
Abstract
A promising vaccine strategy for the treatment of cancer involves the use of vaccines incorporating tumor antigen-derived synthetic peptides that can be coordinately recognized by specific CD4+ and CD8+ T-cells. Previously, we reported that a MAGE-A6-derived peptide (MAGE-A6172–187) and its highly-immunogenic and cross-reactive homolog derived from Mycoplasma penetrans HF-2 permease (HF-2216–229) are promiscuously presented by multiple HLA-DR alleles to responder CD4+ T-cells obtained from healthy donors and melanoma patients. Here, we investigated whether these same peptides could concomitantly stimulate cross-reactive MAGE-A6-specific CD8+ T-cell responses in vitro using cells isolated from HLA-A*0201 (HLA-A2)+ healthy individuals and patients with melanoma. We now show that MAGE-A6172–187 and, even more so, HF-2216–229, induce memory CD8+ T cells that recognize HLA-A2+ MAGE-A6+ tumor target cells. The immunogenicity of these peptides was at least partially attributed to their embedded MAGE-A6176–185 and HF-2220–229 “homologous” sequences. The functional avidity of HF-2216–229 peptide-primed CD8+ T cells for the MAGE-A6172–187 peptide was more than 100-fold greater than that of CD8+ T cells primed with the corresponding MAGE-A6 peptide. Additionally, these 2 peptides were recognized in interferon γ (IFNγ) and granzyme B ELISPOT assays by CD8+ T-cell clones displaying variable T-cell receptor (TCR) Vβ usage. These data suggest that the immune cross-reactivity of the MAGE-A6172–187 and HF-2216–229 peptides extends to CD8+ T cells, at least in HLA-A2+ donors, and supports the potential translational utility of these epitopes in clinical vaccine formulations and for immunomonitoring of cancer patients.
Abbreviations::
- AdV, recombinant adenoviral vector
- APC, antigen presenting cell
- CTL, cytotoxic T lymphocyte
- EBV, Epstein-Barr virus
- FBS, fetal bovine serum
- HD, healthy donor
- HLA, human leukocyte antigen
- HPLC, high-performance liquid chromatography
- iDC, immature dendritic cells
- IVS, in vitro stimulation
- MACS, Magnetic-Activated Cell Sorting
- mDC, mature dendritic cells
- MOI, multiplicity of infection
- PBMC, peripheral blood mononuclear cell
- PFU, plaque forming units
- RT-PCR, reverse transcription polymerase chain reaction
- TAA, tumor associated antigen
- TCM, T cell media
- TCR, T-cell receptor
Introduction
The immune system is capable of mediating the spontaneous regression of solid human tumors, most notably, melanoma and renal cell carcinoma (RCC),Citation1,2 with tumor-specific CD8+ cytotoxic T lymphocytes (CTL) implicated as major antitumor effector cells.Citation3-6 CTL development is typically supported by coordinate helper CD4+ T-cell responses that regulate the quality, magnitude, and durability of CD8+ CTL-mediated immunity in vivo.Citation7-9 In particular, Type-1 helper T-cell responses –distinguishable by virtue of interferon γ (IFNγ) expression– have been shown to facilitate the cross-presentation of tumor-associated antigens (TAAs) by appropriately “licensing” host antigen presenting cells (APCs) and allowing for longitudinal diversification in the evolving antitumor T-cell repertoire.Citation10,11 These studies support the therapeutic need to induce both CD8+ and CD4+ T-cell responses against tumor-associated antigens to successfully induce objective clinical responses in cancer patients.Citation10
In order to achieve immunotherapeutic benefit in the cancer setting using a vaccine approach, it is essential to include appropriate TAAs and/or TAA-derived epitopes. Since most known TAAs are non-mutated “self” antigens, immune tolerance mechanisms or regulatory circuits must be overridden in order to mount robust antitumor responses in vivo. The melanoma antigen (MAGE) gene family of proteins was originally identified as TAAs that could be recognized by CD8+ CTLs, which could then be isolated and expanded from the peripheral blood of cancer patients.Citation12 MAGE proteins are expressed normally by germline cells and in placenta,Citation13,14 however under conditions that favor DNA hypomethylation (e.g. as found in the pro-tumor niche), MAGE protein expression can be induced.Citation15 Expression of MAGE-A family proteins has been reported for more than half of all human cancers.Citation16 One of its members, MAGE-A6, is expressed in more than 60% of melanomas,Citation17 30% of renal cell carcinomas,Citation18 and by numerous other malignancies, such as breast, esophageal, head and neck, bladder, and lung carcinomas.Citation17,Citation19-22 These expression patterns support MAGE proteins/peptides as attractive constituents in the design of therapeutic cancer vaccines.
