore, one possible mechanism to explain the synergy noted with co-inhibition of EZH2 and EGFR is that it is through modulation of autophagy where the colon cancer cells are able to utilize autophagy as a protective mechanism with either EGFR or EZH2 inhibition alone. However, co-inhibition of these 2 pathways dramatically increases autophagy to a point where it may become no longer protective, and instead pushes these cells into a non-proliferative or apoptotic state. In addition, it is also possible that co-inhibition of EGFR and EZH2 synergize through effects on non-autophagy related genes as well, especially those that are epigenetically regulated through H3K27-methylation. The study of these genes is beyond the scope of this paper, but would certainly provide valuable insight into the observed synergy with co-inhibition of EGFR and EZH2.
Of additional interest is that co-inhibition of both EGFR and EZH2 was effective in decreasing cell number and inducing apoptosis in both a KRAS wild type colon cancer cell line (HT-29) as well as a KRAS mutant cell line (HCT-15). EGFR inhibitors have been well known to improve the survival of patients with KRAS wild type metastatic colon cancer, however these inhibitors have the opposite effect on KRAS mutant tumors, where they paradoxically decrease survival.Citation3 KRAS mutant colon cancers are notoriously difficult to treat, yet can represent up to 40% of diagnosed colon cancers.Citation28 In our current study we utilize both a KRAS wild type and a KRAS mutant colon cancer cell line and show that in both cell lines co-targeting EGFR and EZH2 shows similar anti-proliferative effects. Additional work in other KRAS wild type and KRAS mutant cell lines is important to determine the generalizability of these results, and furthermore mouse tumor xenograft models would be important to help elucidate the in vivo effects of this combination. Additionally, these results have logical extension to other types of cancer as well, especially those that depend on EGFR signaling such as non-small cell lung cancer (NSCLC).Citation29,30 NSCLCs often harbor activating EGFR mutations, and small molecule tyrosine kinase EGFR inhibitors are a mainstay of therapy.Citation29,30 Therefore, additional testing of the benefits of co-inhibition of EGFR and EZH2 is warranted in NSCLC.
In summary, we demonstrate that the small molecule UNC1999 effectively inhibits EZH2 in 2 colon cancer cell lines. Furthermore, co-inhibition of EGFR and EZH2 significantly decreases proliferation and induces apoptosis in these cell lines, possibly through increasing autophagy. Ultimately these results demonstrate that inhibiting EZH2 may be an important epigenetic mechanism for enhancing the response of colon cancer to EGFR inhibition, and could also hold potential for the development of new therapeutic regimens to treat metastatic colon cancer.
Materials and Methods
Inhibitors
Gefitinib was obtained from LC Laboratories (#G-4408), UNC1999 was synthesized as previously described,Citation22 and both compounds were prepared as 50 mM stock solutions in DMSO and were stored at −20°C. E64d was obtained from Peptides International (#IED-4321-v), and Pepstatin A was obtained from Santa Cruz Biotechnology (#sc-45036), and both of these compounds were prepared as 20 mg/mL stock solutions in DMSO and were stored at −20°C.
Cell culture
The human colon adenocarcinoma cell lines HT-29 and HCT-15 were obtained from the Cell Culture Core of the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases at the University of Pennsylvania. 293T cells were purchased from American Type Culture Collection. All cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% heat-inactivated fetal bovine serum, 100 Units/mL penicillin, and 100 μg/mL streptomycin, and were maintained at 37°C in a humidified 5% CO2 atmosphere.
TCGA database analysis
Level 3 HiSeq RNASeq data was downloaded from TCGA for 302 colon samples (40 normal, 262 tumor), and raw counts for each gene in each sample were extracted. Raw counts were imported into R (v3.1.1),Citation31 where DESeq2 (v1.4.5)Citation32 was applied to score genes for differential expression between tumor and normal samples. For purposes of visualization, DESeq2-calculated normalized log2-transformed counts for each sample were exported.
