
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Restoration of BECN1/Beclin 1-dependent autophagy and depletion of SQSTM1/p62 by genetic manipulation or autophagy-stimulatory proteostasis regulators, such as cystamine, have positive effects on mouse models of human cystic fibrosis (CF). These measures rescue the functional expression of the most frequent pathogenic CFTR mutant, F508del, at the respiratory epithelial surface and reduce lung inflammation in CftrF508del homozygous mice. Cysteamine, the reduced form of cystamine, is an FDA-approved drug. Here, we report that oral treatment with cysteamine greatly reduces the mortality rate and improves the phenotype of newborn mice bearing the F508del-CFTR mutation. Cysteamine was also able to increase the plasma membrane expression of the F508del-CFTR protein in nasal epithelial cells from F508del homozygous CF patients, and these effects persisted for 24 h after cysteamine withdrawal. Importantly, this cysteamine effect after washout was further sustained by the sequential administration of epigallocatechin gallate (EGCG), a green tea flavonoid, both in vivo, in mice, and in vitro, in primary epithelial cells from CF patients. In a pilot clinical trial involving 10 F508del-CFTR homozygous CF patients, the combination of cysteamine and EGCG restored BECN1, reduced SQSTM1 levels and improved CFTR function from nasal epithelial cells in vivo, correlating with a decrease of chloride concentrations in sweat, as well as with a reduction of the abundance of TNF/TNF-alpha (tumor necrosis factor) and CXCL8 (chemokine [C-X-C motif] ligand 8) transcripts in nasal brushing and TNF and CXCL8 protein levels in the sputum. Altogether, these results suggest that optimal schedules of cysteamine plus EGCG might be used for the treatment of CF caused by the F508del-CFTR mutation.
Abbreviations:
- BECN1/Beclin 1, autophagy-related
- CF, cystic fibrosis
- CFTR, cystic fibrosis transmembrane conductance regulator
- CHX, cycloheximide
- CSNK2, casein kinase 2
- CXCL2, chemokine (C-X-C motif) ligand 2
- CXCL8, chemokine (C-X-C motif) ligand 8
- EGCG, epigallocatechin gallate
- FEV, forced expiratory volume
- PM, plasma membrane
- RPD, rectal potential difference
- SQSTM1, sequestosome 1
- TGM2, transglutaminase 2
- TNF, tumor necrosis factor
Introduction
Cystic fibrosis (CF), the most common lethal monogenic disease in Caucasians, is caused by mutations in the gene coding for cystic fibrosis transmembrane conductance regulator (CFTR), a 1480 amino-acid protein functioning as a chloride channel at apical membrane from epithelial cells.1-Citation3 CF is a systemic disease although the extent of clinical manifestations is highly heterogeneous in distinct organs.Citation4,5 CF-associated bronchopulmonary disease, the principal cause of morbidity and mortality, comprises a compendium of alterations including airway obstruction by viscous mucus and chronic inflammation with recurrent bacterial infections, mainly by Pseudomonas aeruginosa. Other prevalent features of CF are insufficiency of the exocrine pancreas, increased electrolytes in sweat, and male infertility. To date, the clinical management of CF focuses on treating CF symptoms, yet fails to address the primary cause of CF, namely the loss-of-function of CFTR. Therapeutic strategies aimed at correcting the CFTR defect (“CFTR-repair”) have recently emerged.Citation6
In the CFTR gene, more than 1,900 mutations, mostly disease-relevant, have been identified and then categorized in 6 different classes according to their functional impact.Citation6,7 To treat the phenotypic consequences of such genotypes, mutation-specific approaches are focusing on the identification of small molecules capable of correcting the deficient trafficking of the CFTR protein (“correctors”: agents that assure the expression of the mutated protein in the apical plasma membrane) or defective gating (“potentiators”: agents that reinstate the electrolyte pump function of mutated CFTR proteins that are orthotopically expressed). An orally available compound identified by high-throughput screening, the CFTR potentiator VX-770 (Ivacaftor, trade name Kalydeco, Vertex Pharmaceuticals), efficiently reduces chloride levels in sweat and improves lung function in CF patients harboring the CFTRG551D genotype, a rare class III CFTR mutant that affects only 4% to 5% of CF patients worldwide.Citation8
One single mutation, p.phe508delCFTR, commonly known as F508del-CFTR (categorized in class II), accounts for about 70% of CFTR loss-of-function mutations and is present in approximately 90% of CF patients worldwide.Citation3,9 Due to its misfold, F508del-CFTR protein does not reach the plasma membrane (PM) and is prematurely degraded. Although F508del-CFTR can be rescued at the PM by CFTR correctors,Citation6 this mutant CFTR protein is unstable at the cell surface and rapidly redirected from endosomal recycling towards lysosomal delivery and degradation.Citation6,10,11
Despite of the mild gating defect linked to an altered protein conformation, F508del-CFTR acquires some degree of function if rescued and stabilized at the PM.Citation12,13 Restoring even less than 30% of CFTR function in vivo is believed to confer an at least partial clinical benefit to CF patients.Citation14,15 As proof of this concept, rescuing approximately 20% of function of wild-type CFTR largely prevents the CF-associated intestinal manifestations in newborn F508del-CFTR homozygous pigs, a species that, like mice, exhibits a preponderant intestinal phenotype in CFTR-mutated newborns.Citation16 Although correctors have become available to improve the maturation and trafficking of the unstable F508del mutant protein at the PM,Citation6 a CFTR-repairing therapy is not yet clinically available. The investigational F508del corrector VX-809, which is endowed with a high rescuing efficacy in vitro and in primary cultures of lung cells from CF patients,Citation17-19 showed only modest efficacy in a phase II clinical trial in CF patients homozygous for the F508del-CFTR mutant.Citation17 VX-809 Citation20 and another corrector, VX-661,Citation21 are now being evaluated in clinical trials in combination with the potentiator VX-770 for the treatment of homozygous F508del-CFTR CF patients.Citation22, 23
Besides these mutation-specific approaches, more general therapeutic strategiesCitation6 advocating the improvement of proteostasis have emerged.Citation24,25 Thus, a complex derangement of proteostasis takes place in human bronchial F508del-CFTR homozygous epithelial cell lines, as well as in the lungs from F508del-Cftr homozygous (CftrF508del) mice.Citation26-28 Dysfunctional F508del-CFTR protein induces persistent activation of TGM2 (transglutaminase 2),Citation29,30 resulting in cross-linking of several TGM2 substrates, including BECN1,Citation26 a protein essential for autophagy. This leads to functional sequestration of the BECN1 interactome in intracellular aggregates, resulting in defective autophagy with consequent accumulation of the autophagic substrate SQSTM1.Citation26 Transgene-enforced BECN1 overexpression, knockdown of SQSTM1 by means of small interfering RNAs (siRNAs), or addition of autophagy-stimulatory proteostasis regulators, such as cystamine, a known TGM2 inhibitor, can reduce the abundance of inflammatory cytokines both in CFBE41o- bronchial epithelial cells derived from F508del-CFTR homozygous CF patients, as well as in lungs from CftrF508del mice.Citation26,27 These treatments can increase the expression level of F508del-CFTR protein and restore its function at the PM. Citation26,27 Notably, the positive effects of cystamine extend for a while beyond its washout unless the autophagic response is abrogated or CFTR function is inhibited by the functional CFTR inhibitor 172 (CFTRinh-172) during cystamine washout. This indicates that prior re-establishment of autophagy prolongs the persistence at the epithelial surface of a sufficient amount of functional F508del-CFTR to interrupt, for a while, the negative loop that compromises its PM residence and function.Citation31 We have reported that a CFTR-sufficient environment is required to allow F508del-CFTR to traffic to and reside at the PM of bronchial epithelial cells.Citation31 Accordingly, when F508del-CFTR is transfected into cells, the resulting protein is not expressed in the PM of CFTR-deficient HeLa cells, yet is capable of trafficking to and residing at the PM of CFTR-sufficient normal bronchial epithelial cells, unless these latter cells are treated with CFTRinh-172.Citation31 Indeed, the functional inhibition of CFTR in normal bronchial epithelial cells ignites the removal of endogenous wild-type CFTR from the PM by diverting CFTR recycling to lysosomal degradation.Citation31 Proteostasis regulators such as cystamine interrupt this negative feed forward loop (in which a functional CFTR defect entails the destruction of PM-sessile CFTR protein) and create a permissive environment for the restoration of CFTR function.Citation27,31 The ability of cystamine to rescue and conserve F508del-CFTR in mouse and human respiratory cells may also enable the beneficial action of CFTR potentiators.Citation27 These observations highlight cystamine as a potential candidate drug for the therapy of CF patients harboring the F508del-CFTR mutation.
Oral administration of cysteamine, the reduced form of cystamine, has been FDA (Food and Drug Administration) approved for the treatment of cystinosis, a rare genetic disease.Citation32 Morever, clinical trials suggest the efficacy of cysteamine in children suffering from nonalcoholic fatty liver disease.Citation33 Driven by the excellent safety profile of cysteamine, we investigated whether the oral administration of this compound could rescue CFTR function and improve CF symptoms in vivo. Here we report the therapeutic effect of cysteamine on CftrF508del mice as well as the capacity of cysteamine to improve CFTR function of ex vivo cultured airway epithelia from CF patients carrying the F508del-CFTR mutation. Encouraged by these preclinical data, we performed a phase II study revealing an unprecedented clinical efficacy of cysteamine in CF patients homozygous for the F508del mutation.
Results
Therapeutic effects of oral cysteamine on CftrF508del mice
In pigs and mice, loss-of-function mutations of Cftr cause a predominantly intestinal phenotype.Citation16,Citation34-36 Thus 50% to 90% of cftr KO mice die in the first 4 wk after birth, mostly due to intestinal obstruction,Citation34 and up to 40% of F508del-Cftr homozygous (CftrF508del) mice die during the same period,Citation37 unless they are kept under a special diet that reduces the risk of obstruction.Citation38 We evaluated the hypothesis that oral administration of cysteamine might avoid the juvenile overmortality of CftrF508del mice. A total of 297 2-wk-old mice were fed with a normal diet and randomized to receive cysteamine (n = 159, females 70) or vehicle (n = 138, females 65), without prior knowledge of their genotype. Starting from the 15th d of life, mice were treated via gavage with vehicle alone (100 μl saline solution/day) or cysteamine (60 μg/kg in 100 μl saline/day) for 5 wk or until spontaneous death, followed by genotyping. Among the control mice, which were either wild type or heterozygous for the F508del-CFTR mutation, none died. In contrast, the mortality of CftrF508del mice (n = 82) was 49% in the vehicle-treated (n = 47) and 9% in the cysteamine-treated (n = 35) groups, respectively. Kaplan-Meier survival plots revealed significant (Log-Rank test, P = 0.0001) effects of cysteamine with a 2-wk-survival probability of 60% versus 91% for vehicle-versus cysteamine-treated groups, respectively (A and 1B; Fig. S1). Immunoblots of intestinal lysates of CftrF508del mice revealed that CFTR protein was significantly increased after cysteamine treatment as compared to vehicle-treated mice, reaching a mean value of 58% (range 57% to 65%) of that observed in wild-type control mice (C and 1D). To assess CFTR function in vivo, the response of the rectal potential difference (RPD) to the presence of forskolin and luminal amiloride (100 μM, to block Na+-dependent RPD) in Cl− free solutionsCitation39 was determined. In wild-type mice, perfusion with forskolin (20 μM) induced significant RPD hyperpolarization, indicating activation of Cl− conductance in the rectal epithelium, and this ΔRPD was not affected by treatment with cysteamine. In contrast, CftrF508del mice failed to demonstrate a ΔRPD unless they were treated with cysteamine (E and 1F). These results indicate that cysteamine is able to induce forskolin activated RPD in CftrF508del mice.