Most TAA-specific T lymphocytes are believed to undergo thymic and peripheral clonal selection,Citation23 which limits the protective capacity of the TAA-specific T-cell repertoire in cancer patients. One strategy that can break this tolerance is to employ T-cell cross-reactivity against environmental pathogens via the process of epitope mimicry.Citation24,25 Viral myocarditis, Lyme disease, rheumatoid arthritis, multiple sclerosis, and virus-induced autoimmune diabetes are autoimmune diseases that are considered to be initiated or aggravated by immune challenge with microbial pathogens.Citation24-28 Consistent with this model, we have previously shown that a MAGE-A6-derived peptide, MAGE-A6172–187, and a peptide derived from the permease protein of the ubiquitous Mycoplasma penetrans HF-2 bacterium (HF-2216–229) share a high-degree of structural homology and immunologic cross-reactivity in the CD4+ T-cell compartment present in a broad range of healthy donors and melanoma patients.Citation29
We now show that these same MAGE-A6172–187 and HF-2216–229 peptides can induce a cross-reactive, polyclonal CD8+ T-cell repertoire in HLA-A*0201 (HLA-A2)+ healthy individuals and melanoma patients that recognizes MAGE-A6+ tumor cells in vitro. These findings suggest that MAGE-A6172–187 and HF-2216–229 peptides will be able to coordinately promote MAGE-A6-reactive CD8+ and CD4+ T-cell responses in a cohort of HLA-A2+ melanoma patients, making these epitopes useful for immunomonitoring in patients harboring MAGE-A6+ tumors and potential therapeutic vaccine design in the future.
Results
Healthy donor CD8+ T cells stimulated with MAGE-A6172–187 and HF-2216–229 demonstrate cross-reactivity to both peptides and recognize HLA-matched MAGE-A6+ tumor cell lines
MAGE-A6172–187 and HF-2216–229 peptides were tested in vitro for their ability to stimulate cross-reactive CD8+ T-cell responses among T lymphocytes from HLA-A2+ healthy donors (HDs). These peptides were shown to drive expansion of peptide-specific CD8+ T-cell responders from the majority of HDs tested as determinedby IFNγ ELISPOT assays ( and Table SI). In the case of HD6 and 7, cross-reactive Type-1 (Tc1) responses to both peptides were induced using either of the 2 peptides, whereas HD4 T cells primed with both peptides yielded Tc1 responses only against MAGE-A6172–187. In 3 donors, only one of the 2 peptides displayed the capacity to induce cross-reactive CD8+ T-cell responses to their homolog. Only MAGE-A6172–187 was able to induce CD8+ T-cell responses to HF-2216–229 from HD2 and HD3, whereas only HF-2216–229 was able to induce CD8+ T-cell responses to MAGE-A6172–187 from donor HD1. Of the 8 donors tested, only HD8 CTLs were unresponsive to the 2 peptide homologues. These data suggest that these peptides have distinctive, yet interrelated immunogenic properties that may complement one another.