Cell proliferation assays
For the MTS assay, HT-29 cells and HCT-15 cells were plated in 96-well plates at a density of 104 and 5 × 103 cells/well respectively. After attaching overnight, the cells were then treated with DMSO (control), varying concentrations of UNC1999 or gefitinib, or a combination of UNC1999 and gefitinib for 72 hours. The MTS [3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] assay kit (Promega) was utilized to assess cell proliferation and was performed according to the instructions provided by the manufacturer. Absorbance of each well was recorded at 490 nm using an ELISA plate reader, and after subtracting a background reading, these results were normalized to control wells. Each experiment was performed in triplicate, with mean values ± SD reported for each treatment group. For cell counting experiments HT-29 cells and HCT-15 cells were plated in 6-well plates at a density of 2 × 105 and 105 cells/well respectively. After attaching overnight, the cells were then treated with the DMSO (control), UNC1999, gefitinib, or a combination of UNC1999 and gefitinib for 72 hours. The attached cells were trypsinized, stained with tryptan blue and then live cells were counted using a hemocytometer. Each experiment was performed in duplicate, with mean values ± SD reported for each treatment group. P values were calculated using an unpaired 2-tailed t-test.
Clonogenicity assay
HT-29 and HCT-15 cells were plated in 6-well plates at a density of 2 × 103 cells/well and then treated with DMSO (control), UNC1999, gefitinib, or a combination of UNC1999 and gefitinib, with new media/compound(s) changed every 3 days. After 10 days, cells were fixed with 10% formalin and then stained with 0.05% crystal violet. Each experiment was performed in triplicate.
Protein detection by western blotting
HT-29 and HCT-15 cells were plated in 10 cm plates at a density of 2 × 106 and 106 cells/plate respectively and then treated with DMSO (control), varying concentrations of UNC1999 or gefitinib, or the combination of UNC1999 and gefitinib. After 24 or 72 hours, both attached and floating cells were collected and then lysed with SDS buffer containing protease and phosphatase inhibitors. Protein concentrations were determined using a BCA assay kit (Thermo Scientific). Cell lysates were subjected to polyacrylamide gel electrophoresis on Novex gels (Life Technologies), and protein was transferred to PVDF membranes (Life Technologies). Blocking was performed in TBST containing 5% non-fat dry milk. Antibodies for p-EGFR (#3777), EGFR (#4267), Parp (#9542), LC3B (#2775) and EZH2 (#5246) were purchased from Cell Signaling Technology. The antibody for H3K27me3 (#17–622) was purchased from Millipore, the antibody for actin (#A5441) was purchased from Sigma, and the antibody for p62 (#610497) was purchased from BD Biosciences. Anti-rabbit and anti-mouse secondary antibodies were purchased from Bio-Rad. The proteins were visualized by detection with Amersham ECL Western blotting detection reagents (GE Healthcare).
Plasmids and transfections
pBABE-puro mCherry-EGFP-LC3B (referred to as GFP-LC3B) was received as a gift from Dr. Costas Koumenis. Retroviral packing plasmids pVSV-G and pCPG were received as a gift from Dr. Warren Pear. All lentiviral packaging was done with pLKO.1-puromycin derived plasmids. Lentiviral packaging plasmids pMD2.G and psPAX2, vector control plasmid, EZH2 shRNA1 (CCGGGCTAGGTTAATTGGGACCAAACTCGAGTTTGGTCCCAATTAACCTA) and EZH2 shRNA2 (CCGGCCCAACATAGATGGACCAAATCTCGAGATTTGGTCCATCTATGTTG) plasmids both on a pMX-puro backbone, were purchased from Sigma. After transformation using DH5α cells and ampicillin, the plasmids were purified utilizing a GenElute HP plasmid midiprep kit (Sigma). To produce retrovirus, 293T cells were transfected with pVSV-G, pCPG, and GFP-LC3B using calcium phosphate transfection utilizing CaCl2 and HBS. To produce lentivirus, 293T cells were transfected with pMD2.G, psPAX2, and either Vector or EZH2 shRNA using Fugene 6 (Promega) according to the manufacturer's instructions. After collecting and filtering the virus, HT-29 cells were then transduced in the presence of 4 μg/ml polybrene (hexadimethrine bromide). 24 hours after completion of transduction, HT-29 cells were then selected with 2 μg/ml puromycin for 72 hours.
