
Abstract
Autophagy is an evolutionarily conserved biological process involved in an array of physiological and pathological events. Without proper control, autophagy contributes to various disorders, including cancer and autoimmune and inflammatory diseases. It is therefore of vital importance that autophagy is under careful balance. Thus, additional regulators undoubtedly deepen our understanding of the working network, and provide potential therapeutic targets for disorders. In this study, we found that RNF216 (ring finger protein 216), an E3 ubiquitin ligase, strongly inhibits autophagy in macrophages. Further exploration demonstrates that RNF216 interacts with BECN1, a key regulator in autophagy, and leads to ubiquitination of BECN1, thereby contributing to BECN1 degradation. RNF216 was involved in the ubiquitination of lysine 48 of BECN1 through direct interaction with the triad (2 RING fingers and a DRIL [double RING finger linked]) domain. We further showed that inhibition of autophagy through overexpression of RNF216 in alveolar macrophages promotes Listeria monocytogenes growth and distribution, while knockdown of RNF216 significantly inhibited these outcomes. These effects were confirmed in a mouse model of L. monocytogenes infection, suggesting that manipulating RNF216 expression could be a therapeutic approach. Thus, our study identifies a novel negative regulator of autophagy and suggests that RNF216 may be a target for treatment of inflammatory diseases.
Abbreviations:
- Atg, autophagy-related
- BALF, bronchoalveolar lavage fluid
- BMDM, bone marrow-derived macrophage
- CFU, colony-forming unit
- GFP, green fluorescent protein
- HRP, horseradish peroxidase
- i.t., intratracheally
- LPS, lipopolysaccharide
- MAP1LC3A, microtubule-associated protein 1 light chain 3 α
- MOI, multiplicity of infection
- NFKB, nuclear factor of kappa light polypeptide gene enhancer in B-cells
- PBS, phosphate-buffered saline
- RIPK1, receptor (TNFRSF)-interacting serine-threonine kinase 1
- RNF216, ring finger protein 216;TIRAP, toll-interleukin 1 receptor (TIR) domain containing adaptor protein
- TLR, toll-like receptor
- TNF, tumor necrosis factor
- TRAF, TNF receptor-associated factor
- TICAM1/TRIF, toll-like receptor adaptor molecule 1
- TICAM2, toll-like receptor adaptor molecule 2
- shRNA, short hairpin RNA
- Triad, 2 RING fingers and a DRIL (double RING finger linked)
- Ub, ubiquitin
Introduction
Innate immunity is a highly conserved but highly effective first line of defense against pathogens, and plays an indispensable role in organism survival.Citation1-4 The immune cells recognize the invaded pathogens through numerous pattern recognition receptors, among which toll-like receptors (TLRs) are the most-studied family. Upon activation, TLRs initiate NFKB (nuclear factor of kappa light polypeptide gene enhancer in B-cells) signaling and interferon regulatory transcription factor signaling cascade through MYD88 (myeloid differentiation primary response 88)-TIRAP (toll-interleukin 1 receptor [TIR] domain containing adaptor protein) or TICAM1/TRIF (toll-like receptor adaptor molecule 1)-TICAM2 (toll-like receptor adaptor molecule 2), and sequentially results in the production of various cytokines.Citation5-7 Additionally, multiple TLRs induce autophagy, which is considered as an alternative form of innate immunity.Citation8,9
Autophagy is a cellular autonomous process to maintain cellular homeostasis via the degradation of a cell's own cytosolic components or protein aggregates.Citation10-12 Besides starvation, a myriad of extrinsic and intrinsic effectors, including growth factors and intracellular signaling, can initiate autophagy.Citation13-16 Autophagy is involved in diverse physiological events, including survival, differentiation, and development, and contributes to various disorders when it goes awry.Citation12,17-25 So it is of critical importance to maintain appropriate autophagy for the sake of species survival, similar to the elaborated regulation of innate immunity. RNF216/Triad3A (ring finger protein 216)-mediated regulation of innate immunity represents such a mechanism.Citation26
Identified as a RING finger type E3 ubiquitin (Ub) ligase, RNF216 mediates ligation of Ub to diverse TLR, such as TLR4 and TLR9, leading to protein degradation and negative regulation of TLR signaling.Citation26 Subsequent studies demonstrate that RNF216 binds toll-interleukin 1 receptor (TIR) domain-containing adaptor proteins, such as RIPK1 (receptor [TNFRSF]-interacting serine-threonine kinase 1), which is critical for downstream TNF (tumor necrosis factor) production.Citation27 Identification of TRAF3 (TNF receptor-associated factor 3) as a target of RNF216-mediated ubiquitination has revealed RNF216 as an emerging multifunction regulator in innate immunity.Citation28 Further exploration shows that RNF216 negatively regulates the DEAD (Asp-Glu-Ala-Asp) box polypeptide 58 (DDX58/RIG-I) RNA sensing pathway through lysine 48 (K48)-linked, Ub-mediated degradation of the TRAF3 adaptor.Citation28
Due to the widespread crosstalk between autophagy and innate immunity, we hypothesized that RNF216 regulates the autophagy process. To our surprise, RNF216 inhibits starvation-induced autophagy besides TLR4-mediated autophagy, suggesting the existence of unknown common mechanism underlying the inhibition of RNF216 on autophagy. Based on the hypothesis, we elucidate the function of RNF216 on autophagy and the novel mechanism. Our data demonstrate that RNF216 interacts with BECN1/Beclin 1 and promotes its ubiquitination, which accelerates the degradation of BECN1, thereby inhibiting autophagy. We further show that this mechanism is involved in pathogen response, as with the model of L. monocytogenes infection. Thus, our work identifies a novel negative regulator of autophagy that potentially provides a new therapeutic target for infectious and inflammatory diseases.