Figure 1 (See previous page). Effects of 5-wk oral administration of cysteamine on the intestine of 2 wk-old CftrF508del mice. (A) Schematic representation of the effects of oral administration of cysteamine on the mortality of control (wild-type [WT] homozygotes or WT and F508del heterozygotes) and CftrF508del mice. (B) Cumulative survival rate in CftrF508del mice (n = 82) orally administered with either vehicle (n = 47) or cysteamine (n = 35) for 5 wk. Log-rank test, P = 0.0001. (C and D) Effects of cysteamine on CFTR and BECN1 protein levels in the intestine. (C) Mean changes of protein levels in 5 wild-type and 5 CftrF508del mice treated with either vehicle or cysteamine for 5 wk. Mean ± SD of 3 independent measurements; **P < 0.01 versus vehicle-treated CftrF508del mice (ANOVA). (D) Top, representative immunoblot with anti-CFTR (Abcam clone CF3) and BECN1 (Abcam clone Ab55878) in 1 mouse per treatment group. Bottom, densitometric measurement in the CftrF508del mouse, as percentage of vehicle-treated WT mouse normalized to TUBA levels. Mean ± SD of triplicates of independent experiments, **P < 0.01 versus vehicle-treated CftrF508del mice (ANOVA). (E and F) Effects of cysteamine on rectal potential difference (RPD) in response to 20 μM forskolin (Fsk) in 5 WT and 5 CftrF508del mice treated with vehicle or cysteamine. Measurements were conducted during continous perfusion with a Cl− free solution containing 100 μM amiloride. (E) Mean changes of RPD (ΔRPD) in response to Fsk in 5 WT and 5 surviving CftrF508del mice. Responses in CftrF508del mice were significantly smaller than in wild-type mice; responses in cysteamine-treated CftrF508del mice were significantly higher than in vehicle-treated CftrF508del mice. Mean ± SD of 5 measurements in each group; ###P < 0.001 versus vehicle-treated WT mice; ° P < 0.05 versus vehicle-treated CftrF508del mice (ANOVA). (F) Representative response to Fsk (arrow) in one WT and one surviving CftrF508del mouse per treatment group.
![Figure 1 (See previous page). Effects of 5-wk oral administration of cysteamine on the intestine of 2 wk-old CftrF508del mice. (A) Schematic representation of the effects of oral administration of cysteamine on the mortality of control (wild-type [WT] homozygotes or WT and F508del heterozygotes) and CftrF508del mice. (B) Cumulative survival rate in CftrF508del mice (n = 82) orally administered with either vehicle (n = 47) or cysteamine (n = 35) for 5 wk. Log-rank test, P = 0.0001. (C and D) Effects of cysteamine on CFTR and BECN1 protein levels in the intestine. (C) Mean changes of protein levels in 5 wild-type and 5 CftrF508del mice treated with either vehicle or cysteamine for 5 wk. Mean ± SD of 3 independent measurements; **P < 0.01 versus vehicle-treated CftrF508del mice (ANOVA). (D) Top, representative immunoblot with anti-CFTR (Abcam clone CF3) and BECN1 (Abcam clone Ab55878) in 1 mouse per treatment group. Bottom, densitometric measurement in the CftrF508del mouse, as percentage of vehicle-treated WT mouse normalized to TUBA levels. Mean ± SD of triplicates of independent experiments, **P < 0.01 versus vehicle-treated CftrF508del mice (ANOVA). (E and F) Effects of cysteamine on rectal potential difference (RPD) in response to 20 μM forskolin (Fsk) in 5 WT and 5 CftrF508del mice treated with vehicle or cysteamine. Measurements were conducted during continous perfusion with a Cl− free solution containing 100 μM amiloride. (E) Mean changes of RPD (ΔRPD) in response to Fsk in 5 WT and 5 surviving CftrF508del mice. Responses in CftrF508del mice were significantly smaller than in wild-type mice; responses in cysteamine-treated CftrF508del mice were significantly higher than in vehicle-treated CftrF508del mice. Mean ± SD of 5 measurements in each group; ###P < 0.001 versus vehicle-treated WT mice; ° P < 0.05 versus vehicle-treated CftrF508del mice (ANOVA). (F) Representative response to Fsk (arrow) in one WT and one surviving CftrF508del mouse per treatment group.](/cms/asset/5734b372-6b75-4ef4-9f8f-361cad9a0ecb/kaup_a_973737_f0001_c.jpg)
Notably, the oral administration of cysteamine restored BECN1 protein levels in intestinal lysates of CftrF508del mice to levels comparable to those observed in wild-type mice (C and 1D).
While cysteamine had no effect on the weight gain of control mice (either WT or heterozygous for the F508del-CFTR mutation) over the experimental period, cysteamine-treated CftrF508del mice gained significantly (The Student t test, P < 0.01) more weight than did vehicle control CftrF508del mice (A and 2B).
Figure 2. Effects of 5-wk oral administration of cysteamine on weight gain and lung inflammation in 2 wk-old CftrF508del mice. (A and B) Effects of cysteamine on weight gain. (A) Distribution of weight (g) variation of CftrF508del mice (n = 56) orally treated with either vehicle (n = 24) or cysteamine (n = 32) for 5 wk; °°P < 0.01 (Student t test). (B) Representative picture showing difference in body size at the end of treatment in one vehicle-treated (left) and one surviving cysteamine-treated (right) CftrF508del mice. Scale bar: 1 cm. (C) Effects of cysteamine on Tnf (left) and Cxcl2 (right) transcription levels in lung homogenates from 7-wk-old CftrF508del mice treated with either vehicle or cysteamine. Mean ± SD of triplicates of 5 surviving mice per group; °°P < 0.01 (The Student t test). (D to E) Effects of cysteamine on CFTR, and BECN1 protein levels in the lungs. (D) Mean changes of protein levels in 5 wild-type and 5 CftrF508del mice treated with either vehicle or cysteamine for 5 wk. Mean ± SD of 3 independent measurements; **P < 0.01 versus vehicle-treated CftrF508del mice (ANOVA). (E) Top, representative immunoblot with anti-CFTR (Abcam clone) and anti-BECN1 (Abcam clone Ab55878) in one mouse per treatment group. Bottom, densitometric measurement in the CftrF508del mouse, as percentage of vehicle-treated WT mouse normalized to TUBA/α-β tubulin levels. Mean ± SD of triplicates of independent experiments, **P < 0.01 versus vehicle-treated CftrF508del mice (ANOVA).
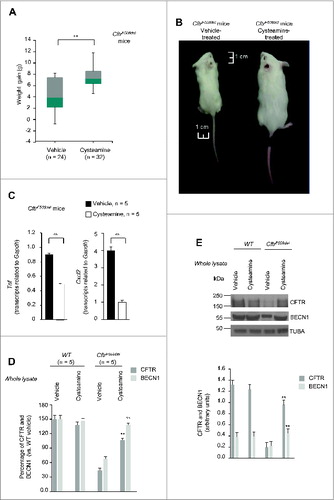
Concomitantly, oral administration of cysteamine was also effective in controlling lung inflammation, as it decreased the abundance of Tnf and Cxcl2 (chemokine (C-X-C motif) ligand 2) transcripts in the lungs of young CftrF508del mice (n = 5, the Student t test, P < 0.01) (C) enrolled in this 5-wk experiment. Furthermore, oral administration of cysteamine rescued CFTR protein in lungs of young CftrF508del mice, and restored BECN1 protein to the levels of wild-type mice (D and 2E).
Gavage of cysteamine for a shorter period (7 d, 100 μl saline solution) also reduced the expression of Tnf and Cxcl2 mRNAs (n = 5, the Student t test, P < 0.001) in the lungs of older (10 to 12 wk-old) CftrF508del mice (Fig. S2).
Together, these data indicate that oral administration of cysteamine has a major positive effect on the intestinal and respiratory tracts of CftrF508del mice.
Effects of combination treatment with cysteamine and epigallocatechin gallate on human airway epithelial cells and mouse lungs
Driven by the positive outcome of the mouse experiments, we next investigated the possibility that cysteamine, like cystamine,Citation28,31,40 would rescue F508del-CFTR at the plasma membrane (PM) and sustain the residence of CFTR mutant after washout in vitro, in polarized CFBE41o- human bronchial epithelial cell lines.Citation27 The cells were transfected with F508del-CFTR at 37 °C, then incubated with cysteamine (250 μM) for 18 h, which was washed out, followed by 24 to 48 h of culture, according to a previously described procedure.Citation27 Surface biotinylation, followed by purification of biotinylated proteins and immunoblot detection of CFTR (to detect its expression at the PM), confirmed that a sizeable fraction of CFTR protein resided at the PM after 24 h of cysteamine washout, as described.Citation27 However, the amount of PM-resident CFTR protein decreased after 48 h of washout to ∼20% of the value observed immediately after cysteamine treatment (A; Fig. S3A). These results prompted us to investigate whether a sequential treatment with natural compounds, with known safety profile, after cysteamine washout would be able to prolong the beneficial effects of cysteamine in conserving F508del-CFTR at the PM after withdrawal. We have previously shown that the CFTR potentiator genistein can increase the beneficial effects of cystamine after washout in controlling lung inflammation in CftrF508del mice.Citation27 Genistein is a member of the well-known class of isoflavones, which are naturally occurring and abundant in soybeans and soy-based foods. However, the use of genistein in CF therapy is limited due to its undesirable effects on the immune and endocrine systems.Citation41,42 In an attempt to avoid the decline of rescued PM-resident F508del-CFTR after cysteamine washout, we tested the effects of another flavonoid, the green tea-derived epigallocatechin gallate (EGCG). This choice was dictated by the consideration that EGCG is authorized as an over-the-counter food additive, with a known safety profile,Citation43 and that EGCG is already being evaluated in clinical trials in CF patients carrying CFTR splice mutants.Citation44 Therefore, we added EGCG (80 μM) for 48 h to the system during cysteamine washout. We found that EGCG was able to maintain the PM residence of rescued F508del-CFTR to ∼80% of the value observed immediately after cysteamine treatment (A). Notably, EGCG alone failed to consistently rescue the PM localization of F508del-CFTR protein and the simultaneous treatment of the cells with cysteamine plus EGCG did not yield a better rescue of the F508del-CFTR protein than did treatment with cysteamine alone (Fig. S4).
Figure 3 (See previous page). Effects of the combination treatment with cysteamine and epigallocatechin gallate (EGCG) on human airway epithelial cells and mouse lungs. (A) CFBE41o-cells were transfected with F508del-CFTR at 37 °C. After transfection, the cells were incubated for 18 h with or without cysteamine (250 μM) and then kept in medium for 24 h or 48 h in the presence or absence of EGCG (80 μM), with or without CFTRinh-172 (20 μM). Cycloheximide (CHX) (100 μg ml−1) was added during the last 8 h of incubation. The lack of CHX toxicity in this model is reported in Fig. S3A. Top, surface biotinylation followed by purification of streptavidin-bound PM proteins and immunoblot with anti-CFTR (clone CF3, Abcam). FLOT1 (clone C-2 Santa Cruz Biotechnology) confirmed cell surface protein-specific localization. Bottom, densitometric measurement of the residual CFTR at the PM expressed as fold increase of the initial amount (medium) normalized to FLOT1 levels, Mean ± SD of triplicates of independent experiments; °°P < 0.01 compared to medium (ANOVA). (B) Effects of EGCG (top) and cysteamine (bottom) on the activity of either the CSNK2A/α subunit or the CSNK2A2-CSNK2B2 (α2β2) holoenzyme acting on the synthetic peptide substrate RRRADDSDDDDD. IC50 values represent the mean of 3 independent experiments with the SD not exceeding 10%. (C) CFBE41o-cells were transfected with F508del-CFTR and incubated with cysteamine and then kept up to 48 h in medium alone as in (A). During cysteamine washout, the cells were incubated with medium or medium added with the CSNK2 inhibitor CX-4945 (5 μM) in the presence or absence of CFTRinh-172 (20 μM). CHX was added to the system as in (A). Top, surface biotinylation followed by purification of streptavidin-bound PM proteins and immunoblot with anti-CFTR (clone CF3, Abcam). FLOT1 (clone C-2 Santa Cruz Biotechnology) confirmed cell surface protein-specific localization. Bottom, densitometric measurement of the residual CFTR at the PM expressed as fold increase of the initial amount (medium) normalized to FLOT1 levels. Mean ± SD of triplicates of independent experiments, °P < 0.05, °°P < 0.01 compared to medium (ANOVA). (D) Tnf (top) and Cxcl2 (bottom) transcription levels in lung homogenates from 10-12 wk-old CftrF508del mice either immediately after treatment with vehicle or cysteamine for 7 d or alternatively after a latency of 10, 20, or 30 d without cysteamine treatment. During this “washout” period, the mice were either left untreated or treated with cysteamine. Mean ± SD of triplicates of 5 mice per group, °P < 0.05, °°°P < 0.001 compared to vehicle and **P < 0.01, ***P < 0.001 compared to 30 d of washout without cysteamine (ANOVA).
