
Abstract
Checkpoint kinase 2 (Chk2) has been implicated in DNA damage signaling. By using BJ human fibroblasts, HCT116 colorectal cancer cells and HeLa cervical cancer cells, we further detailed phosphorylation kinetics of Chk2 under treatment with neocarcinostatin (NCS) or doxorubicin (Dox). After NCS treatment, phosphorylation of Chk2 Thr68 occurs in 3 min, followed by phosphorylation of Ser19 and Ser33/35. In ATM deficient fibroblasts, NCS does not induce phosphorylation of NBS1 Ser343 and Chk2 Ser19 and Ser33/35, however Chk2 Thr68 is still phosphorylated, indicating that ATM is essential for phosphorylation of these residues when treated with NCS. By using Chk2-deficient HCT116 cells re-expressing phospho-mutant Chk2 (T68A), we found that inhibition of Thr68 phosphorylation enhances Ser19 phosphorylation in NCS treated cells. Interestingly, in contrast to NCS, Dox does not induce Ser33/35 phosphorylation in HeLa and HCT116 cells. Phosphorylation of Thr68 is sustained until 3 to 4 hours, and phosphorylation of Ser19 occurs 70 to 80 min after Dox treatment. These results demonstrate that Chk2 s involved in the early stages of DNA damage response. Differential phosphorylation kinetics of these residues suggests that DNA damage determines intermolecular and intramolecular interaction of Chk2, which may regulate phosphorylation.
Keywords:
Abbreviations
| ATM | = | ataxia telangiectasia mutated |
| ATR | = | ATM related kinase |
| Dox | = | doxorubicin |
| NCS | = | neocarzinostatin |
| PAGE | = | poly acryl gel electrophoresis |
Introduction
Chk2, checkpoint kinase 2, is a member of serine/threonine protein kinases that has been implicated in DNA damage response.Citation1,2 It has been well illustrated that DNA damage stimuli induce phosphorylation of different residues of Chk2, including serine 19 (S19), 33/35 (S33/35), and threonine 68 (T68). ATM and ATR1,2,3 phosphorylate Chk2 at these multiple residues in the SQ/TQ-rich region.Citation1-3 The kinase activation is initiated by phosphorylation of T68, followed by dimerization of Chk2 through interaction between the FHA and phosphorylated SQ/TQ-rich region.Citation1-3 Subsequently, autophosphorylation of T383 and T387 occurs in cis and trans.Citation4 In this course of the event, S19, S33 and S35 of the SQ/TQ-rich region are phosphorylated, too.Citation4-6 Although these studies discovered novel phosphorylation sites, the kinetics and inter/intra molecular interaction of each of the phosphorylation sites are largely unknown.
It has been demonstrated that DNA lesions recruit DNA damage response proteins such as γH2AX, MRN complex (Mre11, Rad50 and NBS1) and ATM, creating an amplified signal loop of their phosphorylation.Citation7,8 It is assumed that Chk1/2 and p53 are subsequently phosphorylated, resulting in the regulation of expression of the p53 target genes.Citation9,10 In the present studies, we explored kinetics of the phosphorylation of several residues of Chk2 and studied hierarchy of the signaling. We found that phosphorylation of Chk2 is differentially regulated when cells are treated with NCS or Dox, and that phosphorylation of S33/35 is not detected when treated with Dox throughout the time course we tested. We also found that NBS1 S373 is transiently phosphorylated by Dox, however that phosphorylation is sustained when treated with NCS.