RT-PCR
RNA was extracted from cultured cells with Trizol, and subsequently isolated through the use of an RNeasy Mini Kit (Qiagen). RNA (1.2 μg) was transcribed into cDNA, and real-time PCR (RT-PCR) was performed using a Quantitative SYBR-Green PCR Kit (Qiagen) and a 7500 Fast Real Time PCR System (Applied Biosystems). EZH2 primers included: forward 5′-AGTGTGACCCTGACCTCTGT-3′ and reverse 5′-AGATGGTGCCAGCAATAGAT-3′. Actin primers included: forward 5′-GGTCATCACCATTGGCAATGA-3′ and reverse 5′-GCACTGTGTTGGCGTACA-3′. EZH2 transcript levels were normalized to actin transcript levels, with mean values ± SD reported for each group. All experiments were performed in duplicate.
Fluorescence microscopy
HT-29 cells were plated on sterile coverslips in 6-well plates at a density of 2 × 104 cells/well. After treatment for 48 hours, the cells were fixed with 10% formalin and then mounted using ProLong Gold antifade reagent with DAPI (Life Technologies). These slides were then examined on an inverted Olympus IX81 fluorescent microscope at 200× where pictures were obtained.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
972776_Supplementary_Materials.zip
Download Zip (603.2 KB)Acknowledgments
We would like to thank Dr John Tobias at the University of Pennsylvania Bioinformatics Core for his assistance with the bioinformatics analysis, and Dr Zhenghua Xu at the University of Pennsylvania for his assistance with experimental techniques.
Funding
We would like to acknowledge the following support: NIH/NIDDK T32-DK007066 (BK), NIH R01-DK085121 (XH), NIH R01-GM103893 (JJ), Caring for Carcinoid Foundation-AACR Grant Care for Carcinoid Foundation 11-60-33 (XH), TRP grant from the Leukemia and Lymphoma Society (XH), pilot grant from ITMAT of the University of Pennsylvania (XH), Hubei Province's Outstanding Medical Academic Leader Program–2013 (YL), and NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases (P30DK050306) and its core facilities.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- American Cancer Society. Cancer Facts & Figures 2014. Atlanta, GA: American Cancer Society, 2014.
- Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, Cooper D, Gansler T, Lerro C, Fedewa S, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin 2012; 62:220-41; PMID:22700443
- Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369:1023-34; PMID:24024839; http://dx.doi.org/10.1056/NEJMoa1305275
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008; 358:1160-74; PMID:18337605; http://dx.doi.org/10.1056/NEJMra0707704
- Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533-41; PMID:21878654; http://dx.doi.org/10.1158/1535-7163.MCT-11-0047
- Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, et al. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell 2013; 154:1269-84; PMID:24034250; http://dx.doi.org/10.1016/j.cell.2013.08.015
- Zou Y, Ling YH, Sironi J, Schwartz EL, Perez-Soler R, Piperdi B. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J Thoracic Oncol 2013; 8:693-702; PMID:23575415; http://dx.doi.org/10.1097/JTO.0b013e31828c7210
- Li YY, Lam SK, Mak JC, Zheng CY, Ho JC. Erlotinib-induced autophagy in epidermal growth factor receptor mutated non-small cell lung cancer. Lung Cancer 2013; 81:354-61; PMID:23769318; http://dx.doi.org/10.1016/j.lungcan.2013.05.012
- Bokobza SM, Jiang Y, Weber AM, Devery AM, Ryan AJ. Combining AKT inhibition with chloroquine and gefitinib prevents compensatory autophagy and induces cell death in EGFR mutated NSCLC cells. Oncotarget 2014; 5:4765-78; PMID:24946858
- Dragowska WH, Weppler SA, Wang JC, Wong LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge M, et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PloS One 2013; 8:e76503; PMID:24146879; http://dx.doi.org/10.1371/journal.pone.0076503
- Eimer S, Belaud-Rotureau MA, Airiau K, Jeanneteau M, Laharanne E, Veron N, Vital A, Loiseau H, Merlio JP, Belloc F. Autophagy inhibition cooperates with erlotinib to induce glioblastoma cell death. Cancer Biol Ther 2011; 11:1017-27; PMID:21508666; http://dx.doi.org/10.4161/cbt.11.12.15693