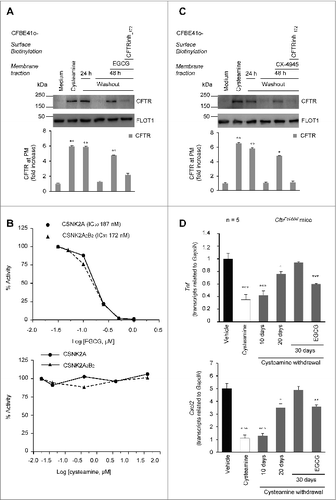
EGCG, similarly to other naturally occurring flavonoids with known beneficial effects on human health,Citation45 has a wide range of pharmacological effects.Citation46,47 EGCG reportedly inhibits several kinases,Citation48 among which protein kinase CSNK2 (casein kinase 2 or protein kinase CK2).Citation49 The inhibition of CSNK2 has been reported to prevent the degradation of CFTR protein, particularly in the case of F508del-CFTR mutant.Citation50 In vitro experiments confirmed that EGCG (80 μM), although not being a specific CSNK2 inhibitor, inhibited both the CSNK2 holoenzyme and its isolated catalytic subunits, whereas cysteamine failed to do so (B). Notably, the capacity of EGCG to sustain the PM residence of rescued F508del-CFTR during cysteamine washout, was mimicked by addition of CX-4945 (5 μM),Citation51 a highly selective CSNK2 inhibitorCitation51 that has been tested in oncological phase I clinical trials Citation52,53 (C). The positive effects of both EGCG and CX-4945 during cysteamine washout were lost if the CFTR inhibitor 172 (CFTRinh-172) was concomitantly added (A and 3C). Therefore, inhibition of CSNK2 could be at least one of the mechanisms by which EGCG prolongs the effects of cysteamine beyond its withdrawal.
The ability of EGCG to prolong the long-term effects of cysteamine adiministration beyond its discontinuation, was confirmed in vivo in CftrF508del mice. We found that short-term oral administration of cysteamine (7 d, 60 μg/kg/day) was effective in significantly reducing lung mRNAs coding for Tnf and Cxcl2 and these anti-inflammatory effects lasted well beyond cysteamine discontinuation. The beneficial effect of cysteamine was lost 30 d after the treatment, unless EGCG (150 μg/kg in 100 μl saline/day for 30 d) was administered during this period. Hence, EGCG extended the efficacy of cysteamine in vivo, significantly reducing the mRNA expression of Tnf (P < 0.001) and Cxcl2 (P < 0.01, the Student t test) (D).
Altogether these results support the possibility that the combinatory treatment with cysteamine and EGCG might be useful for the treatment of CF patients homozygous for the F508del-CFTR mutation.
Effects of cysteamine and EGCG on F508del-CFTR homozygous CF patients ex vivo
Driven by the positive outcome of the mouse experiments, an open-label phase II pilot study (EudraCT number #2013-001258-82 approved by Local Ethics Committee, protocol reference #85/13) was designed to test the efficacy or the combinatory treatment with cysteamine and EGCG in 10 Caucasian CF patients homozygous for the F508del-CFTR mutation ( and ). These patients were in regular follow-up at the Department of Pediatrics, Regional Cystic Fibrosis Care Center, University of Naples Federico II, and were consecutively enrolled according to the eligibility criteria ().
Table 1. Baseline Characteristics of the Patients
Table 2. Study design
Before treatment started, all patients enrolled in the study underwent nasal brushing, following an established procedure.Citation27 We assessed CFTR channel function by measuring the rate of iodide efflux in freshly isolated brushed nasal epithelial cells through a fluorescence-based SPQ assay, to avoid any experimental manipulation of primary epithelial cells.Citation27 The rate of iodide efflux was reduced to a mean of 8.6% (range 6.2 to 12.8; SD 1.9) considering the values obtained from 5 non-CF healthy volunteers as 100% control value (; A; Fig. S5). Brushed nasal epithelial cells belonging to all 10 CF patients were also incubated ex vivo for 18 h with 250 μM cysteamine and then kept in medium for 24 h to 48 h in the presence or absence of 80 μM EGCG. Incubation of these primary CF cells ex vivo for 18 h with 250 μM cysteamineCitation27,31 was effective in rescuing CFTR function to mean levels of 74.4% (range 52.9 to 79.2%, SD 7.9) of the control value (A; Fig. S5). Twenty-four and 48 h after washout of cysteamine, this value dropped to 63.6% (range 48.6 to 72.3, SD 7.4) and 10.7% (range 7.9 to 13.2, SD 1.73), respectively. The addition of EGCG post cysteamine washout sustained (ANOVA, P < 0.001) the function of CFTR ex vivo, which amounted to 61% (range 47.5 to 68.9, SD 7.0) at 48 h (A; Fig. S5). Notably, these effects of EGCG were recapitulated by the specific CSNK2 inhibitor CX-4945 (B).
Table 3. Treatment Effects
Figure 4. Effects of cysteamine and EGCG on surface CFTR in ex vivo cultured primary human nasal epithelial cells belonging to the enrolled F508del-CFTR homozygous CF patients. Freshly isolated brushed nasal epithelial cells were collected from 10 F508del-CFTR homozygous patients and cultured for 18 h with or without cysteamine (250 μM) and then kept for 24 or 48 h in medium or medium added with EGCG (80 μM). Brushed nasal epithelial cells from 5 non-CF healthy controls were cultured with medium alone. (A) Assessment of iodide efflux by a fluorescence assay (SPQ) upon stimulation with forskolin (Fsk) plus 3-Isobutyl-1-methylxanthine (IBMX). Rate of iodide efflux, expressed as percentage of values of 5 healthy controls. The analysis was performed on at least 50 cells per sample and per experiment. Mean ± SD of 3 experiments for each sample. °°°P < 0.001 (ANOVA). (B) Effect of incubation with CX-4549, instead of EGCG, during cysteamine washout. Assessment of iodide efflux. Mean ± SD of 3 experiments for each sample. °°P < 0.01 (ANOVA). (C and D) Effects of ex vivo treatment on CFTR protein levels at the PM of nasal epithelial cells. (C) Mean values of residual CFTR protein at the PM of patients No. 5, 6, and 10 of . The values are expressed as percentage of non-CF healthy control (considered as 100% of value). Mean values of 3 independent experiments for each sample; °°P < 0.01 compared to untreated, ##P < 0.01 compared to non-CF healthy control (ANOVA). (D) Left, representative blot of CFTR protein levels at the PM of nasal epithelial cells from one out of 5 non-CF control and one patient (No. 10 of ) out of 3 patients analyzed. Right, representative blot of CFTR protein levels at the PM of nasal epithelial cells from patient No. 10 cultured ex vivo as indicated. Top, surface biotinylation followed by purification of streptavidin-bound PM proteins and immunoblot with anti-CFTR (clone CF3, Abcam). FLOT1 confirmed cell surface protein-specific localization. Bottom, densitometric measurement of the residual CFTR at the PM expressed as percentage of non-CF healthy control (100% of value) normalized to FLOT1 levels. Mean ± SD of triplicates of independent experiments; °°P < 0.01 compared to untreated, ##P < 0.01 compared to non-CF healthy control (ANOVA).
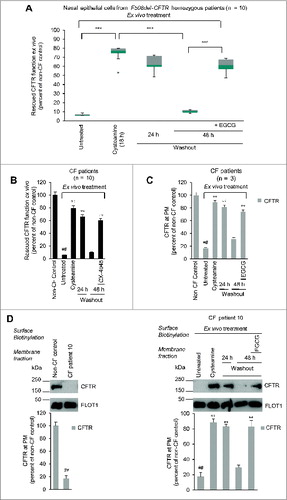
These functional tests could be backed up by measurements of the abundance of the PM-sessile CFTR protein levels in nasal epithelial cells from 3 patients (No. 5, 6 and 10). Surface biotinylation and membrane fractionation revealed that the surface-expressed CFTR was reduced to 17.8% (range 15.1 to 18.3) of the control values from healthy controls (100%) at baseline, yet could be increased by short-term (18 h) culture in the presence of cysteamine to >80% of the control value. EGCG fully avoided the degradation of the F508del-CFTR protein upon cysteamine washout (C and 4D; Fig. S6).
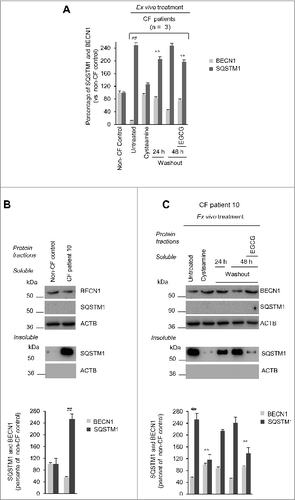
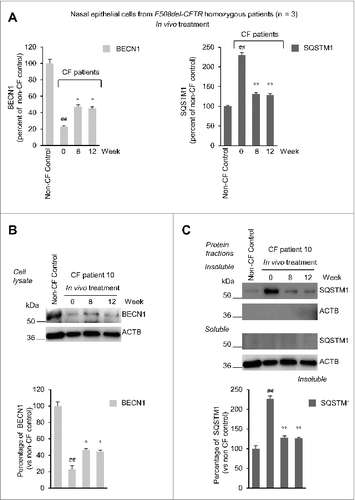
Primary nasal epithelial cells from the 3 CF patients exhibited low BECN1 protein levels together with a 2.5-fold increase of the autophagic substrate SQSTM1 in the insoluble protein fraction, as compared to controls (A to C), indicating an interruption in autophagic flux.Citation26 The incubation with cysteamine restored BECN1 protein, decreased SQSTM1 down to normal levels (A to C) and reduced the activation of TGM2, a key pathogenic player of autophagy inhibition in CF epithelia (Fig. S7).Citation26 These effects were largely lost after 48 h of cysteamine withdrawal, unless EGCG was added to the system (A to C). It should be noted that EGCG did not exert any potentiator activity (Fig. S8A). Furthermore, EGCG alone failed to consistently rescue F508del-CFTR function and the simultaneous treatment ex vivo with cysteamine plus EGCG did not increase F508del-CFTR function as compared to cysteamine alone (Fig. S8B and S8C).
Figure 5. Effects of cysteamine and EGCG on BECN1 and SQSTM1 protein levels in ex vivo cultured primary human nasal epithelial cells belonging to the enrolled F508del-CFTR homozygous CF patients. (A and B) Effects of ex vivo treatment on BECN1 and SQSTM1 protein levels in nasal epithelial cells. (A) Mean values of BECN1 and SQSTM1 of patients No. 5, 6 and 10 of . The values are expressed as percentage of non-CF healthy control (considered as 100% of value). Mean values of 3 independent experiments for each sample; °°P < 0.01 compared to untreated, ##P < 0.01 compared to non-CF healthy control (ANOVA). (B and C) Representative blot of BECN1 and SQSTM1 protein levels in nasal epithelial cells from (B) one out of 5 non-CF control and one patient (No. 10 of ) out of 3 patients analyzed and (C) patient No. 10 cultured ex vivo, as indicated. Top, western blot analysis of insoluble and soluble protein fractions and immunoblot with anti-BECN1 (Abcam) and anti-SQSTM1 (Sigma Aldrich). ACTB was used as negative marker of the insoluble protein fraction and as loading control of the experiment. Bottom, densitometric measurement of BECN1 in the soluble fraction and SQSTM1 level in the insoluble protein fraction expressed as percentage of non-CF healthy control. Mean ± SD of triplicates of independent experiments; °°P < 0.01 compared to untreated, ##P < 0.01 compared to non-CF healthy control (ANOVA).