These studies provide us with an insight of re-evaluation of the Chk2 pathway. Our results implicate that differential regulation of phosphorylation of the Chk2 residues under different conditions of DNA damage could result in the distinct mechanism of Chk2 activation that is determined by the phosphorylation residues and time course. From these results, we propose that integrity of both structure and phosphoresidue is critical for Chk2 tumor suppressor function.Citation11,12
Results and Discussion
Chk2 T68 phosphorylated precedes Chk2 S19 and S33/35 when treated with neocarcinostatin
In the current studies, we further detailed how phosphorylation cascade is regulated when cells are treated with distinct DNA damage agents. BJ cells, normal human fibroblasts, were treated with NCS (xxx mM) for the indicated time course until 6 hours, and phosphorylation of 53BP1 (Ser25/29 (S25/29) and Ser1778 (S1778)), ATM Ser1981 (S1981), NBS1 Ser343 (S343), Chk2 Ser19 (S19), Ser33/35 (S33/35), Thr68 (T68), p53 Ser15 (S15) and γH2AX were studied (). We found that phosphorylation of 53BP1 S25/29 and S1779, ATM S1981 and NBS1 S343 appears within 7 to 10 min after NCS treatment and sustained up to 6 hours. In contrast, induction of the phosphorylation of Chk2 T68 occurs markedly and rapidly within 3 to 5 min after treatment and reaches at the peak in 10 min, and decreased until 5 hours. Phosphorylation of Chk2 S19 appears in 10 to 15 min and is sustained until 6 hours. Phosphorylation of Chk2 S33/35 appears in 15 min, reaches in 25 min, and disappears in 90 to 120 min. Phosphorylation of p53 S15 occurs in 6 to 7 min, increases until 5 hours. Consistent with the previous results indicating the increase in stability of p53 induced by p53 phosphorylation,Citation9 levels of p53 protein increased after NCS treatment. Of note, γH2AX periodically appeared and disappeared in this time course. These results show that, when BJ cells are treated with NCS, phosphorylation of Chk2 T68 precedes the phosphorylation of S19 and S33/35. When phosphorylation of Chk2 S19, S33/35 and T68 was increased, we detected slower migration of these phosphorylated Chk2 by anti-Chk2 immunoblot, suggesting the protein modification after NCS treatment.
Figure 1. Determine the kinetics of phosphorylation of the DNA damage-associated proteins treated with NCS. BJ cells (A), HCT116 cells (B), GM09607 cells (C) and HCT116 Chk2(-) cells (D) were treated with NCS (1 μg/ml) for the indicated time (from 1 min to 6 hours). Cell lysates were collected and protein phosphorylation was studied using the antibodies indicated. Actin serves as a loading control.

Detailed analyses were performed with HCT116 cells, human colorectal cancer cell line (). Compared to BJ cells, similar kinetics of the phosphorylation of 53BP1 S25/29 and S1778, ATM S1981, NBS1 S343, Chk2 S19, S33/35, T68, p53 S15 and γH2AX were detected. Slower migration of the Chk2 protein was detected in immunoblot of Chk2 T68 in HCT116 cells, although it was not obvious in immunoblot of Chk2 S19 and S33/35, compared to the results obtained from BJ cells. γH2AX showed periodic phosphorylation, which is similar to that of BJ cells.
Next, we tested ATM-deficient human fibroblasts, GM09607 (). When these cells were treated with NCS, phosphorylation of NBS1 S343, Chk2 S19 and S33/35 was not detected throughout the time course. However, phosphorylation of Chk2 T68 was similarly induced as detected in both BJ cells and HCT116 cells. These results indicate that ATM is essential for NCS-induced phosphorylation of NBS1 S343, Chk2 S19 and S33/35. It has been demonstrated that ATR also phosphorylates Chk2 T68.2 Thus, it is possible that NCS induces Chk2 T68 phosphorylation by ATR in GM09607 cells.
Roles of Chk2 for phosphorylation of NBS1, p53 and γH2AX were studied using HCT116 deficient for endogenous Chk2 (HCT116 Chk2(-)) in which Chk2 locus was disrupted in vitro,Citation13 when treated with NCS (). NBS1 S343 and p53 S15 are similarly phosphorylated in these cells, indicating that Chk2 is not directly involved for these phosphorylation. γH2AX was also detected, however strong signals were observed in 50 min or later, which was delayed compared to that in the parental HCT116 cells (). Increased protein expression of p53 was also detected in HCT116 Chk2(-) cells after NCS treatment. These results support the previous studies that Chk2 is not essential for phosphorylation of p53 at Ser20 (S20) and subsequent increased stability of the protein,Citation14 demonstrating that p53 S15 and S20 are phosphorylated in the absence of Chk2 under conditions of DNA damage.
