
Abstract
Autophagy provides an important defense mechanism against intracellular bacteria, such as Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis disease (TB). We recently reported that pathogen recognition and antibacterial autophagy are connected by the induction of the DNA damage-regulated autophagy modulator DRAM1 via the toll-like receptor (TLR)-MYD88-NFKB innate immunity signaling pathway. Having shown that DRAM1 colocalizes with Mtb in human macrophages, we took advantage of a zebrafish model for TB to investigate the function of DRAM1 in autophagic host defense in vivo. We found that DRAM1 protects the zebrafish host from infection with Mycobacterium marinum (Mm), a close relative of Mtb. Overexpression of DRAM1 increases autophagosome formation and promotes autophagic flux by a mechanism dependent on the cytosolic DNA sensor TMEM173/STING and the ubiquitin receptor SQSTM1/p62. Here we summarize and discuss the implications of these findings.
DRAM1/DRAM (DNA-damage regulated autophagy modulator 1) is an evolutionarily conserved transmembrane protein. It localizes predominantly to lysosomes, while also colocalizing with the autophagosome marker LC3. DRAM1 overexpression markedly increases LC3-decorated cytoplasmic puncta in model systems ranging from Drosophila to human cells. DRAM1 has drawn considerable attention due to its role in cellular stress and cancer. In the DNA damage response, DRAM1 functions as a target of TP53/p53 (tumor protein p53) and is required for TP53-mediated programmed cell death. More recently, DRAM1 has been shown to function also as a TP53 target during host defense against HIV infection. In contrast, we discovered that DRAM1 functions independently of TP53 in host defense against intracellular mycobacteria. Using zebrafish embryos and human macrophages we found that mycobacterial infection induces DRAM1 by a pathway dependent on the adaptor molecule MYD88 and transcription factor NFKB, which both function downstream of the TLRs that mediate pathogen recognition. DRAM1 is also induced by lipopolysaccharide, a TLR ligand of gram-negative bacteria, suggesting that DRAM1 may function in host defense against a broad range of intracellular pathogens not restricted to mycobacteria.
Infection of zebrafish embryos with Mm recapitulates hallmarks of human TB pathology, including the formation of granulomatous lesions. We found expression of zebrafish dram1 to increase progressively along with the expansion of these lesions. We considered this worthy of further investigation, since patients with active TB have an interferon-inducible transcriptional signature that also includes DRAM1. Mutation of myd88 in zebrafish, but not tp53 mutation, reduces the infection-dependent induction of dram1. Mtb infection of primary human M1 and M2 macrophages also induces DRAM1 expression. Furthermore, both DRAM1 protein and LC3 colocalize with Mtb in these cells. NFKB inhibition abrogates DRAM1 induction in both macrophage types, but MYD88 dependency was observed only in M2 cells. Therefore, in addition to the involvement of MYD88, there is likely a more complex connection between pathogen recognition and autophagy modulation. Furthermore, a regulatory loop between DRAM1-mediated autophagy modulation and inflammation may exist, supported by our observation that knockdown of dram1 in zebrafish strongly increases il1b (interleukin 1, β) expression during mycobacterial infection.
Knockdown of dram1 impairs the ability of zebrafish macrophages to contain Mm growth and leads to severely increased infection at the whole organism level. This knockdown phenotype correlates with a reduction of GFP-Lc3 puncta. In contrast, dram1 overexpression strongly increases the colocalization between GFP-Lc3 and mycobacteria and it reduces TB susceptibility in the zebrafish model. These results indicate that Dram1 functions in defense mechanisms dependent on autophagy or Lc3-associated phagocytosis (LAP). Since both the cytosolic DNA sensor TMEM173 and the ubiquitin receptor SQSTM1 had previously been implicated in autophagic defense against Mtb, we investigated if Dram1 function requires these factors. Knockdown of either tmem173 or sqstm1 can counteract the increase of GFP-Lc3 accumulation due to dram1 overexpression. In agreement with results of others studying Mtb infection in murine cells, it is likely that the TMEM173 homolog of zebrafish is triggered by the escape of Mm bacteria from phagosomes. Escape of Mtb or Mm is dependent on the RD1 virulence locus. Mutation of RD1 prevents GFP-Lc3 accumulation around Mm, and knockdown of dram1 has no effect on infection with RD1 mutant bacteria. Since the escape of virulent mycobacteria will trigger autophagic host defense through TMEM173, SQSTM1, and DRAM1, it is well possible that mycobacteria have evolved strategies to evade this pathway.