Together, these results indicate that the sequential treatment of primary respiratory epithelial cells with cysteamine followed by EGCG improves the expression and function of the F508del-CFTR protein.
Effects of oral combination treatment on CF patients in vivo
In the next step, patients (mean age 15.6 y, range 8.6 to 25.0, 6 F) received cysteamine bitartrate (trade name Cystagon®, Orphan Europe) at the same doses recommended for patients with cystinosis (FDA number NDA020392), and epigallocatechin gallate (EGCG) (trade name Epinerve®, SIFI Pharmaceuticals). Both drugs were orally administered. Cystagon® (150 to 300 mg of cysteamine base every 6 h, based on the short half-life of the agent, Fig. S9A and S9B) was given alone for 4 wk, followed by a combination of Cystagon® (same dose) with Epinerve® (270 mg once daily) for a further 4 wk, followed by Epinerve® (270 mg once daily) only for the final 4-wk period of this 3-mo treatment (). The pharmacokinetic profile of Cystagon® in patients homozygous for the F508del-CFTR mutation recapitulates that observed in patients with cystinosis (Fig. S9A and S9B).
During treatment, the patients remained on their prestudy stable medication, including inhaled antibiotics. Patients were followed up for a further 8 wk beyond the end of treatment (). Minor adverse events were registered as single episodes in different patients within a few days after treatment started. These side effects include mild upper abdominal pain (7 patients), nausea (6 patients), halithosis (5 patients) and altered smell of the skin (2 patients). All of the patients complied with the medication until the end of the study. As primary endpoints, we determined CFTR function using 2 assays, namely, (i) the rate of iodide effluxCitation27 in freshly isolated brushed nasal epithelial cells from the enrolled patients compared to 5 adult non-CF healthy volunteers, and (ii) sweat chloride levels. As secondary endpoints, we monitored sputum and nasal levels of the inflammatory cytokines TNF and CXCL8, and we performed spirometry to determine the forced expiratory volume of the first second (FEV1) and the mid-flow rate of forced expiratory flow occurring between 25% and 75% of the patient's exhaled volume (FEF25-75%), both of which were expressed as percentage of the predicted normal value. All clinical and laboratory parameters were assessed at baseline (wk 0) and after 4, 8 and 12 wk of treatment. Spirometric tests and sweat chloride measurements were also performed 4 and 8 wk after the cessation of experimental medication (wk 16 and 20) ().
Effects of in vivo treatment on chloride function in nasal epithelial cells
The rate of iodide efflux from freshly isolated brushed nasal epithelial cells increased throughout the study in all patients (A; Fig. S10). The percentage of rescued CFTR function augmented from mean 8.6% (range 6.2 to 12.8, SD 1.9) at wk 0 to 18.2% (range 8.8 to 29.2, SD 8.4), 28.2% (range 14.6 to 54.0, SD 10.5) and 24.6% (range 15.1 to 33.2, SD 6.9) of the values of non-CF healthy controls (considered as 100% of function) after 4, 8 and 12 wk of treatment, respectively (P = 0.0003 at wk 8, P = 0.0002 at wk 12, as compared to pretreatment values, ANOVA) (A). The reestablishment of CFTR function was confirmed by immunoblot analyses of nasal epithelia from 3 patients (No. 5, 6, 10). The abundance of CFTR band C raised from mean 9.1% (range 8.4 to 10.9) (considering healthy controls as 100% reference value) pretreatment to 52.8% (44.9 to 66.0) and 31.3% (9.1 to 45.0) after 8 and 12 wk of treatment, respectively (B to 6D; Fig. S11). Concomitantly, the protein BECN1 increased from mean 23.0% (range 20.7 to 25.8; 100% equals the value of healthy controls) before treatment to 47.3% (range 39.6 to 56) and 45.0% (range 40.8 to 50.0) after 8 and 12 wk (A and 7B), and SQSTM1 protein decreased from supranormal values before treatment (∼250% of control values) to close-to-normal values after 8 and 12 wk of treatment (A and 7C). These data indicate that the in vivo treatment effectively rescued the expression and function of respiratory epithelial CFTR protein, as it normalized autophagy-related parameters in CF patients.
Figure 6 (See previous page). Effects of treatment on CFTR function and sweat chloride levels in vivo in F508del-CFTR homozygous patients. (A) Assessment of iodide efflux by a fluorescence assay (SPQ) upon stimulation with Fsk plus IBMX. Rate of iodide efflux in freshly isolated brushed nasal epithelial cells collected from all 10 F508del-CFTR homozygous patients before treatment (wk 0) and after 4, 8 and 12 wk of treatment. Values are expressed as percentage of 5 non-CF healthy controls at each time point (wk 0, 4, 8 and 12). Red markers indicate mean values. Dashed line indicates 20% of rescued function. The analysis was performed on at least 50 cells per sample and per experiment at each time point. Mean ± SD of 3 experiments for each sample, #P = 0.0003 and ##P = 0.0002 versus wk 0. (B and C) Effects of treatment on CFTR protein levels in freshly isolated nasal epithelial cells. (B) Mean values of CFTR Band C in patients No. 5, 6 and 10 of . The values are expressed as percentage of non-CF healthy control (considered as 100% of value). Mean ± SD of triplicates of independent experiments per each patient's sample; ##P < 0.01 compared to non-CF healthy control, °°P < 0.01 versus wk 0 (ANOVA). (C) Left Immunoblot detection of CFTR in whole lysates. ACTB was used as loading control. Representative blot of one non-CF control and 1 out of 3 patients with CF (No. 10 of ) analyzed before and after 8 and 12 wk of treatment in vivo. Right, densitometric measurement of CFTR expressed as ratio of Band C /B and as percentage non-CF healthy control normalized to ACTB levels. Mean ± SD of triplicates of independent experiments; ##P < 0.01 compared to non-CF healthy control, °°P < 0.01 versus wk 0 (ANOVA). (C) Assessment of iodide efflux by a fluorescence assay (SPQ) upon stimulation with Fsk plus IBMX in freshly isolated nasal epithelial cells collected from patient No 10 before treatment and after 8 and 12 wk of treatment in vivo.
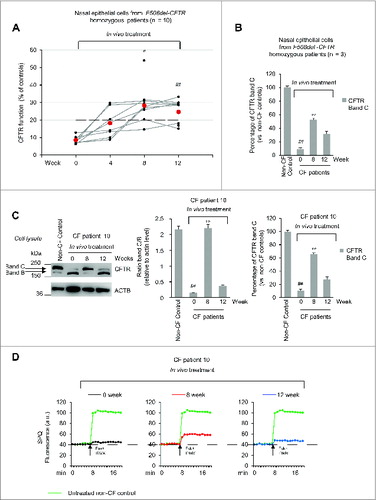
Figure 7. Effects of in vivo treatment on BECN1 and SQSTM1 protein levels in nasal brushing from F508del-CFTR homozygous patients. (A) Effects of treatment on BECN1 (left) and SQSTM1 (tight) protein levels in freshly isolated nasal epithelial cells of patients No. 5, 6 and 10 of . The values are expressed as percentage of non-CF healthy control (considered as 100% of value). Mean ± SD of triplicates of independent experiments per each patient's sample; ##p < 0.01 compared to non-CF healthy control, °P < 0.05 and °°P < 0.01 versus wk 0 (ANOVA). (B and C) Representative blot of 1 patient (No. 10 of ) out of 3 patients analyzed. (B) Western blot analysis of cell lysates and immunoblot with anti-BECN-1 (Abcam); (C) Western blot analysis of insoluble and soluble protein fractions and immunoblot with anti-SQSTM1 (Sigma Aldrich). ACTB was used as negative marker of the insoluble protein fraction and as loading control of the experiment. Bottom, densitometric measurement of (B) BECN-1 and (C) SQSTM1 levels expressed as percentage of non-CF healthy control. Mean ± SD of triplicates of independent experiments; ##P < 0.01 compared to non-CF healthy control, °P < 0.05 and °°P < 0.01 versus wk 0 (ANOVA).
Effects of in vivo treatment on sweat chloride levels
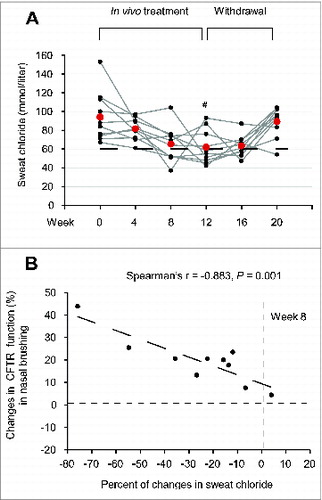
Before treatment (wk 0), all patients exhibited sweat chloride concentrations >60 mMol (mean 94.1, range 67 to 153). As a result of the treatment, sweat chloride levels decreased <60 mMol in 7 patients (No. 1, 2, 5, 6, 7, 8 and 10 of ), who were all under 18 y, corresponding to a decrease by >30% (range 32.3 to 75.8) of the baseline value, and these positive effects lasted beyond the cessation of the treatment. In 2 out of the remaining 3 patients (No. 4 and 9 of ) sweat chloride levels decreased to <60 mmol per liter (54 and 47 mmol per liter, corresponding to ∼36% decrease with respect to the level before treatement) beyond discontinuation of the pharmacological treatment (wk 16 or wk 20) (A; Fig. S12; ). The decrease in sweat chloride was significant after 12 wk of treatment (P = 0.0029, ANOVA) (A; ), and correlated with the amelioration. The decrease in sweat chloride correlated with the amelioration of CFTR function measured in nasal epithelia after 8 wk of treatment (The Spearman r = −0.883, P = 0.001) (B).
Figure 8. Effects of treatment on sweat chloride levels in vivo in F508del-CFTR homozygous patients. (A) Sweat chloride levels (mmol per liter) in all the enrolled CF patients before and after 4, 8 and 12 wk of treatment and after 4 (wk 16) and 8 (wk 20) wk following drug withdrawal. Red markers indicate mean values, #P = 0.0029 vs wk 0. The dashed line indicates 60 mmol/L. (B) Inverse correlation between percent of absolute changes in sweat chloride levels and changes in rate of iodide efflux (expressed as percent of healthy controls) in nasal brushing after 8 wk of treatment in all the enrolled CF patients. The Spearman r = -0.883, P = 0.001.
Effects of in vivo treatment on respiratory inflammation
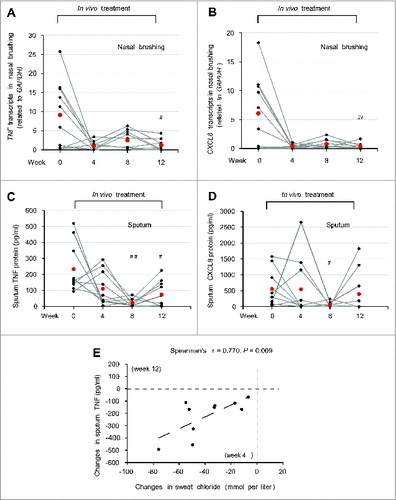
A significant reduction of mRNA levels of TNF and CXCL8 was observed in brushed nasal epithelial cells after 12 wk of treatment (P = 0.026 and P = 0.023 vs baseline values, respectively) (A and 9B). The levels of the inflammatory cytokines in the sputum also significantly decreased during the treatment, as true for both TNF (8 wk: P = 0.0019; 12 wk: P = 0.016) and CXCL8 (8 wk: P = 0.024) (C and 9D). The absolute change from baseline values in TNF protein levels in the sputum after 12 wk of treatment significantly correlated with the decrease of sweat chloride levels early during treatment (The Spearman r = 0.770, P = 0.009) (E). Altogether, these results indicate that all laboratory parameters investigated in this study improved after treatment with cysteamine plus EGCG.