Results
RNF216 negatively regulates autophagy
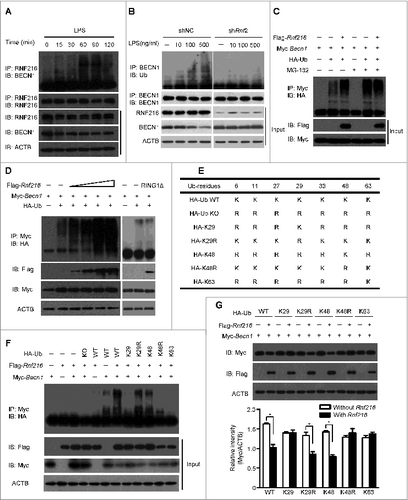
Autophagy is a cellular response upon environment stress to maintain homeostasis. Starvation is a classic means to induce autophagy, and lipopolysaccharide (LPS) treatment induces considerable autophagy in immune cells, such as macrophages.Citation8,9 We used murine macrophage RAW 264.7 cells to investigate the roles of RNF216 in autophagy. First RAW 264.7 cells were transfected with Flag-Rnf216 and subjected to either serum starvation or LPS. The induction of autophagy is assessed by monitoring MAP1LC3A (microtubule-associated protein 1 light chain 3 α) using immunoblotting. MAP1LC3A is a mammalian ortholog of the autophagy-related gene ATG8 in yeast, and is subject to lipidation with phosphatidylethanolamine upon autophagy induction, thus forming MAP1LC3A-II, which associates with the phagophore and autophagosome membrane. This makes MAP1LC3A-II a common readout for autophagy.Citation16,29,30 As shown in , both starvation (Hank's balanced salt solution treatment) and TLR4 activation (LPS stimulation) significantly increased the level of MAP1LC3A-II in RAW 264.7 cells. However, MAP1LC3A-II formation was inhibited by RNF216 overexpression compared with cells transfected with empty vector (). Next we monitored the autophagy formation using confocal imaging. The RAW 264.7 cells were transfected with an expression vector for green fluorescence protein-fused MAP1LC3A (GFP-MAP1LC3A). Upon autophagy initiation, GFP-MAP1LC3A is recruited from the cytosol to phagophore membranes, which can be visualized as puncta by confocal microscopy. As an evolutionarily conserved homeostasis mechanism, the basal level of autophagy is usually pretty low for cells in a resting state. For RAW 264.7 cells in this study, the puncta structure (autophagy) was observed rarely in physiological condition (empty vector group), and we also saw no considerable change even if RNF216 was overexpressed (). BECN1, the mammalian ortholog of yeast Vps30/Atg6, has been commonly regarded as an essential molecule in autophagosome formation, however, there does exist noncanonical autophagy which is independent on BECN1.Citation31 In order to clarify whether it is canonical or noncanonical autophagy that RNF216 inhibited, we monitored autophagy induction under starvation or LPS stimulation in macrophages, following BECN1 knockdown by small interfering RNA against Becn1 (siBecn1), and our data showed that no autophagosomes were observed in BECN1-knockdown RAW 264.7 cells (), demonstrating the inhibition of canonical autophagy by RNF216. However, upon either starvation or TLR4 activation, the featured puncta increased significantly in empty vector-transfected RAW 264.7 cells, while overexpression of RNF216 prevented the increase considerably ().
Figure 1. RNF216 inhibits autophagy in macrophages stimulated with LPS. (A and B) RAW 264.7 cells were transfected with Rnf216 vector or empty vector, and then stimulated without or with LPS (100 ng/mL) for 16 h (A) or were on starvation for 4 h (B). Cell lysates were separated with SDS-PAGE and transferred to polyvinylidene difluoride membranes, following with MAP1LC3A antibody and proper HRP-conjugated secondary antibody. EV, empty vector. The band densitometry was quantified using ImageJ software. The quantitative data were calculated from 3 independent experiments, and were shown as mean ± SEM. (C) Cells grown on coverslips were transiently transfected with GFP-MAP1LC3A and either EV, Rnf216, or siBecn1 overnight, followed by treatment with LPS (100 ng/ml) for 16 h or starvation for 4 h, and then fixed. Digital images were captured with confocal microscopy. Scale bar = 10 μm. (D) Cells with featured puncta were considered as autophagy-positive, and at least 100 cells were quantified. Puncta dots per cell were shown as mean ± SEM. (*P < 0.05).
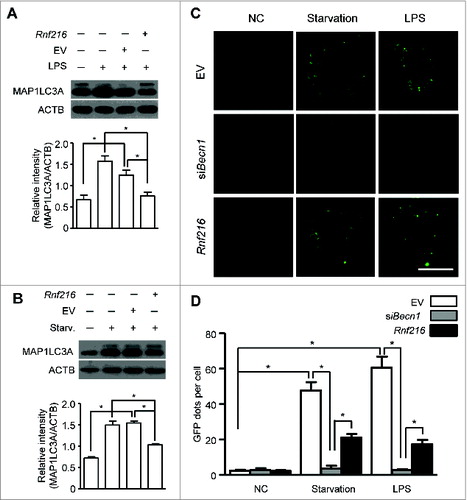
To further confirm the influence of RNF216 on the autophagic process, we knocked down RNF216 in RAW 264.7 cells by expressing 2 different short hairpin RNAs (shRNAs) specific for Rnf216 (shRnf1 and 2). The knockdown efficiency by shRNAs was confirmed via immunoblotting of the endogenous protein levels (). RAW 264.7 cells with RNF216 knockdown were serum starved or treated with LPS, and the induction of autophagy was monitored both by the level of MAP1LC3A-II and the number of GFP-MAP1LC3A puncta. Knockdown of RNF216 in RAW 264.7 cells substantially increased the autophagy initiated by starvation or LPS treatment, as measured by either MAP1LC3A II conversion () or by puncta structure formation (). However, this promotion of autophagy by RNF216 deficiency was obviated if combined with knockdown of BECN1 by siBecn1 (). Taken together, our results demonstrate that RNF216 negatively regulates the BECN1-dependent autophagy upon either starvation or TLR4 activation, suggesting a general mechanism for restricting autophagy.
Figure 2. Knockdown of RNF216 expression abrogates the inhibition of RNF216 on autophagy induction. (A) RAW 264.7 cells were infected with lentivirus with scrambled shRNA (shNC) or Rnf216 shRNA1 and 2 (shRnf1, and 2) (MOI=10), and 48 h later, the cells were lysed and subject to SDS-PAGE followed by being transferred to nitrocellulose membrane. After blotting with RNF216 antibody, the membrane was incubated with HRP-conjugated secondary antibody, and visualized with an ECL chemiluminescence kit. (B and C) RAW 264.7 cells infected with lentivirus with shNC or shRnf2 (MOI=10) were treated with LPS (100 ng/mL) for 16 h (B), or starvation for 4 h (C), then the cells were lysed and subjected to SDS-PAGE followed by being transferred to nitrocellulose membrane. After blotting with MAP1LC3A antibody, the membrane was incubated with HRP-conjugated secondary antibody, and visualized with an ECL chemiluminescence kit. The band densitometry was quantified using ImageJ software. The quantitative data were calculated from 3 independent experiments, and were shown as mean ± SEM. (D) RAW 264.7 cells infected with lentivirus containing scrambled shNC, or shRnf2 (MOI=10) alone or combined with siBecn1 transfection were grown on coverslips, and transiently transfected with GFP-MAP1LC3A overnight, followed by treatment with LPS (100 ng/ml) for 16 h or starvation for 4 h, and then fixed. Digital images were captured with confocal microscopy. Scale bar = 10 μm. (E) Cells with featured puncta were considered as autophagy-positive, and at least 100 cells were quantified. Puncta dots per cell were shown as mean ± SEM. (*P < 0.05).