Distinct DNA damage reagents induce differential phosphorylation of Chk2
We further detailed whether phosphorylation of these DNA damage response proteins is differentially regulated when cells are treated with 2 different reagents, NCS and doxorubicin (Dox, 8.7 μM). HeLa cells were treated with NCS or Dox until 6 hours, and samples were obtained at the indicated time (). When treated with NCS, kinetics of phosphorylation of ATM S1981, NBS1 S343, Chk2 S19, S33/35, T68 was similar with that of BJ and HCT116 cells (). Of note, levels of the Chk2 protein significantly decreased in 40 to 50 min after NCS treat in HeLa cells. When treated with Dox, phosphorylation of ATM S1981 was similarly induced compared to NCS treatment, however, phosphorylated NBS1 S343 was only detected in 30 to 50 min after treatment. Phosphorylation of Chk2 S19 occurred at 70 to 80 min. Interestingly, phosphorylation of Chk2 S33/35 was not detected under treatment with Dox during this time course. Phosphorylated Chk2 T68 appeared in 20 min, and sustained until nearly 5 hours.
Figure 2. Determine the kinetics of phosphorylation of the DNA damage-associated proteins treated with Dox. HeLa cells were treated with NCS (1 μg/ml) or Dox (8.5 μM) for the indicated time (1 min to 6 hours). Cell lysates were collected and protein phosphorylation was studied using the antibodies indicated. Dox does not induce phosphorylation of Chk2 S33/35. Actin serves as a loading control.
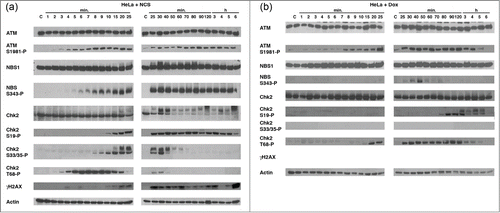
These results suggest at least 2 models, (1) Chk2 is phosphorylated by several kinases including itself and those kinases are differentially regulated by NCS and Dox, (2) NCS and Dox differentially control Chk2 conformation which determines inter- or intra-molecular phosphorylation of the protein.
Antagonistic effect of phosphorylation of Chk2 T68 on Chk2 S19
We next studied whether phosphorylation of the specific residue of Chk2 determines phosphorylation of the other sites. HCT116 Chk2(-) cells were transduced with retroviruses expressing HA-tagged wild type Chk2, Chk2 S19A (Ser19 is changed to Ala), Chk2 S33/35A (Ser33/35 are changed to Ala) and Chk2 T68A (Thr68 is changed to Ala). Among the clones isolated, we chose the cells in which levels of the exogenous Chk2 are similar to those of the parental HCT116 cells (data not shown). These cells were named HA-WT, HA-S19A, HA-S33/35A and HA-T68A cells, respectively.
When these cell lines were treated with NCS (), phosphorylated Chk2 S19 was detected in HA-WT, HA-S33/35A and HA-T68A cells, however, changes of the mobility were observed only in HA-WT and HA-33/35A cells in these blots, suggesting that phosphorylation of Chk2 T68 is necessary for the additional protein modification. Phosphorylation of Chk2 S19, S33/35 and T68 in HA-S19A, HA-S33/35A and HA-T68A cells were similarly induced with that in HA-WT cells, indicating that phosphorylation of each of these residues are independently regulated.