Figure 9. Effects of treatment on respiratory inflammation in vivo in F508del-CFTR homozygous patients. (A and B) TNF and CXCL8 transcript levels in nasal brushing from all the enrolled CF patients before (wk 0) and after 4, 8 and 12 wk of treatment. (A) TNF, ##P = 0.026 and (B) CXCL8, ##P = 0.023 vs wk 0. (C and D) TNF and CXCL8 protein levels (pg/ml) in the sputum before treatment (wk 0) and after 4, 8 and 12 wk of treatment. (C) TNF, ##P = 0.0019, #P = 0.016 as compared to wk 0. (D) CXCL8, #P = 0.024 vs wk 0. (E) Correlation between absolute changes in TNF protein levels (pg/ml) in the sputum after 12 wk of treatment and in sweat chloride levels (mmol/L) after 4 wk of treatment. The Spearman r = 0.770, P = 0.009.
In addition, although the spirometric values (FEV1 and FEF25-75%) failed to reach a significant increase over the observation period, the average changes in FEV1 and FEF25-75% were 5.1% (range -14.3; 35.9) and 3.8% (range -6.6; 36.7), respectively, at the end of treatment (wk 12), and tended to increase (mean 5.4% (range -13.1; 43.0) and 10.9% (range -6.9; 64.4) during the observation period after discontinuation of the treatment (wk 16) (Fig. S13; ).
Discussion
CFTR repairing strategies are an emerging option in CF therapy.Citation54 Rescuing a functional F508del-CFTR mutant at the PM of epithelial cells is the focus of a series of drug discovery programs. This strategy is usually based on a protein-specific approach by means of small molecules (pharmacological chaperones) that would ideally either promote protein folding and correct the mislocalization of the mutant protein (CFTR correctors) or increase the defective CFTR chloride channel function (CFTR potentiators).Citation6,7,55 In addition, an increasing number of small organic molecules can mimic the activity of pharmacological chaperones by modulating the folding machinery or protein stability (chemical chaperones).Citation56 Besides these approaches that target the F508del-CFTR protein, alternative approaches aim at modulating CFTR interacting proteins, such as intracellular chaperones, or generally target the proteostasis network.Citation6 Recently, we have demonstrated that restoring disabled autophagy is a new strategy to circumvent CFTR defect. We discovered that cystamine, a small molecule that acts as a modulator of proteostasis, is able to rescue and stabilize F508del-CFTR at the PM of CFBE41o- airway epithelial cell lines as well as in primary human nasal epithelial cells.Citation27,28,31 In this study we evaluated the therapeutic potential of cysteamine, the reduced form of cystamine, stimulated by the fact that cysteamine is an FDA-approved drug with a known safety profile and good bioavailability upon oral administration.Citation57 In a preclinical animal model, the CftrF508del mouse, we observed that the oral treatment for 5 wk with cysteamine significantly reduced mortality, improved weight gain, and increased the expression of functional CFTR protein at the intestinal level, at the same time that it restored BECN1 protein expression to wild-type levels.
Ideally, compounds used for lifetime therapy should be endowed with the ability to mediate long-term benefits after short-term pulses of treatment to avoid undesirable effects. The effects of cysteamine in reducing respiratory inflammation extend for a short period beyond its withdrawal and rely on the cell's ability to conserve a functional F508del-CFTR at the PM and to restore autophagy, as they are abrogated if autophagy is inhibited by 3-methyladenine or if CFTR function is inhibited by CFTRinh-172 during the washout period.Citation27,31 Therefore, the identification of natural compounds with known safety profile, capable of prolonging the beneficial effects of cysteamine after withdrawal, could be of great value in CF therapy. Here we show that the effects of cysteamine after withdrawal can be further sustained by the addition of a natural compound, the green tea-derived flavonoid epigallocatechin gallate.Citation58 Although not effective on its own in rescuing mutant CFTR protein, EGCG can help conserving F508del-CFTR rescued at the PM when added during the period in which cysteamine was removed from the system. EGCG is a multifunctional compound that can exert beneficial effects on human health by modulating a panel of intracellular pathways.Citation43,45,46 Here we selected EGCG on the basis of its ability to modulate the activity of one particular Ser/Thr kinase, CSNK2, a “master protein kinase” involved in multiple cellular processes.59 CSNK2 activity is reportedly involved in the proteolytic degradation of F508del-CFTRCitation50,60 in a self-supported loop where CSNK2 ultimately promotes fragmentation/degradation of CFTR such that the resulting CFTR fragments allosterically activate CSNK2.Citation61 Thus, targeting CSNK2 could represent an alternative approach to improve F508del-CFTR stability, in accord with the finding that a highly selective inhibitor of CSNK2 mimicked the capacity of EGCG to sustain F508del-CFTR function beyond cysteamine washout.Citation50,60
Notably, the ex vivo treatment of nasal epithelial cells with EGCG during cysteamine washout, stabilized the protein levels of BECN1 and SQSTM1. Disabled autophagy correlating with the sequestration of BECN1 interactome ultimately results in reduced availability of PtdIns3-phosphate at the early endosomes, thereby impairing CFTR recycling at PM and resulting in its degradation.Citation26,27,31 Moreover, accumulating SQSTM1 can bind ubiquitinated CFTR through its ubiquitin-binding domain, thus targeting PM CFTR to lysosomal degradation.Citation27,31 Maintaining appropriate autophagic flux therefore could help conserving a sufficient amount of rescued F508del-CFTR at the PM to interrupt the feed forward loop that ignites CFTR disposal.
We predicted the effects of combined treatment ex vivo in freshly isolated primary nasal epithelial cells collected from the patients enrolled in the study before in vivo therapy. Primary respiratory cell cultures derived from CF patients are considered the gold standard to validate compound efficacyCitation27,62 and might help as an ex vivo test to predict the propensity of each individual patient to benefit from the in vivo administration of the candidate drug. We observed that cysteamine could rescue >70% of the expression of functional CFTR mutant in ex vivo cultures of freshly isolated patients’ nasal epithelial cells. To our knowledge, the combination of 3 different classes of correctors previously restored in vitro mutant CFTR protein level only to 40% to 50% of the control levels observed in healthy subjects bearing wild-type CFTR.Citation63
Encouraged by these preclinical results, we performed a phase II open-label pilot clinical trial in 10 CF patients homozygous for the F508del-CFTR mutant. Our data revealed that the oral administration of the combination therapy was effective in rescuing iodide efflux in nasal epithelia to up to 28% of the wild-type values, correlating with an increase in the expression level of the F508del-CFTR protein. Thus, rescuing >70% of functional F508del-CFTR in vitro in primary respiratory cells collected before in vivo treatment started, might predict a significant (>25%) therapeutic effect in vivo.
The combination therapy was effective in significantly reducing sweat chloride levels, a well-representative surrogate markerCitation64 of CFTR function. Sweat chloride levels fell below 60 mmol per liter in 9 out of 10 patients, either during treatment (which was the case for 7 patients) or shortly thereafter (which was the case in 2 patients). Importantly, the reduction in sweat chloride concentrations correlated with the improvement of iodide efflux measured in nasal epithelial cells.
Our previous preclinical data indicated that cystamine controlled both signs of inflammation and hyper-responsiveness to lipopolysaccharide from Pseudomonas aeruginosa in CF mice through restoration of autophagy.Citation27 Accordingly, it has been reported that restoring autophagy can mediate bacterial clearance and depleting SQSTM1 can improve the delivery of bacteria to the autophagic machinery, ultimately favoring their clearance from CF macrophages.Citation65,66 The anti-inflammatory effects of cysteamine lasted for one wk beyond its removal from the experimental system.Citation27 Notably, these effects were not due to putative intrinsic antiinflammatory properties of cystamine and rather relied on the ability of cystamine to sustain CFTR function, because the functional inhibition of CFTR abrogated the antiinflammatory effects of cystamine during washout.Citation27,31 Here, we show that cysteamine, alone and then combined with EGCG, reduced signs of inflammation, both in lungs, as revealed by the decrease of TNF and CXCL8 in the sputum, and in nasal epithelia. A significant correlation between early reductions in sweat chloride and later (wk 12) improvements in inflammatory parameters in the sputum supports the notion that the antiinflammatory effects of the treatment are secondary to the normalization of CFTR function.
Our data demonstrate that the combination therapy reverted the key pathogenic mechanisms causing mismanaged proteostasis in CF epithelia.Citation26,27,31 Indeed, the in vivo treatment increased BECN1 protein and reduced SQSTM1 levels to the values of non-CF controls. Since restoration of BECN1 and autophagy with reduction of SQSTM1 accumulation are key events in sustaining the residence of CFTR at the PM of respiratory epithelial cells,Citation26,27,31 our data suggest that re-establishment of autophagy could be a predictive marker of efficacy of CFTR-repairing strategies.
Our pilot trial enrolled a small number of patients without the inclusion of placebo-treated control groups. Moreover, our trial was too short to measure possible significant effects on lung function. Given these limitations, these data must be confirmed in larger clinical trials. Prompted by the encouraging results obtained in this first pilot trial, we are now in the process of launching a new long-term trial that will enroll a much larger cohort of patients, as well as adequate control groups.
Materials and Methods
Human subjects
Study design. Ten patients with CF, homozygous for F508del-CFTR mutation, were consecutively enrolled for an open-label phase II pilot study (EudraCT number #2013-001258-82 approved by Local Ethics Committee, Protocol reference #85/13) at the Department of Pediatrics, Regional Cystic Fibrosis Care Center, University of Naples Federico II. The patients and their parents provided written informed consent. Clinical characteristics of patients, study design and inclusion criteria are reported in and .
Exclusion criteria were as follows: i) treatment with glucocorticoids per os or via inhalation at screening or within 4 wk before screening visit; ii) treatment with oxygen or with other experimental drugs; iii) referred hypersensitivity (local or general) to cysteamine or penicillamine prior study; iv) modifications of therapeutic regimens in patients assuming macrolides, antiasthmatic, mucolytic drugs, Dornase alfa, and/or FANS, within 28 d before screening visit; v) organ transplantation; vi) kidney or hepatic alterations at screening visit: vii) pregnancy or nursing at screening visit; viii) refusing to employ contraceptive methods during the study; ix) psychiatric pathologies or neurological diseases.
Spirometry was performed in all CF patients according to American Thoracic Society guidelines.Citation67 Sweat chloride values were obtained by quantitative pilocarpine iontophoresis according to the standardized protocol of Gibson and Cooke.Citation68
Five adult non-CF healthy volunteers underwent nasal brushing at each time at which nasal epithelial cells were collected by nasal brushing from CF patients (wk 0, 4, 8, 12) and used as control for the iodide efflux and biochemical studies in nasal epithelial cells. These subjects provided written informed consent.
Pharmacokinetic and pharmacodynamic analyses
The methodologies of the analyses for plasma cysteamine levels were performed69 as described in Supplemental Materials.
Nasal brushing
Freshly isolated brushed nasal epithelial cells were collected by nasal brushing, as previously described,Citation27 from 10 F508del-CFTR homozygous CF patients at wk 0, 4, 8, 12 and from 5 non-CF healthy volunteers recruited from staff at the same time points. Both patients and healthy volunteers gave written informed consent.