RNF216 promotes proteasomal degradation of BECN1
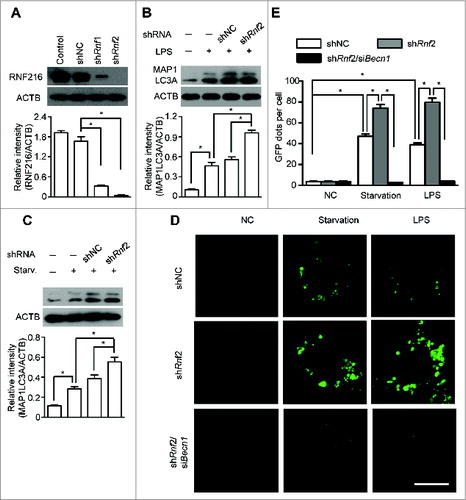
Previous studies have demonstrated that RNF216 dampens TLR4-mediated signaling by interacting, directly or indirectly, with TLR4, TICAM1, TIRAP, and RIPK1 and stimulating the degradation of these molecules.Citation26-28 Because RNF216 negatively regulates autophagy induced through LPS treatment or starvation, we hypothesized that RNF216 interacts with other molecules that are important for autophagy. We constructed various Myc-tagged plasmids containing the sequences of genes that are essential for autophagy, including Atg5, Atg14, and Becn1. After cotransfecting the Flag-Rnf216 and different Myc-tagged molecules (with Tirap as a positive control), we monitored the expression of the molecules, and the results showed that there was no visible change for ATG5 and ATG14, while the expression of BECN1 was markedly decreased (). In order to confirm the effects of degradation of BECN1 by RNF216, we cotransfected Myc-Becn1 and different doses of Flag-Rnf216 into 293T cells and found that BECN1 levels decreased with increased expression of RNF216 (), suggesting that RNF216 downregulates BECN1. During protein turnover, there are 2 pathways involved in protein degradation, utilizing the proteasome and the lysosome. To further investigate the localization of BECN1 degradation, we treated 293T cells with MG132, an irreversible proteasome inhibitor, or E64d, a lysosomal protease inhibitor. Degradation of BECN1 was blocked by MG132 treatment, while E64d treatment did not modify RNF216-mediated degradation of BECN1 (). Collectively, our results demonstrate that RNF216 promotes BECN1 degradation through a proteasome-dependent pathway.
Figure 3. Inhibition of autophagy by RNF216 was mediated through BECN1. (A) Flag-Rnf216 and Myc-tagged plasmid (Atg5, Atg14, Tirap, Becn1) were used to transfect the 293T cells. Then the cells were lysed and subjected to immunoblotting, and the bands were visualized with an ECL chemiluminescence kit. (B) 293T cells were transfected with Becn1 combined with increased Rnf216, and subjected to immunoblotting for the corresponding proteins. (C) 293T cells transfected with Becn1 and Rnf216 were treated with MG132 or E64d, and lysed before being subjected to SDS-PAGE, followed by transferring to nitrocellulose membrane. Then the membrane was blotted with anti-Flag or -Myc antibody, and then incubated with HRP-conjugated secondary antibody. The bands were visualized with an ECL chemiluminescence kit. The images displayed were representatives from 3 independent experiments.
RNF216 interacts with BECN1 through the triad domain
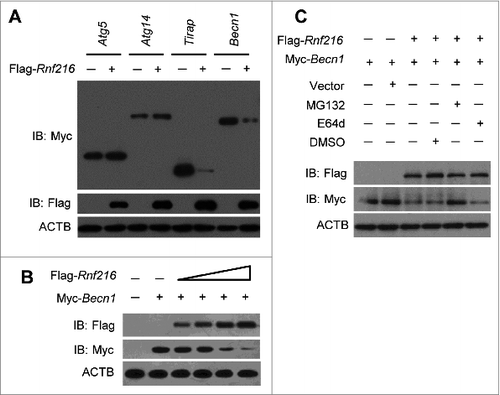
To further explore the mechanism by which RNF216 degrades BECN1 expression, we first investigated the existence of a physical interaction between these proteins. Plasmids Myc-Becn1 and Flag-Rnf216 were cotransfected into 293T cells, and immunoprecipitation experiments were performed with Myc or Flag antibodies. As shown in , RNF216 was coprecipitated with BECN1, and vice versa. To probe for an interaction between endogenous RNF216 and BECN1, RAW 264.7 cells were subject to starvation or LPS stimulation, followed by immunoprecipitation assays with BECN1 or RNF216 antibodies. We observed interactions between BECN1 and RNF216 constitutively, however, autophagy induction initiated stronger binding between these proteins (). We next performed double-immunofluorescence staining of RNF216 and BECN1 to study their colocalization in RAW 264.7 cells grown on coverslips. There was no visible colocalization between these proteins in the basal state, however the colocalization was considerably increased in perinuclear regions after starvation or LPS stimulation ().
Figure 4. RNF216 interacts with BECN1 through the triad domain. (A) 293T cells transfected with plasmids of Flag-Rnf216 and Myc-Becn1 were immunoprecipitated with IgG, Myc (left panel) or Flag (right panel) antibodies. The precipitates were subjected to blotting with the indicated antibodies, and the bands were visualized with an ECL chemiluminescence kit. (B) RAW 264.7 cells subjected to starvation or LPS treatment were immunoprecipitated with an antibody against RNF216 (left panel) or BECN1 (right panel), followed by immunoblotting with the indicated antibodies, and the bands were visualized with an ECL chemiluminescence kit. The images displayed are representative of 3 independent experiments. The densitometry of the bands was quantified using ImageJ software, and is shown as mean ± SEM. (C) RAW 264.7 cells grown on coverslips were subjected to starvation or LPS (100 ng/mL) treatment, followed by staining with BECN1, RNF216, and DAPI. The images were captured with a confocal microscope. Colocalization of BECN1 and RNF216 was processed using LaserSharp 2000 software (Bio-Rad), is presented as a percentage of RNF216, and is shown as mean ± SEM calculated from 3 independent experiments (*P < 0.05). Scale bar = 10 μm. (D) Schematic diagram of RNF216 and truncation mutants used in this study. TIM, TRAF6 interacting domain; IBR, ‘in-between-RING’ domain. (E) Myc-tagged full-length RNF216 or truncates were transiently transfected into 293T cells. Cell lysates were analyzed by immunoblotting with anti-Myc antibody for the expression of RNF216 protein or by immunoprecipitation with anti-BECN1 antibody following by immunoblotting with the anti-Myc antibody for the association between RNF216 and BECN1 proteins.