Figure 3. Phosphorylation of the specific residues of Chk2 regulates other phosphorylation sites dependent on the DNA damage. HCT116 Chk2(-) cells were transduced by retrovirus expressing Chk2 cDNA of HA-tagged wild type, S19A, S33/35A or T68A. Stable clones were treated with NCS (A), 1 μg/ml) or Dox (B), 8.5 μM) for the indicated time. Cell lysates were collected and subjected to immunoblot analysis of the indicated antibodies. Actin serves as a loading control.
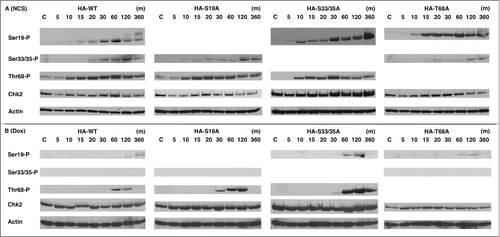
Dox-induced Chk2 phosphorylation was also studied in these generated cell lines (). As shown before, phosphorylated Chk2 S33/35 was not detected in HA-WT, HA-S19A and HA-T68A cells after Dox treatment. Compared to HA-WT cells, HA-19A and S33/35A cells showed enhanced phosphorylation of Chk2 T68 in 60 to 120 min after treatment. These results suggest that phosphorylation of Chk2 T68 is to some extent negatively regulated with Chk2 S19 and S33/35. We also observed that phosphorylation of Chk2 S19 is enhanced in S33/35A cells, suggesting that phosphorylation of Ser33/35 is inhibitory for phosphorylation of Ser19.
Taken together, the present studies indicate that distinct DNA damage determines the site and kinetics of Chk2 phosphorylation. Given the previous studies of Chk2's dimerization after DNA stress, the protein conformation is differentially regulated dependent on the stress. Furthermore, phosphorylation of the specific residues affects the additional protein modification. Functional interaction of phosphorylated residues of Chk2 in response to anti-cancer drugs is in progress.
Materials and Methods
Cell culture and DNA damage treatment
BJ (human fibroblasts), HeLa (human cervical cancer cell line) and HCT116 (human colorectal cancer cell line) cells were purchased from ATCC and maintained in Dulbecco's Modified Eagle Medium (GIBCO, Grand Island, NY, USA), supplemented with fetal bovine serum (10%, Invitrogen, Grand Island, NY, USA). HCT116 Chk2(-) cells were kindly provided from Dr. Bert Vogelstein at Johns Hopkins University.Citation13 To induce DNA stress, 1 μg/ml of neocarzinostatin (Kayaku, Tokyo, Japan) or 8.5 μM of doxorubicin (Sigma, St. Louis, MO, USA) was added to the cell culture media.
Establishment of stable cell lines
cDNA encoding HA-tagged human Chk2 was subcloned into pBabepuro vector. To generate the point mutant forms of Chk2 S19A, S33/35A and T68A, in vitro mutagenesis was preformed using the following oligos.
S19A: 5′- GGC AGC AGT GCC TGT GCA CAG CCC CAT GGC AGC -3′
S33/35A: 5′- CAG TCC CAA GGC TCC TCC GCA CAG GCC CAG GGC ATA TCC AGC TCC-3′
T68A: 5′- TCC TTA GAG ACA GTG TCC GCT CAG GAA CTC TAT TCT ATT-3′
HCT116 Chk2(-) cells were transduced with retrovirus generated from these constructs using Phoenix packaging cell line as described previously,Citation15 followed by puromycin (Sigma) selection (1 μg/ml) for 10 days. Immunoblot was performed to confirm that levels of exogenous Chk2 proteins in the isolated clones are similar with those in the parental HCT116 cells.