After nostril washing to remove mucus, cytological brushes (Robimpex, MO11157) were used to scrape the mid part of the inferior turbinate from both nasal nostrils. Brushes with cells were immediately transferred into RPMI 1640 medium (Invitrogen) containing 1% penicillin-streptomycin (Lonza, 17-602E), in 15-ml sterilized tubes. The tubes were incubated at 37 °C for 2 h on a thermo shaker, to remove all cell from brushes, the brushes were then removed and the cells centrifuged at 800 x g (2000 rpm) for 20 min. The supernatant fractions were discarded and the cell pellet treated with 150 μL of trypsin-versene (EDTA) solution (Lonza, 17-161) for 4 min at 37 °C to disaggregate possible cell clusters. Trypsin solution was inactivated by adding 3 ml of serum-free Bronchial Epithelial cell Growth Medium BEGM (Clonetics, Lonza, CC3170). After centrifugation at 800 x g (2000 rpm) for 10 min, cells were placed in CELL+ T 25 flasks (Sarstedt Ltd, CS300) with 10 ml of BEGM medium. Nonspecific epithelial cells were removed during the daily cell washing and medium changes. Counterstaining with anti-KRT18/cytokeratin-18 (CK-18; Abcam, ab52948) antibody was used to confirm epithelial cell purity, and with anti-CD3+, (Abcam, ab5690), CD4+ (Abcam, ab51312) or CD19+ (Abcam, ab25232) 1:500 antibodies to exclude the presence of limphocytes and inflammatory cells. At 60% of confluence the cells were passed in new flasks and Trypan blue staining exclusion test was used to establish total viable cell number. Cells were also stained with anti-Pan-cytokeratin (Sigma Aldrich, C5992) 1:500 and MUC5AC (Abcam, ab3649) or MUC3B (Abcam, ab85006) 1:200 antibodies to exclude mucipar differention. The analysis of freshly isolated nasal cells was performed after 48 h of seeding to ensure the depletion of nonspecific debris and minimal contamination from other mixed cell populations.
In vivo analysis
Samples were assessed for iodide efflux, cytokine expression, and biochemical analysis before and after treatments at each point through the study.
Ex vivo analysis
Nasal epithelial cells collected at the baseline time point (wk 0) were also cultured ex vivo until 60% confluence and then split. The cells (maximum 3 passages) were treated with cysteamine (250 μM; Sigma Aldrich, M9768) for 18 h and finally kept untreated until 48 h in presence of medium alone or of medium added with EGCG (80 μM; Sigma Aldrich, E4143) or with CX-4945 (5 μM; SYNthesis Med Chem, SYN-1109).
Cell lines and treatments
CFBE41o-cells (F508del/F508del-CFTR) (kindly provided by DC Gruenert) were grown in Transwells (Corning, 3470 or 3460, to seed 8 × 104 or 3 × 105 cells, respectively), and transfected with F508del-CFTR at 37 °C.
CFBE41o-cells were cultured in Minimum Essential Medium Earle's salt (200 mM L-glutamine, 10% fetal bovine serum, and penicillin/streptomycin). CFBE41o- cells were grown in Transwells under the normal condition. Briefly, 8 × 104 or 3 × 105 cells were seeded in 6.5-mm diameter or 12-mm diameter collagen-coated Transwells, respectively, and grown until the RT reached 800 to 1,000 Ω・cm2. Transwells with a pore size of 0.4 μm were used. Medium in both the apical and basolateral chambers was changed every other day. Monolayers were immunostained for OCLN/occludin (Abcam, ab31721) to confirm polarization.Citation27
Cells were incubated with cysteamine (250 μM) for 18 h and then kept for the following 48 h with medium alone or medium added with either EGCG (80 μM) or CX-4945 (5 μM) in the presence or absence of CFTRinh-172 (20 μM; Sigma Aldrich, C2992). Cycloheximide (CHX, 100 μg ml−1; Sigma Aldrich, C4859) was added during the last 8 h of incubation to inhibit protein neosynthesis, as previously described.Citation27,31 The MTT (5 mg ml−1; Sigma Alrich, M5665) assay was performed to verify the CHX toxicity, as previously described.Citation27
Plasmids and transfection
The pcDNA3.1ΔF508-CFTR expression vector (PRIMM, Milan), was used for transfection experiments in CFBE41o- cell lines, as described.Citation27 Cells were transfected with pcDNA3.1 ΔF508-CFTR or expression vectors, by Lipofectamine 2000 (Invitrogen, 11668-027) according to the manufacturer's instructions.
Mice and treatments
CF mice homozygous for the F508del-Cftr in the 129/FVB outbred background (Cftrtm1EUR, F508del, FVB/129, abbreviated CftrF508del) were obtained from Bob Scholte, Erasmus Medical Center Rotterdam, The Netherlands (CF-coordinated action program EU FP6 LSHM-CT-2005-018932). These studies and procedures were approved by the local Ethics Committee for Animal Welfare (IACUC N° 553 and 582) and conformed to the European Community regulations for animal use in research (2010/63 UE). Young adult heterozygous mice were housed in static isolator cages at the animal care specific pathogen-free facility of Charles River (Calco, Italy). The newborn mice aged 15 d old (n = 297 mice) were administered via gavage with cysteamine (60 μg/kg in 100 μl saline/day) or 100 μl saline/d for 5 consecutive d/wk for 5 wk. Mice were weighed on the first and last d of the treatment.
Seventy adult (10- to 12-wk-old) CftrF508del mice were orally administered with vehicle alone (n = 35) or cysteamine (n = 35) (7 d, via gavage, 60 μg/kg in 100 μl saline/d). Thirty of the CftrF508del mice cysteamine-treated group of mice were subsequently randomized as following. Mice (n = 5 per group) were administered via gavage with vehicle or EGCG (150 μg/kg in 100 μl saline/day) for 10, 20, or 30 d after the cessation of cysteamine treatment.
At the end of the treatment, mice were anesthetized with Avertine (tribromoethanol, 250 mg/kg; Sigma Aldrich, T48402) and a segment of tail was collected for genotyping. Mice were then sacrificed and lungs and intestines collected for analysis.
Immunoblot analysis
The proteins were obtained from both treated and untreated nasal epithelial cell and CFBE41o- cell lines as well as from either mouse intestinal or lung homogenates and the amounts of proteins were determined by a Bio-Rad protein assay to ensure equal protein loading before immunoblot analysis. For freshly isolated cell, after 48 h seeding, a large amount (120 μg) of protein was loaded to detect CFTR protein. An amount of 70 μg of protein from ex-vivo nasal epithelial cells, CFBE41o- cells and samples from mouse tissue homogenates were loaded in each lane. Western blot analysis was performed with antibodies against the following proteins: SQSTM1, (Sigma Aldrich, 108k4767) 1:1000, BECN1 (Abcam, ab58878) 1:1000, CFTR clone CF3 (Abcam, ab2784) 1:1000, CFTR clone M3A7 (Abcam, ab4067) 1:500, CFTR clone H-182 (Santa Cruz Biotechnology, sc-10747) 1:500, FLOT1/Flotillin-1 clone C-2 (Santa Cruz Biotechnology, sc-74576) 1:1000, ACTB/β-actin (Cell Signaling Technology, 4970) 1:1000, TUBA/α-β tubulin (Cell Signaling Technology, 2148) 1:1000. The densitometric analysis was performed by ImageJ software and each data point was expressed as the mean ± SD of triplicate of independent experiments.
Soluble and insoluble protein fractions
Cells were lysed in buffer containg 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Nonidet P40 (Sigma Aldrich, NP405), 5 mM EDTA, 1 mM phenylmethylsulphonyl fluoride (Sigma Aldrich, P7626), 50 mM NaF (Sigma Aldrich, S1504), 10 μg ml−1 leupeptin (Sigma Aldrich, L5793) and 10 μg ml−1 aprotinin (Sigma Aldrich, A3428) supplemented with protease inhibitors (Sigma Aldrich, P8340) and centrifuged at 9,000 x g at 4 °C for 20 min. After centrifugation the soluble (supernatant) and insoluble (pellet) fractions were used in western blot analysis with anti-SQSTM1 antibody. The pellet insoluble in Nonidet P40 was dissolved 5 times in sample buffer, boiled at 95 °C for 5 min and resolved on a polyacrylamide gel. The densitometric analysis was performed by ImageJ software and each data point was expressed as the mean ± SD of triplicate of independent experiments.
Cell surface biotinylation assay and membrane fractionation
Cell-surface proteins were biotinylated and the plasma membrane fraction was collected as previously described.Citation26, 27 Cell-surface proteins were biotinylated using sulfosuccinimidyl-6-(biotinamido) hexanoate (sulfo-NHS-LC-Biotin; Pierce, 21335, dissolved at 1 mg ml−1 in PBS (GIBCO, 18912-014, pH 8.2), as described.Citation27 Cells were homogenized with a Potter-Elvehjem pestle and centrifuged at 2300 x g for 15 min at 4 °C. Supernatant fractions that contain the cytoplasmic and plasma membrane fractions were centrifuged 1 h at 16,000 x g at 4 °C; the pellet was the intact membrane and was solubilized in Buffer A (20 mM Tris-HCl, pH 7.4, 2 mM EDTA, 20 mM 2-mercaptoethanol, 1X PMSF, 1 μg ml−1 inhibitor protease cocktail (Sigma Aldrich, P8340) +1% Triton X-100 (Sigma Aldrich, X-100-RS) and centrifuged 1 h at 60,000 x g in the ultracentrifuge. The supernatant fractions were collected as plasma membrane fraction. Equivalent amounts of protein (500 μg) were used for streptavidin-agarose affinity isolation (Pierce, 20349). Biotinylated proteins of plasma membrane fraction were immunoblotted against CFTR (clone CF-3, or M3A7) or FLOT1. The densitometric analysis was performed by ImageJ software and each data point was expressed as the mean ± SD of triplicate of independent experiments.
Measurement of rectal potential difference
RPD was measured with a protocol similar to that for nasal potential difference measurements in humans, as described.Citation39,70 Mice were anesthetized with tribromoethanol (250 mg/kg). RPD was sensed with a digital volthometer inserted for about 2 cm in the rectum. Potentials were measured with respect to a subcutaneous 1 M NaCl-filled needle. A second rectal tube was used for continouous perfusion of drug adiministration into the rectum. The Cl− containing solution had the following composition: 145 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES (Sigma Aldrich, H3375), 0.1 mM amiloride (Sigma Aldrich, 1019701), at a final pH 7.4. The Cl− free solution had the following composition: 145 mM sodium gluconate (Sigma Aldrich, 52054), 4 mM potassium gluconate (Sigma Aldrich, 1550001), 4 mM calcium gluconate (Sigma Aldrich, C0300000), 1 mM magnesium gluconate (Sigma Aldrich, G9130), 10 mM HEPES, 0.1 mM amiloride, at a final pH 7.4.
Assessment of CFTR function in freshly isolated nasal epithelial cells from F508del-CFTR homozygous patients
Freshly isolated nasal cell were allowed to seed for 48 h and then analyzed before and after ex vivo culture. The analysis of iodide efflux was performed in nasal cells, by the iodide-sensitive fluorescent indicator, SPQ (Molecular Probes/Invitrogen, M440), as previously described.Citation27
Iodide efflux
The iodide-sensitive fluorescent indicator SPQ (Molecular Probes/Invitrogen, M440) was introduced into the cells in a hypotonic solution of iodide buffer (in mM: 130 NaI [Sigma Aldrich, 229911], 4 KNO3 (Sigma Aldrich, P6030), 1 Ca(NO3)2 (Sigma Aldrich, 31218), 1 Mg(NO3)2 (Sigma Aldrich, 6384), 10 glucose and 20 HEPES, pH 7.4) diluted 1:1 with water and containing a final concentration of 10 μM SPQ. Cells were loaded for 20 min at 37 °C in a humidified chamber with 5% CO2. The SPQ-loaded cells were then mounted on a LSM510 Meta confocal microscope (Zeiss, Milan, Italy) with a 37 °C heated stage and perfused with iodide buffer for 5 to 8 min. Changes in CFTR-mediated SPQ fluorescence were monitored at the 445 nm wavelength in response to excitation at 340 nm during perfusion at 37 °C in nitrate buffer replaced with 130 mM NaNO3 (Sigma Aldrich, S8170) with 20 μM forskolin (Fsk; Sigma Aldrich, F6886) plus 100 μM IBMX (Sigma Aldrich, I5879) and fluorescence intensity measured for a further 10 to 12 min. Signals were collected at 30-sec interval. For each minute the average of the fluorescence intensity was measured from 50 cells for population per coverslip and the peak of iodide efflux rate (usually after Fsk plus IBMX adding) of cells was calculated in accordance with the Stern-Volmer relationship as follows:where F is the observed fluorescence, Fo is the fluorescence in the absence of a quenching anion, CQ is the concentration of the quenching anion, and K is the Stern-Volmer quench constant. The rates were calculated using SigmaPlot Version 7.1 for each mean fluorescence trace for each time point generated from the 50 cells examined per population per coverslip. Citation71-75
Real-time and reverse-transcription PCR analysis
The analysis was performed as previously described.Citation29,76 Total RNA was extracted with the RNeasy Mini Kit (Qiagen, 74104) from mouse lung homogenates and from nasal brushing of CF patients.