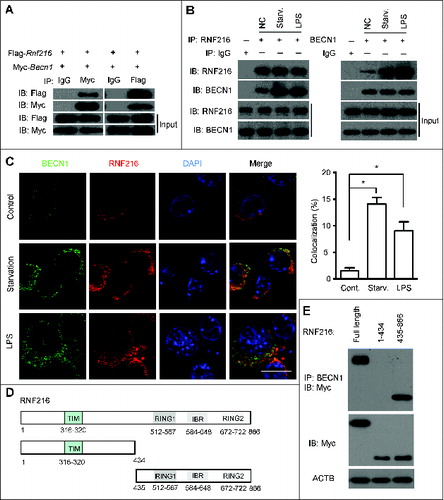
To clarify the binding site between RNF216 and BECN1, we generated truncation mutants of RNF216. As RNF216 contains 2 distinct domains, we created truncations of each domain, as shown in . The first truncation eliminates the TIM domain (RNF216 [1 to 434]), and the other eliminates the triad (2 RING fingers and a DRIL [double RING finger linked]) domain (RNF216 [435 to 866]). The full-length and truncated RNF216 proteins were expressed in 293T cells and analyzed by coimmunoprecipitation and immunoblotting to determine their binding with BECN1. The results showed that triad domain is indispensable for binding to BECN1, while TIM domain is not necessary (). All together, these data demonstrate the physical interaction between RNF216 and BECN1 through the triad domain.
RNF216 promotes K48-linked ubiquitination of BECN1
Before being transported to the proteasome for degradation, proteins usually are subjected to ubiquitination.Citation32-34 Thus we tested whether ubiquitination of BECN1 increased during autophagy induction. We stimulated RAW 264.7 cells with LPS, there was slight ubiquitination of BECN1. However, immunoprecipitation using an antibody against RNF216, followed by immunoblotting for BECN1, shows increasing ubiquitination with longer LPS stimulation (), suggesting a specific ubiquitination of BECN1 by RNF216. Treatment of RAW 264.7 cells with a higher concentration of LPS promotes the ubiquitination, while knockdown of RNF216 with specific shRnf2 abolishes the ubiquitination (), implying the potential role of RNF216 in BECN1 ubiquitination. In order to confirm this mechanism, we first assayed in vivo ubiquitination of Myc-tagged BECN1 in 293T cell after transfection with plasmids for RNF216 and HA-Ub. Immunoprecipitated BECN1 had increased levels of polyubiquitination when RNF216 and MG132 were expressed simultaneously compared to BECN1 and Ub alone (). Increasing the transfected amount of RNF216 led to even more readily detectible ubiquitination (, left panel), supporting the essential role of RNF216 in ubiquitination of BECN1. The RING1 domain of triad E3 ligases mediates the interaction with E2 conjugase,Citation35 and thus plays an indispensible role to position Ub in preparation for its transfer to substrate proteins. So we constructed a RNF216 truncate with a RING1 domain (512 to 567) deletion (RNF216RING1Δ), which lacks the ability of transferring Ub. After cotransfection RAW 264.7 cells with the indicated plasmids, we found that in contrast to wild-type RNF216 transfection, the ubiquitination of BECN1 in the RNF216RING1Δ transfection remained elusive to detect (, right panel), confirming the essential role of RNF216 in BECN1 ubiquitination. We then expressed different mutants of Ub combined with RNF216 and BECN1, and found that RNF216 promoted K48-linked, but not K29 or K63-linked polyubiquitination of BECN1 (). Using suboptimal levels of Flag-tagged RNF216 limited BECN1 ubiquitination (). Lastly, the different Ub mutants were transfected in 293T cells, and the degradation of BECN1 was observed in intact K48 Ub-expressing cells (as in WT, K29R, and K48), but not in K48-mutant cells (), further confirming that the RNF216-mediated modification of BECN1 is a K48-linked ubiquitination. Collectively, these data demonstrate that RNF216-mediated degradation of BECN1 occurs through modification of K48-linked ubiquitination.
Figure 5 (see previous page). K48 ubiquitination mediated by RNF216 is necessary for BECN1 degradation. (A) RAW 264.7 cells were stimulated with LPS (100 ng/mL) for different periods of time, followed by immunoprecipitation with anti-RNF216 antibody, and then blotted with the indicated antibodies. Meanwhile, lysates were immunoblotted with different antibodies, followed by band visualization. (B) RAW 264.7 cells infected with lentivirus with scrambled shNC or shRnf2 (MOI=10) were treated with or without LPS (at the indicated concentration) for 1 h, followed by immunoprecipitation with anti-RNF216 antibody, and then blotted with the indicated antibodies. Meanwhile, lysates were immunoblotted with different antibodies, followed by band visualization. (C) RAW 264.7 cells transfected with the indicated plasmids were subjected to treatments in the absence or presence of MG-132, and followed by immunoprecipitation with anti-Myc antibody, then blotted with anti-HA antibody. The bands were visualized with an ECL chemiluminescence kit. (D) RAW 264.7 cells transfected with the indicated plasmids (RING1Δ represents Rnf216RING1Δ) were subjected to immunoprecipitation with anti-Myc antibody, then blotted with anti-HA antibody. The bands were visualized with an ECL chemiluminescence kit. (E) Schematic illustration of HA-Ub wild type (WT) and mutants. (F) RAW 264.7 cells transfected with the indicated plasmids were subjected to immunoprecipitation with anti-Myc antibody, then blotted with anti-HA antibody. The bands were visualized with an ECL chemiluminescence kit. (G) RAW 264.7 cells transfected with the indicated plasmids were subjected to immunoblotting with indicated antibodies. The bands were visualized with an ECL chemiluminescence kit. The band densitometries were quantified using ImageJ software. The quantitative data were calculated from 3 independent experiments, and are shown as mean ± SEM. (*P < 0.05).
