
Abstract
A growing number of innovative mAb therapeutics are on the global market, and biosimilar versions have now also been approved, including in India. Although efficacy and safety is demonstrated prior to approval, targeted pharmacovigilance is essential for the identification and assessment of risk for any mAb products. We analyzed the ADR data related to mAbs reported to the NCC-PvPI through the spontaneous reporting system Vigiflow during April 2011 to February 2014 to identify mAbs with the highest number of ADR including fatal/serious ADR. Only 0.72% reports were related to mAbs. Although 15 mAbs are approved in the country, only 6 mAbs were reported through Vigiflow. Rituximab was highly reported, and no fatal/serious ADR related to any mAbs were reported during the study period. Our study shows that PvPI is effective and robust system in the detection and assessment of risks associated with the use of mAbs.
Abbreviations
| ADR/E | = | adverse drug reactions/event |
| ADR | = | adverse drug reactions |
| AMCs | = | ADR monitoring centers |
| CDSCO | = | Central Drugs Standard Control Organization |
| CLL | = | chronic lymphocytic leukemia |
| DCGI | = | Drug Controller General of India |
| EMA | = | European Medicines Agency |
| EU | = | European Union |
| GEAC | = | Genetic Engineering Appraisal Committee |
| HBV | = | hepatitis B virus |
| ICSRs | = | Individual Case Safety Reports |
| IPC | = | Indian Pharmacopoeia Commission |
| mAb | = | monoclonal antibody |
| mAbs | = | monoclonal antibodies |
| MedDRA | = | Medical Dictionary for Regulatory Activities |
| MHRA | = | Medicines and Healthcare Products Regulatory Agency |
| MoEF | = | Ministry of Environment and Forests |
| MoHFW | = | Ministry of Health and Family Welfare |
| NCC-PvPI | = | National Coordination Centre-Pharmacovigilance Program of India |
| NHL | = | non Hodgkin lymphoma |
| NRA | = | National Regulatory Authority |
| PML | = | progressive multifocal leukoencephalopathy |
| PSUR | = | Periodic Safety Updates Report |
| RCGM | = | Review Committee on Genetic Manipulation |
| rDNA | = | recombinant DNA |
| SOC | = | System Organ Class |
| US | = | Unites States of America |
| USFDA | = | United States Food and Drug Administration |
| WHO | = | World Health Organization |
Introduction
Biologics are drugs derived from living cells, and have high molecular weight and complexity in their structures compared to small molecule drugs. Biologic medicines include recombinant molecules such as mAbs and fusion proteins, as well as other protein therapeutics.Citation1 Compared to small molecule drugs, these biologics are highly specific and they have revolutionized the treatment for the patients with solid tumors or hematological cancers, immune-mediated disorders and neurological diseases.
A number of mAb products have been on the market for over 15 years, and patents for some of these products have expired already or will expire soon. Thus, manufacturers are producing similar biologic products, which, as the name implies, are similar but not identical to the original product due to variations that can occur in the manufacturing process. A similar biologic in India is defined as,’ A biological product/ drug produced by genetic engineering techniques and claimed to be “similar” in terms of safety, efficacy and quality to a reference biologic, which has been granted a marketing authorization in India by DCGI on the basis of a complete dossier, and with a history of safe use in India’.Citation2
In India, new biologics and similar biologics undergo stringent regulatory processes to gain marketing approval. The process includes approval from the RCGM,Citation2,Citation3 which functions in the Department of Biotechnology, Ministry of Science and Technology; the GEAC,Citation2,Citation3 which functions under the MoHFW; and the CDSCO, which is headed by the DCGI and is the apex regulatory body under MoHFW.Citation2 The DCGI is responsible for the approval of new drugs, including biologics, in India.
Approximately 40 biologics and similar biologics have been approved for marketing or import in India by DCGI since 1999, among which 15 were mAbs. Citation4 To ensure the safety of drugs in India, the IPC was given the mandate to act as the NCC for the PvPI starting on April 15, 2011.Citation5The aim of this study was to review ADRs associated with therapeutic mAbs reported to NCC-PvPI, with a focus on the severe complications that are associated with the use of therapeutic mAbs and that require critical care.