Mouse: The mRNA was reverse transcribed with a SuperScriptTM III First Strand Synthesis System (Promega, A5001). Quantitative RT-PCR was performed with an iCycler iQ Multicolor Real-Time PCR Detector (Bio-Rad, Milan, Italy) with iQ TM SYBR Green supermix (Five Prime, 2900217). The relative amounts of mRNA were calculated by using the comparative Ct method. Real-time RT-PCR analyses were executed for evaluating the efficiency of expression. Thermocycling consisted of an initial polymerase activation step at 98°C for 5 min, and amplification was performed with 35 cycles of 95°C for 15 sec, 68°C for 10 sec and 72°C for 20 sec with data acquisition at this stage and the reaction finished by the built in melt curve.
The sequences of mouse primers were: Tnf forward 5′-CCACCACGCT CTTCTGTCTA-3′ and reverse 5′-AGGGTCTGGG CCATAGAACT-3′;
Cxcl2 forward 5′-GCTGGCCACC AACCACC-3′ and reverse 5′AGCGAGGCAC ATCAG GTACG-3′; Gapdh forward 5′-GTGATGCTGG GTG-3′ and reverse 5′-CAGTCTTCTG AG-3′. Expression levels of genes were normalized to Gapdh levels in the same sample.
Human: The mRNA was reverse transcribed with a SuperScriptTM III First Strand Synthesis System. Quantitative RT-PCR was performed with an iCycler iQ Multicolor Real-Time PCR Detector (Bio-Rad) with iQ TM SYBR Green supermix. Thermocycling consisted of an initial polymerase activation step at 95°C for 5 min, amplification was performed with 40 cycles of 95°C for 15 sec, 60°C for 10 sec and 72°C for 20 sec with data acquisition at this stage and the reaction finished by the built in melt curve.
The sequences of human primers were: TNF forward 5′-CGAGTCTGGG CAGGTCTACT TT-3′ and reverse 5′-AGAGGTTGAG GGTGTCTGAAG G-3′; CXCL8 forward 5′-ATG ACTTCCAAGC TGGCCGTGGC T-3′ and reverse 5′-TCTCAGCCCT CTTCAAAAAC TTCT-3′; for ACTB forward 5′-CCGATCCACA CGGAGTACTT G-3′ and reverse: 5′-GGCACCCAGC ACAATGAAG-3′. Expression levels of genes were normalized to ACTB levels in the same sample.
ELISA
Sputum samples were diluted 1:2 with PBS and digested with 1 U/μl of DNAse (Sigma Aldrich, D4263) for 4 h at 37°C. The supernatant fractions were collected and stored at -80 ºC until usage. The samples were then centrifuged at 800 x g for 10 min and the supernatants used to measure TNF and CXCL8 levels by ELISA. ELISA analysis was performed using standard ELISA kits (R&D Systems, STA00C for TNF and S8000C for CXCL8), according to the manufacturer's instructions, as previously reported.Citation26,Citation77 Samples were read in triplicate at 450 nm in a Microplate Reader (BioRad, Milan, Italy) using Microplate Manager 5.2.1 software. Values were normalized to protein concentration evaluated by Bradford analysis.
Inhibition of protein kinase CSNK2
Recombinant CSNK2, either CSNK2A, the α subunit or the CSNK2A2-CSNK2B2 (α2β2) holoenzyme, was obtained as described,Citation50 and incubated for 10 min at 37°C in a final volume of 25 μl containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 12 mM MgCl2, 100 μM synthetic peptide substrate RRRADDSDDDDD (in house synthesized) and 20 μM [33P-ATP] (1000 to 2000 cpm/pmol). The reaction was stopped by ice cooling and absorption on phospho-cellulose p81 paper (Whatman, 369-8915). Papers were washed 3 times with 75 mM phosphoric acid, dried, and counted in a scintillation counter. IC50 values represent the mean of 3 independent experiments with the SD not exceeding 10%.
In situ detection of TGM2 enzyme activity
TGM2 activity in nasal epithelial cells was detected as previously reported.Citation26,30 Detailed methods are reported in Supplemental Materials.
Statistical analysis
Categorical variables are presented as number (%) and continuous variables as mean (SD) when normally distributed or median (IQR) when not.
Mice. All laboratory tests on mice were performed at least in triplicate.
Survival analysis was performed using the Kaplan-Meier method to evaluate the effect of cysteamine on mouse survival. Log-rank test was used to check statistical significance of differences in survival.
Between-group comparisons were evaluated by the unpaired Student t test or one-way analysis of variance (ANOVA), and post hoc comparisons were made using Tukey-Kramer test, when appropriate. We set the level of significance at P < 0.05. Analyses were carried out with SPSS 12 software.
Humans. The effects of treatment in CF patients were analyzed by repeated measures ANOVA to compare the means of the laboratory and clinical variables measured at different times of treatment and during the follow-up period: wk 8 vs baseline (wk 0), wk 12 vs wk 0 for each variable; wk 16 vs wk 12 for sweat test, FEV1% and FEF25-75%; wk 20 vs wk 12 and wk 20 vs wk 0 only for sweat test.
According to Šidák correction for multiple comparisons,Citation78 each comparison was considered significant if P values (for the 2-tailed test) were less than , were α is the overall type I error probability and n is the number of comparisons. Therefore, with an overall α = 0.05 and n = 5, the significance threshold for each comparison is 0.01; when n = 3, the threshold for each comparison is 0.017 and when n = 2, the threshold for each comparison is 0.025; with an overall α = 0.01 the thresholds for each comparison are 0.002, 0.003, 0.005 respectively.
All data processing and analyses were carried out with SAS statistical software (version 9.2; SAS Institute, Cary, NC, USA).
In vitro experiments
Data are analyzed as previously reported.Citation31 Data distribution was analyzed for normality and statistical analysis performed using the one-way ANOVA. Significant differences are indicated in the figures. All data were obtained from independent measurements. Data manipulations were carried out with SPSS 12 software.
Conflict of Interest
V.R., L.M. and G.K. are listed as inventors on a patent application describing the use of cysteamine for the treatment of CF.
Supplemental_DOC.doc
Download MS Word (46 KB)Supplemental_Figs.pdf
Download PDF (176.5 KB)Acknowledgments
We thank Bob Scholte who provided Cftrtm1eur (F508del (FVB/129) mice (European Economic Community European Coordination Action for Research in Cystic Fibrosis program EU FP6 SHM-CT-2005-018932), Dieter C. Gruenert (California Pacific Medical Center Research Institute, San Francisco, CA) who provided CFBE41o- and 16HBE14o- cell lines, and Romina Monzani and Ilenia Sana, European Institute for Research in Cystic Fibrosis, and Sara Lusa, Department of Pharmacy, Federico II University of Naples for technical assistance.
Funding
This study was supported by the European Institute for Research in Cystic Fibrosis (IERFC), Italian Cystic Fibrosis Association (LIFC) and Regional Cystic Fibrosis Associations of Sicilia, Lazio, Lucania, Friuli-Venezia-Giulia (L.M., V.R., V.R.V, D.D.S, S.E), Cancéropôle Ile-de-France, Ligue Nationale contre le Cancer (équipe labellisée), Agence Nationale pour la Recherche, Association pour la Recherche sur le Cancer, European Research Council (Advanced Investigator Award), Fondation Bettencourt-Schueller, Fondation pour la Recherche Médicale, Institut National du Cancer, and the LabEx Onco-Immunology (G.K.), the Wellcome Trust, grant numbers 088929 (L.A.P. and A. M.) and 069150 (A.M.).
References
- Riordan JR, Rommens JM, Kerem B-s, Alon N, Rozmahel R, Grzelczak Z,, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245:1066-73.
- Riordan JR, Chang X-B. CFTR, a channel with the structure of a transporter. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1992; 1101:221-2.
- Ratjen F, Döring G. Cystic fibrosis. Lancet 2003; 361:681-9.
- De Boeck K, Derichs N, Fajac I, de Jonge HR, Bronsveld I, Sermet I, Vermeulen F, Sheppard DN, Cuppens H, Hug M, et al. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cystic Fibrosis 2011; 10:S53-S66.
- Kerem B, Kerem E. The molecular basis for disease variability in cystic fibrosis. Eur J Hum Genet 1996; 4:65.
- D Amaral M, M Farinha C. Rescuing Mutant CFTR: A Multi-task Approach to a Better Outcome in Treating Cystic Fibrosis. Curr Pharm Des. 2013; 19:3497-508.
- Amaral MD, Kunzelmann K. Molecular targeting of CFTR as a therapeutic approach to cystic fibrosis. Trends Pharmacol Sci 2007; 28:334-41.
- Harrison MJ, Murphy DM, Plant BJ. Ivacaftor in a G551D Homozygote with Cystic Fibrosis. N Engl J Med. 2013; 369:1280-2.
- Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem 2008; 77:701-26.
- Farinha CM, Matos P, Amaral MD. Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi. FEBS J 2013; 280:4396-406.
- Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010; 329:805-10.
- Lukacs G, Chang X-B, Bear C, Kartner N, Mohamed A, Riordan J, Grinstein S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem 1993; 268:21592-8.
- Lukacs GL, Verkman AS. CFTR: folding, misfolding and correcting the DeltaF508 conformational defect. Trends Mol Med 2012; 18:81-91.
- Ramalho AS, Beck S, Meyer M, Penque D, Cutting GR, Amaral MD. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am J Respir Cell Mol Biol 2002; 27:619-27.
- Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pulm Med. 2004; 10:547-52.
- Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, Samuel MS, Reznikov LR, Rector MV, Gansemer ND, et al. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clinic investigation 2013; 123:2685.
- Clancy J, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012; 67:12-8.
- Van Goor F, Straley KS, Cao D, González J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006; 290:L1117-30.
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011; 108:18843-8.
- Boyle MP BS, Konstan M, McColley SA, Kang L, Patel N. The investigational CFTR corrector, VX-809 (lumacaftor) co-administered with the oral potentiator ivacaftor improved CFTR and lung function in F508del homozygous patients: Phase II study results [Abstract 260]. Pediatr Pulmonol Suppl. 2012;47(S35): 315., 2012:315.
- S. Donaldson JP, M. Griese, Q. Dong, P-S. Lee, for the VXII-661-101 Study Group. VX-661, an investigational CFTR corrector, in combination with ivacaftor, a CFTR potentiator, in patients with CF and homozygous for the F508del CFTR mutation: interim analysis;. Abstract from the 36th European Cystic Fibrosis Congress; June 12-15, 2013; Lisbon, Portugal, 2013.
- http://investors.vrtx.com/ReleaseID=757597. Treatment with VX-661 and Ivacaftor in a Phase 2 Study Resulted in Statistically Significant Improvements in Lung Function in People with Cystic Fibrosis Who Have Two Copies of the F508del Mutation. 2013.