RNF216 regulates TLR-mediated antimicrobial responses via control of autophagy
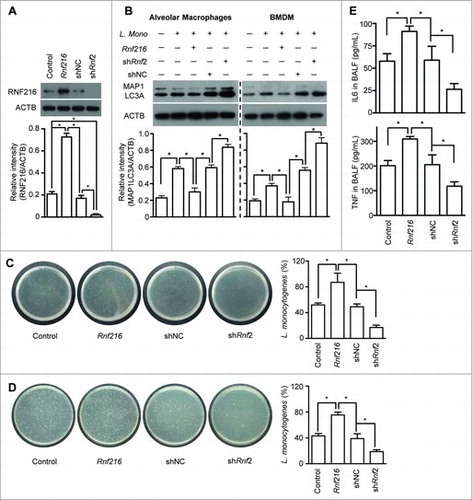
Autophagy has been proposed to be a mechanism of innate immunity against various intracellular pathogens,Citation8,11,15,36,37 such as L. monocytogenes, especially in the early phase of infection.Citation38,39 We further investigated the function of RNF216 in controlling TLR-mediated uptake of bacteria into autophagosomes and restriction of their growth. We inoculated mice intratracheally (i.t.) with the gram-negative bacteria, L. monocytogenes, which invades alveolar macrophages and rarely lung epithelial cells.Citation40 Before the infection of L. monocytogenes, some of mice were inoculated i.t. with lentivirus containing Rnf216 (overexpression) or shRnf2 (knockdown). After 48 h, alveolar macrophages were isolated from the mice. Immunoblot analysis demonstrates that RNF216 was successfully overexpressed or knocked down (). Induction of autophagy was assessed by MAP1LC3A-II immunoblotting. L. monocytogenes infection strongly promoted autophagy in alveolar macrophages, and RNF216 overexpression dampens this response drastically (, left panel). Knockdown of RNF216 had the opposite effect, suggesting a role for RNF216 in autophagy. Accordingly, the growth of L. monocytogenes was much higher when RNF216 was overexpressed, while RNF216 knockdown inhibited proliferation of the bacteria (). Infection of bone marrow-derived macrophages (BMDM) with L. monocytogenes in vitro induces autophagy, and modification of RNF216 expression in BMDM produce similar effects on autophagy to alveolar macrophages (, right panel), emphasizing the crucial role of RNF216 in regulation of L. monocytogenes-induced autophagy. Likewise, the recovered bacteria from BMDM with RNF216 overexpression showed stronger growth, in contrast to RNF216 knockdown (), showing the vital part of RNF216 in host response to L. monocytogenes infection. Bronchoalveolar lavage fluid (BALF) was collected from mice with L. monocytogenes infection, and the levels of inflammatory cytokines such as TNF and IL6. TNF and IL6 were assayed, which were significantly higher when RNF216 was overexpressed, and much lower when RNF216 was knocked down (). These data support the idea that RNF216 can control the progression of infections. Together, our results demonstrate the essential role of RNF216 in TLR-mediated antimicrobial responses through control of autophagy.
Figure 6. Inhibition of RNF216 contributes to elimination of intracellular pathogens. Mice were infected with lentivirus i.t., followed by infection with Listeria monocytogenes i.t.. Twenty-four h later, the mice were sacrificed. The alveolar macrophages were isolated and lysed, then subjected to immunoblotting with RNF216 antibody (A) or MAP1LC3A antibody (B, left panel). BMDM with the indicated treatment also were lysed and immunobloted to detect MAP1LC3A expression (B, right panel). The band densitometries were quantified using ImageJ software. Meanwhile Listeria monocytogenes used for infection and recovered from alveolar macrophages (C) or BMDM (D) were cultured, then the CFU was calculated, and the percentage represents the ratio of CFU of recovered bacterial with that of the bacteria used for infection (C, D). (E) BALF was prepared, and ELISA was used to detect the inflammatory cytokines IL6 and TNF. All quantified data were calculated from 3 independent experiments, and presented as mean ± SEM. (*P < 0.05).
Discussion
A myriad of biological stimuli and environment stresses contribute to autophagy, an ancient biological process for maintaining cellular homeostasis. Following induction, autophagy will go through steps of phagophore elongation, and maturation into an autophagosome, which subsequently fuses with a lysosome where the cargoes are delivered for degradation and recycling.Citation41-43 During the process, numerous homologs and orthologs of products of autophagy-related genes have been identified, that signal to the autophagic machinery.Citation16,22,30,44,45 Among them, BECN1 is arguably the most-studied molecule regulating the process, and is central to health and disease processes such as tumor, inflammation, and aging.Citation14,22,46,47 BECN1 is one of the key initiators of the autophagic process, which associates with PtdIns3K, thereby mediating biogenesis and dynamics of subcellular membranes involved in autophagy.Citation48,49
Due to its multiple functions and diverse roles in cellular survival and death, it is no surprise that there are many mechanisms regulating BECN1. It has been reported that binding partners of BECN1, such as the autophagy/Beclin 1 regulator 1 (AMBRA1),Citation50 UV radiation resistance associated (UVRAG),Citation51 and ATG14/BarkorCitation52,53 positively regulate autophagy, whereas B-cell CLL/lymphoma 2 (BCL2)Citation54 and KIAA0226/Rubicon (1700021K19Rik in mice)Citation53 inhibit autophagy. Furthermore, a recent study demonstrated that K63-linked ubiquitination of BECN1 is regulated by TRAF6 and tumor necrosis factor α-induced protein 3 (TNFAIP3/A20). TRAF6 promotes the K63-linked ubiquitination of BECN1 to induce TLR4-mediated autophagy, and tumor necrosis factor α-induced protein 3 deubiquitinates the K63-linked Ub chains.Citation55 However, we found in previous studies that BECN1 experienced K48-linked ubiquitination,Citation8 the E3 Ub-protein ligase mediated the modification remains to be identified. In addition, the function of K48-linked ubiquitination of BECN1 is also unknown. In this study, we identified RNF216 as an E3 Ub ligase to mediate the K48-linked ubiquitination of BECN1, which represents a negative pathway for autophagy regulation.