Figure 1. Healthy donor CD8+ T cells stimulated in vitro with MAGE-A6172–187 and HF-2216–229 peptides cross-react to both peptides and recognize HLA-matched MAGE-A6+ tumor cells. CD8+ T cells purified from healthy donor (HD) peripheral blood underwent a single round of in vitro stimulation with the following autologous mature dendritic cell (mDC) groups: unpulsed (negative control), MAGE-A6172–187 (M6.172; M6) or HF-2216–229 (HF2.216; HF) peptide-pulsed. The relative frequencies of peptide- and tumor-specific CD8+ T-cell responders primed with the indicated peptide were measured in interferon γ (IFNγ) and granzyme B ELISPOT assays. (A) CD8+ T cells were screened for their ability to produce IFNγ in response to autologous antigen presenting monocytes pulsed with the indicated peptides, and (B) for their ability to produce IFNγ and granzyme B in response to HLA-A2+ MAGE-A6+ Mel526, SLM2, and WS-LCL cell lines. Background levels of IFNγ and granzyme B produced by CD8+ T cells stimulated with unpulsed mDC and induced by autologous monocytes or allogeneic tumor cell targets were subtracted from the presented peptide- and tumor-specific responses, respectively. Spot counts greater than 10 over the control were considered to be antigen-specific (dotted lines in (A) and (B) represent the cut-off) and the number of reactive spots per 105 CD8+ T cells are shown. Eight healthy HLA-A2+ donors (HD) were evaluated. Note: for HD5, insufficient numbers of cells were obtained after MAGE-A6172–187 stimulation to allow for testing of peptide cross-recognition; for HD2, insufficient cell numbers were obtained after peptide stimulations to allow for testing of WS-LCL recognition. Unmanipulated ELISPOT data presented in this figure are presented in Table SI and Figure S2.
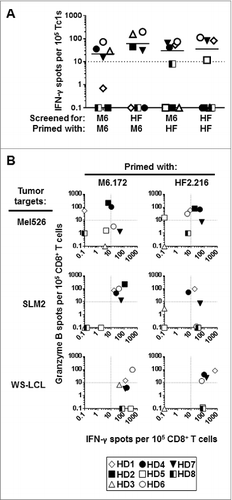
Next, we tested the ability of peptide-stimulated CD8+ T cells to recognize naturally-processed MAGE-A6 epitopes presented on HLA-A2+ MAGE-A6+ melanoma (Mel526 and SLM2) and EBV-transformed B (WS-LCL) cell targets (Fig. S1A and S1B). Both MAGE-A6172–187 and HF-2216–229 peptide-primed CD8+ T effector cells coordinately recognized all 3 tumor cell lines as evinced by IFNγ and granzyme B ELISPOT assays ( and Fig. S2). The quality and magnitude of responses against tumors varied with respect to the peptide used in IVS, the T cell donor, and the tumor cell target tested. MAGE-A6172–187 peptide elicited detectable T-cell responsiveness against Mel526 tumor cells in 6/8 donors tested. Specifically, 2 responders secreted only granzyme B (HD1 and HD2), one secreted both IFNγ and granzyme B (HD4), and 2 secreted IFNγ only (HD6 and HD7). In contrast, HF-2216–229 peptide promoted mixed IFNγ/granzyme B responses among T lymphocytes derived from 2 donors (HD2 and HD4), and uniquely induced granzyme B secretion from HD1 and HD6 T cells. HD5-derived T cells were minimally responsive to either peptide. Regarding T-cell responses against the SLM2 tumor, MAGE-A6172–187-based IVS induced mixed IFNγ/granzyme B secretion from T cells isolated from 5/8 donors tested (HD1, HD2, HD4, HD6, HD7). On the other hand, HF-2216–229-based IVS induced mixed IFNγ/granzyme B secretion from HD1-derived T lymphoctes, IFNγ-only responses from T cells from 4 donors (HD2, HD6–8), and granzyme B-only production from HD4 donor T cells. IVS-activated Tc1 responses to WS-LCL also displayed unique profiles. MAGE-A6172–187-sensitized CD8+ T-cells reacted against WS-LCL target cells based on secretion of IFNγ in 7/7 donors tested (HD1, HD3-HD8) and granzyme B production in 2/7 donors tested (HD1 and HD6). Conversely, HF-2216–229-sensitized CD8+ T cells responded to WS-LCL in 6/7 donors tested, including secretion of mixed IFNγ/granzyme B from 4/7 donor T cells tested (HD1, HD4, HD6 and HD7), and production of only IFNγ from T cells derived from donors HD5 and HD8. Overall, these data suggest that both the MAGE-A6172–187 and HF-2216–229 peptides can elicit tumor-specific CD8+ T-cell responses important for recognition of naturally-processed MAGE-A6 epitopes in HLA-A2+ individuals, and that they may stimulate an at least partially-overlapping, “complementary” Tc1 repertoire from a range of HLA-A2+ donors evaluated.