Results
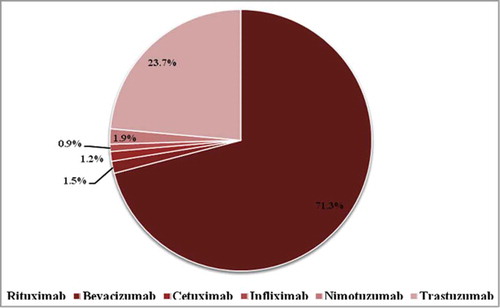
Data from India reported to Vigiflow from April 15, 2011 to February 28, 2014 were analyzed. Of 58827 ICSRs pertaining to drugs of chemical and biological origin reported to Vigiflow during the study period, 424 (0.72%) ICSRs were related to therapeutic mAbs. The 15 mAbs approved for marketing in India by DCGI are shown in . Of these, both innovator and similar biologics of rituximab, trastuzumab and infliximab are approved; however, the infliximab biosimilar product was approved after data collection for this report had concluded. The Vigiflow database with respect to India showed only the reports of 6 therapeutic mAbs: rituximab (71.3%), infliximab (0.9%), bevacizumab (1.5%), cetuximab (1.2%), nimotuzumab (1.9%) and trastuzumab (23.7%) (), including innovator and similar biologic products.
Table 1. Status of biologic and similar biologic mAbs in various countries and their therapeutic indications
Figure 1. Adverse drug reactions of therapeutic mAbs reported to NCC-PvPI through Vigiflow.
ADRs reported as per the SOC of the MedDRA are shown in . Data collected from Vigiflow through February 28, 2014 showed 473 ADRs related to rituximab from India, including reports with concomitant drugs with reaction level preferred. When classified by system organ, ADRs over 5% were skin and appendages disorders (17.1%; n = 81), including alopecia (8.8%; n = 42); central and peripheral nervous system disorder (6.5%; n = 31), including peripheral neuropathy (1.8%; n = 9) and fever convulsions (1.4%; n = 7); psychiatric disorders (5.8%; n = 28), including anorexia (4.2%; n = 20); gastro-intestinal disorders (19.6%; n = 93), including constipation (5%; n = 24), nausea (2.7%; n = 13) and vomiting (4.6%; n = 22); white cell and reticuloendothelial system disorders (6.3%; n = 30), including leucopenia (2.3%; n = 11); ‘body as a whole – general’ disorders (25.8%; n = 122), including allergic reaction (2.9%; n = 14), asthenia (2.9%; n = 14), fever (4%; n = 19), and rigors (11.7%; n = 56) were reported.
Table 2. ADR of therapeutic mAbs with respect to primary SOC of MedDRA reported to NCC-PvPI through Vigiflow
The ADR reporting data from India showed that 10 (1.5%) and 153 (23.07%) ADRs were reported for bevacizumab and trastuzumab, respectively. Maximum ADRs reported for both bevacizumab and trastuzumab were related to gastro-intestinal system disorders (40%; n = 4 and 32.6%; n = 50, respectively; ), in particular diarrhea. ADR reports on bevacizumab and trastuzumab to NCC-PvPI did not show any fatal/serious events.
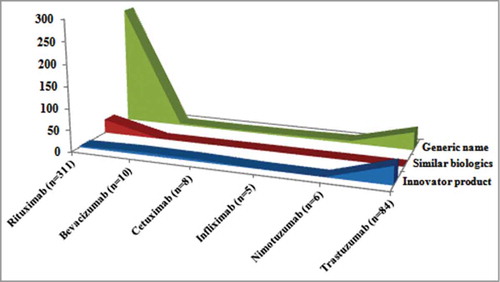
shows that 99–100% of the reports indicated that the relation to drug was suspected. As 5 of the 6 mAbs are treatments for cancer, some of the ADRs may also be due to concomitant use of chemotherapeutic agents. Out of 473 ADRs reported for rituximab, 98.9% (468) were possible and no report was fatal/serious. Pharmacovigilance data collected from NCC-PvPI during the study period showed that, out of 473 ADRs reported, 1% (n = 5) and 6.7% (n = 32) were specifically reported as related to exposure to innovator and similar biologic rituximab, respectively (). The remaining 92% (n = 436) were reported only with the international non-proprietary name as rituximab, which thus may include ADRs from either innovator or similar biologic rituximab.
Table 3. Summary of reported ADRs due to the use of therapeutic mAbs by profile and gender
Table 4. Pharmacovigilance data of rituximab
shows that the maximum ADR reports (79%) were reported by the generic name of the drug, and only 13.4% reports were classified as innovator products and 7.5% as similar biologics. The data clearly justifies the need for including the brand name of the drug while reporting the ADRs to NCC-PvPI. The ADR reporting form of NCC-PvPI (especially for biologics) needs to be amended to include proprietary name (brand) name, international non-proprietary name, batch/lot number, manufacturer's name, country of origin, lack of efficacy, less effective and over effective,Citation6,Citation7 so that safety and efficacy difference among the products and manufacturing batch can be monitored and actions can be taken specifically.