RNF216 is known to mediate the ubiquitination of various TLRs (TLR4 and TLR9) and downstream signaling molecules, including RIPK1 and TICAM1, causing their subsequent degradation via the proteasome and dampening of NFKB signaling.Citation26,27 In addition, RNF216 interacts with TRAF3, induces ubiquitination and degradation of TRAF3 and negatively regulates DEAD (Asp-Glu-Ala-Asp) box polypeptide 58/ mitochondrial antiviral signaling protein (MAVS)-mediated NFKB activation, thereby modulating innate immune receptor responses toward several invading pathogens.Citation28 It is no wonder that RNF216 interferes with the TLR4-induced autophagy in macrophages. However, when we tried to test whether RNF216 has regulatory effects in autophagy induced by starvation, we observed that RNF216 still has strong inhibition ability on autophagy, which prompted us to focus on common molecules other than those of TLR signaling cascade, such as BECN1 or ATG5 (data not shown). For macrophages in resting state, we observed elusive interaction between RNF216 and BECN1, and there was no considerable inhibition on autophagy by RNF216 overexpression, putatively due to the low basal level of autophagy.
Our study further demonstrates that RNF216 physically interacted with BECN1 for macrophages under starvation or treated with LPS. Unlike the interaction between RNF216 and TRAF3 through TIM domain, RNF216 binds to BECN1 through a triad domain consisting of 2 RING domains and an ‘in-between-RING’ domain. The detailed mechanism by which RNF216 ubiquitinates the protein with different domains remains to be elucidated.
We sought to evaluate the in vivo function of RNF216 on controlling pathogen growth. L. monocytogenes has been widely used as a model to investigate the pathogenesis of an intracellular microorganisms and the regulation of cellular immunity. When the mice are infected i.t. with L. monocytogenes, most pathogens invade alveolar macrophages.Citation40 L. monocytogenes infection of macrophages induces strong autophagic response, theoretically triggered by activation of TLRs that inhibit the growth of the bacteria and cell-to-cell spreading.Citation38 In this study, we found that overexpression of RNF216 in alveolar macrophages inhibited autophagy, which contributed to L. monocytogenes growth and spread, while knockdown of RNF216 had the opposite effects. Accordingly, the pulmonary inflammation in mice with RNF216 overexpression was much more severe than that without RNF216 overexpression, while that in mice with RNF216 knockdown was much less severe.
In summary, we found RNF216 could potently downregulate autophagy through promoting proteasome-dependent degradation of BECN1 via K48-linked ubiquitination. Given the importance of autophagy in the innate immune responses during bacterial and viral infection, our study identifies a novel regulator of autophagy and represents a potential target for drug development in the control of various diseases such as infectious disease, where autophagy has gone awry.
Materials and Methods
Reagents and antibodies
LPS (Re595) from Salmonella minnesota R595, MG132 and E64d were purchased from Sigma (L9764, M7449, and E8640). Anti-Myc, anti-Flag, anti-HA, anti-MAP1LC3A monoclonal antibodies (mAbs) were purchased from Cell Signaling Technology (2287, 8146, 2367, and 12741). Ub K48-specific mAb and Ub K63-specific mAb were purchased from Millipore (05–1307, 05–1313). BECN1 mAb was purchased from Santa Cruz Biotechnology (sc-49341). Rabbit polyclonal anti-RNF216 Ab was from Abcam (ab25961). HRP-conjugated anti-mouse and anti-rabbit secondary antibodies were purchased from Jackson ImmunoResearch (115–035–003, 111–035–003). siBecn1 targeting mouse Becn1 was obtained from Life Technologies (s80168).
cDNA and plasmid constructs
The GFP-MAP1LC3A construct was a kind gift from Dr. Tamotsu Yoshimori (Osaka University, Osaka, Japan). HA-Ub wild-type, knockout, K29, K29R, K48, K63 and K48R Ub mutants were from Addgene (Plasmid # 17608, 17603, 22903, 17602, 17605, 17606, 17604, respectively). Full-length Becn1, Bcl2 and Rnf216 cDNAs were PCR amplified from first-strand cDNA libraries, as described previously.Citation8,26 cDNAs for Rnf216 truncations were generated by PCR amplification with the full-length Rnf216 cDNA as template. These cDNAs were subcloned into PRK5 mammalian expression vectors containing an N-terminal Myc or Flag epitope tag. The cDNA of mouse Flag-Rnf216 in a pRK5 vector was also cloned into pLVX-IRES-EGFP. Rnf216 shRNA1 (shRnf1) targeting nucleotide sequence (1532 to 1551) 5′-GAGCAGGAGTTCTATGAGCA-3′, Rnf216 shRNA2 (shRnf2) targeting nucleotide sequence (1195 to 1214) 5′-GGACACTATGCAATCACCCG-3′ and scrambled shRNA control (shNC) have been previously described.Citation28
Cell culture and transfection
RAW 264.7 cells and 293T cells were cultured in DMEM medium supplemented with 10% FBS. 293T cells were transfected by using Lipofectamine 2000 (Invitrogen, 11668019). BMDM were obtained as previously describedCitation56 with modification. Briefly, cells were prepared by flushing the bone marrow from femurs and tibias and then maintained in DMEM medium containing 10% FBS and supplemented with 10 ng/ml M-CSF (Peprotech, 315–02). Four to 5 d later, adherent cells were dissociated and cultured in DMEM supplemented with 10% FBS and growth factor. RAW 264.7 and BMDM cells were transfected with TransIT-Jurkat (Mirus Bio, MIR2125), according to the manufacturer's instruction.
Immunoblotting
Cell lysates were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% fat-free milk in phosphate-buffered saline (PBS, Gibco, 21600–044) with 0.1% Tween-20 (Sinopharm Chemicals, 30189328) for 2 h and then incubated with the indicated antibody in PBS plus 0.5% fat-free milk overnight. After washing, the membranes were incubated with HRP-conjugated goat anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch Laboratories, 115–035–003, 111–035–003) for 1 h. After subsequent washes, the immunoreactive bands were visualized with ECL Plus Western blotting detection reagents (Millipore, 2650). For some immunoblots, the band densitometry was quantified using ImageJ software (National Institutes of Health).
Coimmunoprecipitation
Cells were lysed in lysis buffer [50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 0.5% Nonidet P-40 (Sangon Biotech, NE109), plus complete protease inhibitor mixture (Roche Applied Science, 04693116001)]. Cell lysates were clarified by centrifugation and incubated with the indicated antibody plus protein G-Sepharose (Amersham Biosciences, 17–0618–02) at 4°C overnight to form immunocomplexes. After extensive washing with lysis buffer, the immunocomplexes were analyzed by immunoblotting as described.
Fluorescence confocal microscopy
Cells grown on coverslips were fixed in BD Cytofix/Cytoperm solution (BD Biosciences, 554714) at room temperature for 15 min. These coverslips were incubated with the primary antibody, followed by fluorochrome-conjugated secondary antibody, before mounting. For fluorescence analysis, cell samples were visualized on an Olympus Fluoview confocal microscope with appropriate emission filters (Olympus, Tokyo, Japan).