- http://investors.vrtx.com/ReleaseID=583683. Interim Phase 2 Data Showed a Combination of VX-770 and VX-809 Improved Function of the Defective Protein that Causes Cystic Fibrosis in People With the Most Common Form of the Disease. 2011.
- Hutt D, Balch WE. Cell Biology. The proteome in balance. Science 2010; 329:766-7.
- Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol 2010; 6:25-33.
- Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 2010; 12:863-75.
- Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, Guido S, Pettoello-Mantovani M, Carnuccio R, Scholte B, et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012; 8: 1657-72.
- Villella VR, Esposito S, Bruscia EM, Maiuri MC, Raia V, Kroemer G, Maiuri L.. Targeting the Intracellular Environment in Cystic Fibrosis: Restoring Autophagy as a Novel Strategy to Circumvent the CFTR Defect. Front Pharmacol 2013; 4:1-8.
- Maiuri L, Luciani A, Giardino I, Raia V, Villella VR, D’Apolito M, Pettoello-Mantovani M, Guido S, Ciacci C, Cimmino M, et al. Tissue transglutaminase activation modulates inflammation in cystic fibrosis via PPARgamma down-regulation. J Immunol 2008; 180:7697-705.
- Luciani A, Villella VR, Vasaturo A, Giardino I, Raia V, Pettoello-Mantovani M, D’Apolito M, Guido S, Leal T, Quaratino S, Maiuri L. SUMOylation of tissue transglutaminase as link between oxidative stress and inflammation. J Immunol 2009; 183:2775-84.
- Villella VR, Esposito S, Bruscia EM, Vicinanza M, Cenci S, Guido S, Pettoello-Mantovani M, Carnuccio R, De Matteis MA, Luini A, et al. Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ 2013; 20:1101-15.
- Gahl WA. Early oral cysteamine therapy for nephropathic cystinosis. Eur J Pediatr 2003; 162:S38-S41.
- Dohil R, Meyer L, Schmeltzer S, Cabrera BL, Lavine JE, Phillips SA. The effect of cysteamine bitartrate on adiponectin multimerization in non-alcoholic fatty liver disease and healthy subjects. J Pediatr 2012; 161:639-45.e1.
- Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science 1992; 257:1083-8.
- O’Neal WK, Hasty P, McCray PB, Casey B, RiveraPérez J, Welsh MJ, Beaudet AL, Bradley A. A severe phenotype in mice with a duplication of exon 3 in the cystic fibrosis locus. Hum Mol Genet 1993; 2:1561-9.
- Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, Colledge WH. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat Genet 1993; 4:35-41.
- van Doorninck JH, French PJ, Verbeek E, Peters R, Morreau H, Bijman J, Scholte BJ. A mouse model for the cystic fibrosis delta F508 mutation. EMBO J 1995; 14:4403.
- Legssyer R, Huaux F, Lebacq J, Delos M, Marbaix E, Lebecque P, Lison D, Scholte BJ, Wallemacq P, Leal T. Azithromycin reduces spontaneous and induced inflammation in Delta F508 cystic fibrosis mice. Respir Res 2006; 7.
- Illek B, Fischer H. Flavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivo. American Journal of Physiology-Lung Cellular and Molecular Physiology 1998; 275:L902-L10.
- Villella VR, Esposito S, Maiuri MC, Raia V, Kroemer G, Maiuri L. Towards a rational combination therapy of cystic fibrosis: How cystamine restores the stability of mutant CFTR. Autophagy 2013; 9.
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs Jr DR, Lee D-H,, Shioda T, Soto AM, vom Saal FS, Welshons WV, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev 2012; 33:378-455.
- Patisaul HB. Effects of Environmental Endocrine Disruptors and Phytoestrogens on the Kisspeptin System. Kisspeptin Signaling in Reproductive Biology: Springer, 2013:455-79.
- Renouf M, Marmet C, Guy PA, Beaumont M, Lepage M, Williamson G, Dionisi F. Dose-response plasma appearance of green tea catechins in adults. Mol Nutr Food Res. 2013: 57:833-9.
- Kerem Eithan HMO. Efficacy and Safety Study of EGCG/Tocotrienol in 18 Patients With Splicing-mutation-mediated Cystic Fibrosis (CF). ClinicalTrials.gov:NCT00889434, Last updated: November 11, 2012.
- Kim HS, Quon MJ, Kim JA. New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol. 2014; 2:187-195.
- Huo C, Wan S, Lam W, Li L, Wang Z, Landis-Piwowar KR, Chen D, Dou QP, Chan TH.. The challenge of developing green tea polyphenols as therapeutic agents. Inflammopharmacology 2008; 16:248-52.
- Yang H, Landis-Piwowar K, Chan TH, Dou QP. Green tea polyphenols as proteasome inhibitors: implication in chemoprevention. Curr Cancer Drug Targets. 2011;11:296-306.
- Van Aller GS, Carson JD, Tang W, Peng H, Zhao L, Copeland RA, Tummino PJ, Luo L. Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res Commun 2011; 406:194-9.
- Lolli G, Cozza G, Mazzorana M, Tibaldi E, Cesaro L, Donella-Deana A, Meggio F, Venerando A, Franchin C, Sarno S, et al. Inhibition of protein kinase CK2 by flavonoids and tyrphostins. A structural insight. Biochemistry 2012; 51:6097-107.
- Venerando A, Franchin C, Cant N, Cozza G, Pagano MA, Tosoni K, Al-Zahrani A, Arrigoni G, Ford RC, Mehta A, et al. Detection of phospho-sites generated by protein kinase CK2 in CFTR: mechanistic aspects of Thr1471 phosphorylation. PloS one 2013; 8:e74232.
- Cozza G, Pinna LA, Moro S. Kinase CK2 inhibition: an update. Curr Med Chem 2013; 20:671-93.
- Pharmaceuticals Cylene. Study of CX-4945 in Patients With Relapsed or Refractory Multiple Myeloma. ClinicalTrials.gov :NCT01199718, Last updated: June 13, 2011.
- Pharmaceuticals Cylene. Dose-escalation Study of Oral CX-4945. ClinicalTrials.gov Identifier:NCT00891280, Last updated: June 13, 2011.
- Amin R, Ratjen F. Emerging drugs for cystic fibrosis. Expert Opin Emerg Drugs 2014.
- Amaral MD. Targeting CFTR: how to treat cystic fibrosis by CFTR-repairing therapies. Curr Drug Targets 2011:683-93.
- Welch WJ, Brown CR. Influence of molecular and chemical chaperones on protein folding. Cell Stress Chaperones 1996; 1:109-15.
- Bouazza N, Tréluyer JM, Ottolenghi C, Urien S, Deschenes G, Ricquier D, Niaudet P, Chadefaux-Vekemans B. Population pharmacokinetics and pharmacodynamics of cysteamine in nephropathic cystinosis patients. Orphanet J Rare Dis 2011; 6:86.
- Hendrich AB. Flavonoid-membrane interactions: possible consequences for biological effects of some polyphenolic compounds. Acta Pharmacol Sin 2006; 27:27-40.
- Venerando A, Ruzzene M, Pinna LA. Casein kinase: the triple meaning of a misnomer. Biochem J. 2014; 460:141-56.
- Cesaro L, Marin O, Venerando A, Donella-Deana A, Pinna LA. Phosphorylation of cystic fibrosis transmembrane conductance regulator (CFTR) serine-511 by the combined action of tyrosine kinases and CK2: the implication of tyrosine-512 and phenylalanine-508. Amino Acids 2013; 45:1423-9.
- Venerando A, Cesaro L, Marin O, Donella-Deana A, Pinna LA. A “SYDE” effect of hierarchical phosphorylation: possible relevance to the cystic fibrosis basic defect. Cell Mol Life Sci. 2014;71:2193-6.
- Masvidal L, Igreja S, Ramos MD, Alvarez A, de Gracia J, Ramalho A, Amaral MD, Larriba S, Casals T. Assessing the residual CFTR gene expression in human nasal epithelium cells bearing CFTR splicing mutations causing cystic fibrosis. Eur J Hum Genet 2013.
- Wang Y, Loo T, Bartlett M, Clarke D. Additive effect of multiple pharmacological chaperones on maturation of CFTR processing mutants. Biochem J 2007; 406:257-63.
- Accurso FJ, Van Goor F, Zha J, Stone AJ, Dong Q, Ordonez CL, Rowe SM, Clancy JP, Konstan MW, Hoch HE, et al. Sweat chloride as a biomarker of CFTR activity: proof of concept and ivacaftor clinical trial data. J Cyst Fibros. 2014; 13:139-47.
- Abdulrahman BA, Khwk AA, Akhter A, Caution K, Tazi M, Hassan H, Zhang Y, Rowland PD, Malhotra S, Aeffner F, et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/p62 from macrophages harboring cftr ΔF508 mutation improves the delivery of Burkholderia cenocepacia to the autophagic machinery. J Biol Chem. 2013; 288:2049-58
- Assani K, Tazi MF, Amer AO, Kopp BT. IFN-γ Stimulates Autophagy-Mediated Clearance of Burkholderia cenocepacia in Human Cystic Fibrosis Macrophages. PLoS One. 2014; 9:e96681.
- Standardization of Spirometry, 1994 Update. American Thoracic Society. Am J Respir Crit Care Med 1995; 152:1107-36.
- Gibson LE, Cooke RE. A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 1959; 23:545-9.
- Perna AF, Castaldo P, De Santo NG, di Carlo E, Cimmino A, Galletti P, Zappia V, Ingrosso D. Plasma proteins containing damaged L-isoaspartyl residues are increased in uremia: implications for mechanism. Kidney Int 2001; 59:2299-308.
- Fischer H, Fukuda N, Barbry P, Illek B, Sartori C, Matthay MA. Partial restoration of defective chloride conductance in ΔF508 CF mice by trimethylamine oxide. American Journal of Physiology-Lung Cellular and Molecular Physiology 2001; 281:L52-L7.
- Verkman AS, Galietta LJV. Chloride channels as drug targets. Nature Reviews Drug Discovery 2009; 8:153-71.
- Jayaraman S, Teitler L, Skalski B, Verkman A. Long-wavelength iodide-sensitive fluorescent indicators for measurement of functional CFTR expression in cells. American Journal of Physiology-Cell Physiology 1999; 277:C1008-C18.
- Munkonge F, Alton EW, Andersson C, Davidson H, Dragomir A, Edelman A, Farley R, Hjelte L, McLachlan G, Stern M, et al. Measurement of halide efflux from cultured and primary airway epithelial cells using fluorescence indicators. Journal of Cystic Fibrosis 2004; 3:171-6.
- Stern M, Munkonge F, Caplen N, Sorgi F, Huang L, Geddes D, Huang L, Geddes DM, Alton EW. Quantitative fluorescence measurements of chloride secretion in native airway epithelium from CF and non-CF subjects. Gene therapy 1995; 2:766-74.
- Mansoura MK, Biwersi J, Ashlock MA, Verkman A. Fluorescent chloride indicators to assess the efficacy of CFTR cDNA delivery. Human gene therapy 1999; 10:861-75.
- Amano H, Yamamoto H, Senba M, Oishi K, Suzuki S, Fukushima K, Mukaida N, Matsushima K, Eguchi K, Nagatake T. Impairment of endotoxin-induced macrophage inflammatory protein 2 gene expression in alveolar macrophages in streptozotocin-induced diabetes in mice. Infect Immun 2000; 68:2925-9.
- Paine R, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, Thannickal VJ, Burnham EL, Brown MB, Hyzy RC. A randomized trial of recombinant human granulocyte-macrophage colony stimulating factor for patients with acute lung injury. Crit Care Med 2012; 40:90-7.
- Abdi H. Bonferroni and Šidák corrections for multiple comparisons. In NJ Salkind (ed.). Encyclopedia of Measurement and Statistics. Thousand Oaks, CA: Sage. 2007.