Figure 2. Pharmacovigilance data of reference and similar biologics mAb reported to NCC-PvPI.
Discussion
Indian biopharmaceutical companies started establishing a solid foothold in the biologics market in 1997 after the introduction of recombinant hepatitis B vaccine in the Indian market.Citation8 Biologics are heterogeneous and may contain aggregated, oxidized, deamidated molecules, as well as various glycoforms, that could affect the safety and efficacy of the product.Citation9 Pharmacovigilance of biologics and similar biologics is thus prudent to ensure positive outcomes for patients.
Therapeutic mAbs have been available to Indian patients since 1999, and these products have revolutionized the treatment for various life-threatening disorders. The mAbs provide targeted therapy and are generally considered to be safe, but the risk-benefit can be effectively analyzed only by using an effective post-marketing pharmacovigilance system in the country. ADR data reported to NCC-PvPI until February 2014 were analyzed. During the study period, only 0.72% (n = 424) of ICSRs submitted to PvPI related to therapeutic mAbs, and these were only with respect to 6 mAbs, although 15 mAbs are approved in India. Among 424 ICSRs, ADRs due to rituximab were highly reported. The data were analyzed and summarized in the study for relatedness to product, gender and primary SOC.
The originator and similar rituximab products approved for marketing in India target CD20 on B-lymphocytes and cause depletion of these immune system cells.Citation4 Rituximab is therefore used for the treatment of diseases mediated by abnormal functioning of B cells, e.g., hematological cancers, rheumatoid arthritis. The reports for the 6 mAbs that we examined were predominantly about rituximab, but there were no reports on virus reactivation. Various studies have reported ADRs such as listeriosis, reactivation of latent tuberculosis, hepatitis B or C or opportunistic infectionsCitation10,Citation11 and PMLCitation12 on treatment with rituximab. The USFDA issued a revision in the label of rituximab to have new ‘Boxed Warning’ information about the risk of reactivation of HBV infection in 2013.Citation13 It also notes that viral reactivation of JC virus leads to PML and reactivation of HBV leads to fulminant hepatitis, hepatic failure and death.Citation14 The UK's MHRA also recommended screening for hepatitis B infection in all patients before starting treatment with rituximab.Citation15
According to Schedule Y of the Drugs and Cosmetics Act 1940, post-marketing surveillance should be conducted and the PSURs should be submitted to regulatory authority every 6 months initially for a period of 2 y and then annually for another 2 years, serious and unexpected ADRs should be reported within 15 d of its report.Citation20 Post-marketing pharmacovigilance plans should include reporting of ADRs to NCC-PvPI, apart from submission of PSURs to the NRA, which would allow amalgamation of PSUR submission with PvPI.
Similar biologics of rituximab, trastuzumab and infliximab have been approved by DCGI, and these products require stringent pharmacovigilance due to potential differences in quality attributes. As per the Guidelines on Similar biologics, preclinical and clinical data can be reduced by showing comparability with reference products,Citation2 but a notice can be issued by the drug regulator for additional safety monitoring to assess the risk-benefit ratio of biologic/similar biologic mAbs in the country.
Our study highlighted that therapeutic mAbs may be under-reported to the national pharmacovigilance system. Because there is limited understanding about the safety of new molecules, particularly in case of similar biologic mAbs, targeted pharmacovigilance is needed. Analysis of individual risk-benefit through targeted spontaneous reporting is highly recommended. Ultimately, this may also help manufacturers to produce safe and effective therapeutic mAbs for patients and help physicians to understand the risks associated with mAbs e.g., in prescribing to patients with histories of latent infections. This study underlines the role of AMCs in identifying local factors that may increase the reporting of ADRs. Instructions issued by the regulator regarding targeted spontaneous reporting and additional transparent safety monitoring are required to ensure the safe and effective use of therapeutic mAbs in the country.
NCC-PvPI and the CDSCO should work together to initiate a culture of reporting of ADR in patients treated with similar biologics, which are under-reported. PSURs on therapeutic mAbs submitted to CDSCO are to be reviewed, and regulatory intervention can be made, or, as with other NRA, drug alerts or warnings may be issued. As NCC-PvPI has already started targeted spontaneous reporting of certain drugs, these biologics and similar biologics also need to be included, so that regulatory intervention on similar biologics based on an Indian database would be more meaningful. NCC-PvPI and CDSCO should sensitize the stakeholders to the fact that intensive monitoring is required to collect ADR/AE on similar biologics, particularly mAbs.