Autophagy analyses
Autophagy was analyzed by immunoblotting or fluorescence microscopy, as described previously.Citation8 In the immunoblotting analysis, cells were treated as indicated, and cell lysates were immunoblotted with anti-MAP1LC3A antibody to monitor the MAP1LC3A-II generated during the formation of autophagosomes. In the fluorescence confocal microscopy analysis, cells were transfected with a GFP-MAP1LC3A construct and treated as indicated. These cells were imaged by fluorescence confocal microscopy, with single-line excitation at 488 nm for GFP, for the formation of puncta in autophagic cells. A minimum of 100 cells was analyzed for each treatment, and each experiment was performed at least 3 independent times.
Ubiquitination assay
For analysis of ubiquitination of endogenous BECN1, whole-cell extracts were immunoprecipitated with anti-BECN1 and analyzed by western blot with anti-Ub Ab. For analysis of the ubiquitination of overexpressed BECN1, 293T cells were transfected with Myc-Becn1, HA-Ub wild type, or HA-Ub mutants and Flag-Rnf216; then whole-cell extracts were immunoprecipitated with anti-Myc and analyzed by immunoblotting with anti-Ub Ab.
Intratracheal administration of lentivirus
Lentivirus suspensions (100 μl) with a dosage of 5 × 108 plaque-forming units was injected i.t. into mouse lungs, using a Hamilton syringe with a sterile 30-gauge needle. Three d later, mice were used for bacterial infection. Female C57BL/6 mice (8 to 12 wk) used in this study were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences, and were kept under specific pathogen-free conditions in the animal center of Shanghai Jiao Tong University School of Medicine (Shanghai, China). All mouse experiments were approved by the Animal Welfare & Ethics Committee of the Shanghai Jiao Tong University School of Medicine.
L. monocytogenes infection
L. monocytogenes used in this study is strain 10403S. Before using for infection, the bacteria were grown in Brain Heart Infusion (BD Biosciences, 237400) broth for 16 h or until mid-log phase growth was reached with shaking. After pellet with centrifugation at 5000 g for 15 min and washing with PBS twice, the bacteria density was determined by spectrophotometry at 600 nm, and diluted with sterile PBS to prepare the inoculum for the mice. For in vivo infection, 1 ×103 (50 μl) bacteria in PBS was used to inoculate the mice i.t.. Twenty-four h later, the mice were sacrificed, and the lungs were removed from the mice. After homogenization, the homogenates were cultured in Brain Heart Infusion agar plates (BD Biosciences, 237400), and bacterial colony forming units (CFU) were determined after 16 h growth at 37°C. For infection of cells, L. monocytogenes were used to infect cells at a multiplicity of infection (MOI) of 2:1. After 1 h after infection, the cells were washed with PBS to remove extracellular bacteria. The cells were cultured in fresh medium for 12 h more, and then washed 3 times with PBS and treated with 0.1% Triton X-100 (Sigma-Aldrich, X100) to harvest the surviving intracellular bacteria. We grew in parallel the original and the recovered bacteria on plates, and the percentage represents the ratio of CFU of recovered bacteria relative to the CFU of the original bacteria used for infection.
BALF collection and isolation of alveolar macrophages
After 2 d of bacterial infection, BALF was collected as previously described.Citation57 Briefly, the mice were euthanized and the lungs were lavaged with 0.5 ml sterile PBS for 4 times, and the lavage fluid was centrifuged, followed by collection for detection of cytokines or stored at −80°C until use. After suspending the pellet with PBS, the cells were counted and subjected to alveolar macrophages isolation as described.Citation58
Statistical analysis
The 2-tailed Student t test or one-way analysis of variance followed by the Turkey post-hoc test was used for all statistical analyses in this study using SPSS 18.0. A P value less than 0.05 is considered as statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Rennolds S. Ostrom (University of Tennessee Health Science Center in Memphis, TN) for the constructive suggestions and critical review of this manuscript.
Funding
This work was supported by the Ministry of Science and Technology of China 2011CB966200 (to Y.Z.), Strategic Priority Research Program of the Chinese Academy of Sciences XDA01040000 (to Y.Z.), National Natural Science Foundation of China 81130057, 81071748 (to Y.Z.), 81190133 (to C.X.), 31300708 (to H.S.), Natural Science Foundation of Shanghai Municipality 12ZR1415900 (to C.X.), 11JC1411400, 11431920900 (to Y.Z.), and Shanghai Municipal Education Commission J50207 (to Y.Z. and C.X.), 14YZ036 (to C.X.), Shanghai Bureau of Public Health 2012187 (to C.X.).