Overexpression of dram1 in zebrafish not only increases GFP-Lc3 accumulation but also dramatically increases lysosomal acidification surrounding Mm. Electron microscopy revealed that dram1-overexpressing embryos frequently contain electron-dense compartments of 2–5 μm in size that are surrounded by a single membrane and contain multiple Mm bacteria as well as many fragments of membranes, suggesting that these compartments result from multiple vesicle fusion events. We also observed the fusion of a double-membrane autophagosome with such a larger Mm-containing compartment. This led us to propose that these compartments have the characteristics of late endosomes and that Dram1 promotes their maturation by facilitating multiple fusion events between lysosomes and autophagosomes (). The work of others has highlighted the unique bactericidal properties of autophagosomes, and thus the Dram1-mediated vesicle fusion events might serve to deliver neo-antimicrobial peptides to the Mm-containing compartments. Identification of the interaction partners of Dram1 will be important to elucidate the precise molecular mechanism by which Dram1 facilitates these vesicle fusion events.
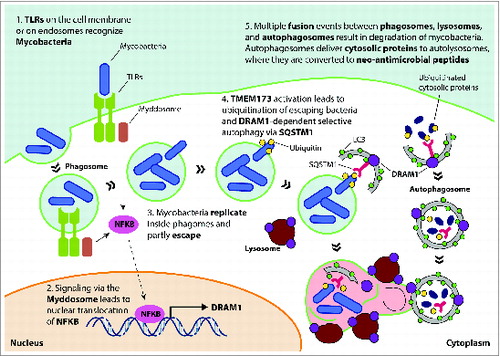
Figure 1. Model for DRAM1 function in host defense against mycobacteria. TLR recognition of mycobacterial ligands leads to formation of the myddosome (a complex consisting of MYD88-IRAKs-TRAF6), which induces DRAM1 transcription via nuclear translocation of NFKB. Mycobacteria replicate in phagosomes and eventually use RD1-dependent virulence factors to partly escape into the cytoplasm. Release of mycobacterial DNA is thought to trigger activation of TMEM173, ubiquitination of mycobacteria, and recognition by the selective autophagy receptor SQSTM1. DRAM1 protein, localizing on lysosomes and autophagosomes, stimulates formation of autophagosomes and promotes maturation of mycobacteria-containing compartments by facilitating their fusion with lysosomes and autophagosomes that may deliver neo-antimicrobial peptides derived from ubiquitinated cytosolic proteins. DRAM1, DNA-damage regulated autophagy modulator 1; TLR, toll-like receptor; MYD88, myeloid differentiation primary response 88, IRAKs, interleukin-1 receptor-associated kinases; TRAF6, TNF receptor-associated factor 6, E3 ubiquitin protein ligase; NFKB, nuclear factor of kappa light polypeptide gene enhancer in B-cells; SQSTM1/p62, sequestosome 1; TMEM173/STING, transmembrane protein 173.
There is currently much interest in the TB field for novel host-directed therapeutic strategies that may complement antibiotic interventions and provide a solution for emerging antibiotic resistances. That Dram1 overexpression is protective against mycobacterial infection in the zebrafish TB model is especially interesting because general autophagy-inducing drugs like Ar-12 and rapamycin worsen TB disease in the same model, probably due to broad side effects of these drugs. Our study suggests that the Dram1-mediated selective autophagy pathway is a potential target for host-directed anti-TB therapy, but it remains to be tested whether DRAM1 overexpression can also reduce bacterial burdens in mammals during progressive or reactivated TB disease. Furthermore, the question is how DRAM1 activity can be stimulated in patients, other than by approaches that rely on adjunctive treatment with TLR ligands to overactivate the entire innate immune response.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank all our co-authors for their valuable contributions to this study.
Funding
AHM and MV were funded by the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Ministry of Education, Culture and Science.