- Xu L, Beckebaum S, Iacob S, Wu G, Kaiser GM, Radtke A, Liu C, Kabar I, Schmidt HH, Zhang X, et al. MicroRNA-101 inhibits human hepatocellular carcinoma progression through EZH2 downregulation and increased cytostatic drug sensitivity. J Hepatol 2014; 60:590-8; PMID:24211739; http://dx.doi.org/10.1016/j.jhep.2013.10.028
- Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 2008; 32:491-502; PMID:19026780; http://dx.doi.org/10.1016/j.molcel.2008.10.016
- McCabe MT, Creasy CL. EZH2 as a potential target in cancer therapy. Epigenomics 2014; 6:341-51; PMID:25111487; http://dx.doi.org/10.2217/epi.14.23
- Shen L, Cui J, Liang S, Pang Y, Liu P. Update of research on the role of EZH2 in cancer progression. OncoTargets Ther 2013; 6:321-4; PMID:23589697; http://dx.doi.org/10.2147/OTT.S42453
- Kristensen LS, Nielsen HM, Hansen LL. Epigenetics and cancer treatment. Eur J Pharmacol 2009; 625:131-42; PMID:19836388; http://dx.doi.org/10.1016/j.ejphar.2009.10.011
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6:38-51; PMID:16397526; http://dx.doi.org/10.1038/nrc1779
- Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours–lessons from the past. Nat Rev Clin Oncol 2013; 10:256-66; PMID:23546521; http://dx.doi.org/10.1038/nrclinonc.2013.42
- Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beausejour M, Beaulieu JF, Lipkin SM, Gudas LJ. Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res 2013; 319:1463-70; PMID:23588203; http://dx.doi.org/10.1016/j.yexcr.2013.04.006
- Benoit YD, Laursen KB, Witherspoon MS, Lipkin SM, Gudas LJ. Inhibition of PRC2 histone methyltransferase activity increases TRAIL-mediated apoptosis sensitivity in human colon cancer cells. J Cell Physiol 2013; 228:764-72; PMID:23001792; http://dx.doi.org/10.1002/jcp.24224
- Fussbroich B, Wagener N, Macher-Goeppinger S, Benner A, Falth M, Sultmann H, Holzer A, Hoppe-Seyler K, Hoppe-Seyler F. EZH2 depletion blocks the proliferation of colon cancer cells. PloS One 2011; 6:e21651; PMID:21765901; http://dx.doi.org/10.1371/journal.pone.0021651
- Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol 2013; 8:1324-34; PMID:23614352; http://dx.doi.org/10.1021/cb400133j
- Barth S, Glick D, Macleod KF. Autophagy: assays and artifacts. J Pathol 2010; 221:117-24; PMID:20225337; http://dx.doi.org/10.1002/path.2694
- Komatsu M, Kageyama S, Ichimura Y. p62/SQSTM1/A170: physiology and pathology. Pharmacol Res 2012; 66:457-62; PMID:22841931; http://dx.doi.org/10.1016/j.phrs.2012.07.004
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005; 1:84-91; PMID:16874052; http://dx.doi.org/10.4161/auto.1.2.1697
- Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, Kroemer G. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta 2009; 1793:1524-32; PMID:19371598; http://dx.doi.org/10.1016/j.bbamcr.2009.01.006
- Chen Y, Klionsky DJ. The regulation of autophagy–unanswered questions. J Cell Sci 2011; 124:161-70; PMID:21187343; http://dx.doi.org/10.1242/jcs.064576
- Han CB, Li F, Ma JT, Zou HW. Concordant KRAS mutations in primary and metastatic colorectal cancer tissue specimens: a meta-analysis and systematic review. Cancer Invest 2012; 30:741-7; PMID:23075074; http://dx.doi.org/10.3109/07357907.2012.732159
- Remon J, Moran T, Reguart N, Majem M, Carcereny E, Lianes P. Beyond EGFR TKI in EGFR-mutant non-small cell lung cancer patients: main challenges still to be overcome. Cancer Treat Rev 2014; 40:723-9; PMID:24759598; http://dx.doi.org/10.1016/j.ctrv.2014.03.006
- Mok TS, Lee K, Leung L. Targeting epidermal growth factor receptor in the management of lung cancer. Semin Oncol 2014; 41:101-9; PMID:24565584; http://dx.doi.org/10.1053/j.seminoncol.2013.12.010
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2014. http://www.R-project.org/
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. BioRxiv 2014. http://dx.doi.org/10.1101/002832