References
- O'Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors– redefining innate immunity. Nat Rev Immunol 2013; 13:453–60; PMID:23681101; http://dx.doi.org/10.1038/nri3446
- Moussion C, Sixt M. A conduit to amplify innate immunity. Immunity 2013; 38:853-4; PMID:23706666; http://dx.doi.org/10.1016/j.immuni.2013.05.005
- Ayres JS, Vance RE. Cellular teamwork in antibacterial innate immunity. Nat Immunol 2012; 13:115-7; PMID:22261960; http://dx.doi.org/10.1038/ni.2212
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124:783-801; PMID:16497588; http://dx.doi.org/10.1016/j.cell.2006.02.015
- Kawai T, Akira S. TLR signaling. Seminars in Immunology 2007; 19:24-32; PMID:17275323; http://dx.doi.org/10.1016/j.smim.2006.12.004
- Lee CC, Avalos AM, Ploegh HL. Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol 2012; 12:168-79; PMID:22301850
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol 2003; 21:335-76; PMID:12524386; http://dx.doi.org/10.1146/annurev.immunol.21.120601.141126
- Xu C, Liu J, Hsu L-C, Luo Y, Xiang R, Chuang T-H. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J 2011; 25:2700-10; PMID:21543763; http://dx.doi.org/10.1096/fj.10-167676
- Xu Y, Jagannath C, Liu X-D, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27:135-44; PMID:17658277; http://dx.doi.org/10.1016/j.immuni.2007.05.022
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009; 5:527-49; PMID:19527881; http://dx.doi.org/10.1016/j.chom.2009.05.016
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333:1109-12; PMID:21868666; http://dx.doi.org/10.1126/science.1201940
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011; 147:728-41; PMID:22078875; http://dx.doi.org/10.1016/j.cell.2011.10.026
- Chen Y, Klionsky DJ. The regulation of autophagy–unanswered questions. J Cell Sci 2011; 124:161-70; PMID:21187343; http://dx.doi.org/10.1242/jcs.064576
- Deretic V. Multiple regulatory and effector roles of autophagy in immunity. Curr Opin Immunol 2009; 21:53-62; PMID:19269148; http://dx.doi.org/10.1016/j.coi.2009.02.002
- Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev 2011; 25:1999-2010; PMID:21979913; http://dx.doi.org/10.1101/gad.17558811
- Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol 2008; 3:427-55; PMID:18039129; http://dx.doi.org/10.1146/annurev.pathmechdis.2.010506.091842
- Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009; 46:210-40; PMID:19552522; http://dx.doi.org/10.1080/10408360903044068
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/10.1126/science.1193497
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell 2011; 146:682-95; PMID:21884931; http://dx.doi.org/10.1016/j.cell.2011.07.030
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B-G, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1[bgr] production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/10.1038/nature07383
- Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell 2010; 140:798-804; PMID:20303871; http://dx.doi.org/10.1016/j.cell.2010.02.015
- Chuang T-H, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol 2004; 5:495-502; PMID:15107846; http://dx.doi.org/10.1038/ni1066
- Fearns C, Pan Q, Mathison JC, Chuang T-H. Triad3A regulates ubiquitination and proteasomal degradation of RIP1 following disruption of Hsp90 binding. J Biol Chem 2006; 281:34592-600; PMID:16968706; http://dx.doi.org/10.1074/jbc.M604019200
- Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog 2009; 5:e1000650; PMID:19893624; http://dx.doi.org/10.1371/journal.ppat.1000650
- Murrow L, Debnath J. Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease. Annu Rev Pathol 2012; 8:105-37; PMID:23072311; http://dx.doi.org/10.1146/annurev-pathol-020712-163918
- Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol 2012; 30:611-46; PMID:22449030; http://dx.doi.org/10.1146/annurev-immunol-020711-074948
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol 2011; 13:7-12; PMID:22166994
- Kish-Trier E, Hill CP. Structural biology of the proteasome. Ann Rev Biophys 2013; 42:29-49; http://dx.doi.org/10.1146/annurev-biophys-083012-130417
- Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell 2010; 143:682-5; PMID:21111229; http://dx.doi.org/10.1016/j.cell.2010.11.012
- Finley D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 2009; 78:477-513; PMID:19489727; http://dx.doi.org/10.1146/annurev.biochem.78.081507.101607
- Spratt DE, Walden H, Shaw GS. RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J 2014; 458:421-37; PMID:24576094; http://dx.doi.org/10.1042/BJ20140006
- Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and cellular immune responses. Immunity 2013; 39:211-27; PMID:23973220; http://dx.doi.org/10.1016/j.immuni.2013.07.017
- Munz C. Enhancing immunity through autophagy. Annu Rev Immunol 2009; 27:423-49; PMID:19105657; http://dx.doi.org/10.1146/annurev.immunol.021908.132537
- Py BF, Lipinski MM, Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 2007; 3:117-25; PMID:17204850; http://dx.doi.org/10.4161/auto.3618
- Ogawa M, Yoshikawa Y, Mimuro H, Hain T, Chakraborty T, Sasakawa C. Autophagy targeting of Listeria monocytogenes and the bacterial countermeasure. Autophagy 2011; 7:310-4; PMID:21193840; http://dx.doi.org/10.4161/auto.7.3.14581
- Munder A, Zelmer A, Schmiedl A, Dittmar KE, Rohde M, Dorsch M, Otto K, Hedrich HJ, Tümmler B, Weiss S, et al. Murine pulmonary infection with Listeria monocytogenes: differential susceptibility of BALB/c, C57BL/6 and DBA/2 mice. Microbes Infect 2005; 7:600-11; PMID:15820148; http://dx.doi.org/10.1016/j.micinf.2004.12.021
- Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 2009; 46:210-40; PMID:19552522; http://dx.doi.org/10.1080/10408360903044068
- Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. New Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Liu G, Bi Y, Wang R, Wang X. Self-eating and self-defense: autophagy controls innate immunity and adaptive immunity. J Leukoc Biol 2013; 93:511-9; PMID:23271703; http://dx.doi.org/10.1189/jlb.0812389
- Rubinstein AD, Kimchi A. Life in the balance–a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci 2013; 125:5259-68; http://dx.doi.org/10.1242/jcs.115865
- Deretic V. Autophagy: an emerging immunological paradigm. J Immunol 2012; 189:15-20; PMID:22723639; http://dx.doi.org/10.4049/jimmunol.1102108
- Terman A, Gustafsson B, Brunk UT. Autophagy, organelles and ageing. J Pathol 2007; 211:134-43; PMID:17200947; http://dx.doi.org/10.1002/path.2094
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, Packer M, Schneider MD, Levine B. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011; 147:223-34; PMID:21962518; http://dx.doi.org/10.1016/j.cell.2011.08.037
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839-49; PMID:17893711; http://dx.doi.org/10.1038/cr.2007.78
- Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007; 447:1121-5; PMID:17589504
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh B-H, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8:688-98; PMID:16799551; http://dx.doi.org/10.1038/ncb1426
- Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 2008; 105:19211-6; PMID:19050071; http://dx.doi.org/10.1073/pnas.0810452105
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385-96; PMID:19270696; http://dx.doi.org/10.1038/ncb1846
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/10.1016/j.molcel.2008.06.001
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3:ra42; PMID:20501938
- Cai W, Du A, Feng K, Zhao X, Qian L, Ostrom RS, Xu C. Adenylyl cyclase 6 activation negatively regulates TLR4 signaling through lipid raft-mediated endocytosis. J Immunol 2013; 191:6093-100; PMID:24218452; http://dx.doi.org/10.4049/jimmunol.1301912
- Nagatani K, Dohi M, To Y, Tanaka R, Okunishi K, Nakagome K, Sagawa K, Tanno Y, Komagata Y, Yamamoto K. Splenic dendritic cells induced by oral antigen administration are important for the transfer of oral tolerance in an experimental model of asthma. J Immunol 2006; 176:1481-9; PMID:16424176; http://dx.doi.org/10.4049/jimmunol.176.3.1481
- Xu J, Xu F, Barrett E. Metalloelastase in lungs and alveolar macrophages is modulated by extracellular substance P in mice. Ame J Physiol–Lung Cell Mol Physiol 2008; 295:L162-L70; http://dx.doi.org/10.1152/ajplung.00282.2007