Methodology
The study was a retrospective analysis of ICSRs from India related to therapeutic mAbs reported through spontaneous online reporting system Vigiflow, developed by the Uppsala Monitoring Centre, the World Health Organization's collaborating center for international drug monitoring. ADR/Es reported to NCC-PvPI, IPC, Ghaziabad, India were studied for the period of one year and 9 months, from 15th April 2011 to 28th February 2014. The NCC has been associated with 150 multi-specialty hospitals as AMCs throughout India to monitor the safety of drugs. The data were collected from the reports with/without concomitant drugs and with reaction level preferred. The data were analyzed with respect to the primary SOC as per the MedDRA for age and gender of the patients, the severity of the report and the relatedness of the product using ‘Search and statistics’ tool in Vigiflow.Citation21
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- World Health Organization Consultation. Quality control of biologicals produced by recombinant DNA techniques. Bull World Health Organ 1983; 61(6): 897-911; PMID:6609009
- Guidelines on Similar Biologics. Regulatory Requirements for Marketing Authorization in India. Central Drugs Standard Control Organisation; 2012. Accessed on 30 January 2014. Available from: www.cdsco.nic.in
- Rules for the manufacture, use, import and storage of hazardous microorganisms, Genetically engineered organisms or cells. 1989. Ministry of Environment and Forestry, Government of India, GSR 1037 (E), Environment (Protection) Act, 1986.
- Central Drugs Standard Control Organization. Accessed on 11 February 2014. Available from: www.cdsco.nic.in
- Kalaiselvan V, Jai Prakash, Gyanendra Nath Singh. Pharmacovigilance Programme of India. Arch Pharm Pract 2012; 3(3):229-232; http://dx.doi.org/10.4103/2045-080X.116605
- Shanthi N Pal, Chris Duncombe, Dennis Falzon, Sten Olsson.WHO Strategy for Collecting Safety Data in Public Health Programmes: Complementing Spontaneous Reporting Systems. Drug Saf 2013; 36(2): 75-81; PMID:23329541; http://dx.doi.org/10.1007/s40264-012-0014-6
- WHO Expert Committee on Biological Standardization. Sixtieth report. WHO Technical Report Series 977. Geneva, Switzerland: WHO Press.
- Sarah E Frew, Rahim Rezaie, Stephen M Sammut, Monali Ray, Abdallah S Daar, Peter A Singer. India's health biotech sector at a crossroads. Nat Biotechnol 2007; 25:403-17; PMID:17420744; http://dx.doi.org/10.1038/nbt0407-403
- Hui F Liu, Junfen Ma, Charles Winter, Robert Bayer. Recovery and purification process development for monoclonal antibody production. MAbs 2010; 2(5): 480-99; PMID:20647768; http://dx.doi.org/10.4161/mabs.2.5.12645
- Rubbert-Roth A. Assessing the safety of biologic agents in patients with rheumatoid arthritis. Rheumatology (Oxford) 2012; 51(5):38-47; http://dx.doi.org/10.1093/rheumatology/kes114
- Bodro M, Paterson DL. Listeriosis in patients receiving biologic therapies. Eur J Clin Microbiol Infect Dis 2013; 32(9):1225-30; PMID:23568606; http://dx.doi.org/10.1007/s10096-013-1873-1
- Ruderman EM. Overview of safety of non-biologic and biologic DMARDs. Rheumatology (Oxford) 2012; 51(6):37-43; PMID:22075064; http://dx.doi.org/10.1093/rheumatology/kes283
- U.S. Food and Drug Administration. Accessed on 14 February 2014. Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformation-forPatientsandProviders/ucm109106.htm
- U.S. Food and Drug Administration. Accessed on 14 February 2014. Available from: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformation-forPatientsandProviders/ucm109106.htm
- Pal S. WHO Pharmaceuticals Newsletter 2014; 1:8.
- European Medicines Agency. Accessed on 11 February 2014. Available from: www.ema.europa.eu
- U.S. Food and Drug Administration. Accessed on 12 February 2014. Available from: www.fda.gov.
- Australian Government Department of Health. Therapeutic Goods Administration. Accessed on 12 February 2014. Available from: www.tga.gov.au.
- Pharmaceuticals and Medical Devices Agency. Accessed on 16 February 2014. Available from: www.pmda.go.jp
- Drug and Cosmetic Act, 1940 and Rules there under. Ministry of Health and Family Welfare, Government of India.
- Uppsala Monitoring Centre. Accessed on 17 February 2014. Available from: www.who-umc.org.