Immunoblot analysis
Following antibodies were used for this study: 53BP1, phospho-53BP1 S25/29, phospho-53BP1 S1778, NBS1, phospho-NBS1 S343, Chk2, phosphor-Chk2 S19, phosphor-Chk2 S33/35, phosphor-Chk2 T68, phosphor-p53 S15 (Cell Signaling), ATM (GeneTex), phosphor-ATM S1981 (Rockland), p53, Actin (Santa Cruz), γH2AX (Novus). Cell lysates were prepared on ice with EBC buffer (50 mM Tris-HCl, pH 8.0, 120 nM NaCl, 0.5% Nonidet P-40, 100 mM NaF, 200 μM sodium orthovanadate, 100 μg/ml phenylmethylsulfonyl fluoride, 2 μg/ml leupeptin, 2 μg/ml aprotinin). Protein concentration was determined by Bio-Rad protein assay kit. Twenty to 30 micrograms of proteins were loaded and separated in the 6% or 10% SDS-PAGE. Proteins were transferred to Immobilon-P membrane (Millipore, Billercia, MA) by using a semi-dry device (Trans-Blot; Bio-Rad, Hercules, CA) in 25mM Tris base, 192mM glycine and 10% methanol for 60 min at 15 V. Membranes were blocked with 1% nonfat dried milk in phosphate buffered saline/0.05% Tween and incubated with primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Jackson Laboratory). Signals were detected by ECL kit.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author Contribution
MO and TO planned and generated all the data, discussed the results and wrote the paper.
Acknowledgment
We thank all the members of the Ouchi Laboratory for helpful discussion. We particularly thank Dr Bert Vogelstein at Johns Hopkins University for providing us with HCT116 Chk2(-) cell line.
Funding
This work is supported by grants R01CA79892, R01CA90631, and 5P30CA16056 from National Institutes of Health, Breast Cancer Research Grant from Susan G. Komen Foundation.
References
- Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol 2013; 14:563-80; PMID:23969844; http://dx.doi.org/10.1038/nrm3640
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73-112; PMID:21034966; http://dx.doi.org/10.1016/B978-0-12-380888-2.00003-0
- Buscemi G, Carlessi L, Zannini L, Lisanti S, Fontanella E, Canevari S, Delia D. DNA damage-induced cell cycle regulation and function of novel Chk2 phosphoresidues. Mol Cell Biol 2006; 26:7832-45; PMID:16940182
- Oliver AW, Paul A, Boxall KJ, Barrie SE, Aherne GW, Garrett MD, Mittnacht S, Pearl LH. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J 2006; 25:3179-90; PMID:16794575
- Olsen BB, Larsen MR, Boldyreff B, Niefind K, Issinger OG. Exploring the intramolecular phosphorylation sites in human Chk2. Mutat Res 2008; 646:50-9; PMID:18812180; http://dx.doi.org/10.1016/j.mrfmmm.2008.09.002
- Gabant G1, Lorphelin A, Nozerand N, Marchetti C, Bellanger L, Dedieu A, Quéméneur E, Alpha-Bazin B. Autophosphorylated residues involved in the regulation of human chk2 in vitro. J Mol Biol 2008; 380:489-503; PMID:18538787; http://dx.doi.org/10.1016/j.jmb.2008.04.053
- Dimitrova N, de Lange T. Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol Cell Biol 2009; 29:5552-63; PMID:19667071; http://dx.doi.org/10.1128/MCB.00476-09. Epub 2009 Aug 10
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007; 26:7741-8; PMID:18066086
- Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA Repair (Amst) 2004; 3:1039-47; PMID:15279791
- Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39-85; PMID:15189136
- Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006; 25:5912-9; PMID:16998506
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73-112; PMID:21034966; http://dx.doi.org/10.1016/B978-0-12-380888-2.00003-0
- Jallepalli PV1, Lengauer C, Vogelstein B, Bunz F. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 2003; 278:20475-9; PMID:12654917
- Ahn J, Urist M, Prives C. Questioning the role of checkpoint kinase 2 in the p53 DNA damage response. J Biol Chem 2003; 278:20480-9; PMID:12654916
- Grignani F1, Kinsella T, Mencarelli A, Valtieri M, Riganelli D, Grignani F, Lanfrancone L, Peschle C, Nolan GP, Pelicci PG. High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res 1998; 58:14-9; PMID:9426049