MAGE-A6176–185 and HF-2220–229 epitopes are recognized by CD8+ T cells stimulated with homologous long peptides
We evaluated the MAGE-A6172–187 and HF-2216–229 peptides using an epitope prediction algorithm for HLA-A2-binding peptides. Two homologous decamers with maximal predicted HLA-A2 binding affinity (MAGE-A6176–185 and HF-2220–229) were synthesized () and analyzed for their ability to stabilize HLA-A2 complexes on the surface of T2 cells in vitro. The binding affinities of these 2 peptides were comparable, but modest when compared to the high-affinity HLA-A2-binding peptide FluM158–66 (). CD8+ T cells primed with MAGE-A6172–187 and HF-2216–229 peptides were subsequently tested for their native ability to recognize the 2 decamers using IFNγ ELISPOT assays. While the magnitude of responses to shorter MAGE-A6176–185 and HF-2220–229 peptides varied among the donors evaluated, the recognition patterns generally matched those determined for the longer MAGE-A6172–187 and HF-2216–229 sequences ( and S2; Table SI). This is particularly true in the case of recognition by MAGE-A6172–187-stimulated CD8+ T cells isolated from donors HD3 and HD4–7. Likewise, MAGE-A6172–187-stimulated Tcells that recognized the long HF-2216–229, also recognized the HF-2220–229 decamer, albeit at different frequencies (HD2–3, HD6–7). Similar peptide recognition trends were observed for HF-2216–229–stimulated CD8+ T cells. Of note, we observed MAGE-A6176–185 decamer-specific responses in 2 donors that previously did not respond to MAGE-A6172–187 (HD2 and HD5), though at the lower end of the detection range. Interestingly, we observed HF-2220–229 decamer-specific responses within the HD8 T cell repertoire, which previously failed to respond to HF-2216–229.
Table 1. MAGE-A6172–187 and HF-2216–229 long peptides encode for HLA-A2-restricted homologous epitopes
Figure 2. Short homologous MAGE-A6176–185 and HF-2220–229 epitopes are recognized by CD8+ T cells stimulated with the corresponding long peptide variants. Homologous MHC Class I-binding epitopes within these peptides (MAGE-A6176–185 and HF-2220–229; ) were identified using the SYFPEITHI epitope prediction algorithm. The peptides were chemically synthesized and tested for HLA binding and immunogenicity. (A) MAGE-A6176–185 (M6.176) and HF-2220–229 (HF2.220) binding affinities for HLA-A2 were determined using the T2-based HLA-A2-binding assay. T2 cells were loaded with titrated amounts of peptide (≤50 μg /mL, as indicated). Cells were stained with anti-HLA-A2 antibody and fluorophore-conjugated secondary. Mean fluorescence index (MFI) was determined by fluorescence cytometry. (B) CD8+ T cells purified from healthy donor (HD) peripheral blood primed with long peptides and evaluated in were screened for their ability to recognize autologous monocytes ± short M6.176 (sM6) and HF2.220 (sHF) using the interferon γ (IFNγ) ELISPOT assay. Data presented were adjusted for non-specific IFNγ production by CD8+ T cells, and spot counts greater than 10 over control were considered to be antigen-specific (dotted line represents the cut-off). (C-D) To further confirm the immunogenicity of the MAGE-A6176–185 and HF-2220–229 peptides, the peptides were compared to their long counterparts for their ability to sensitize CD8+ T cells against naturally-processed MAGE-A6. (C) MAGE-A6 protein expression of immature dendritic cells (iDCs) transduced with AdV.LacZ (DC.LacZ) or AdV.MAGE-6 (DC.MAGE-A6; MOI 250) shown by Western blot. Recombinant MAGE-A6 (rMAGE) was used as the positive control. Results are reflective of data obtained using 2 healthy donors. (D) CD8+ T-cells, which underwent a single round of in vitro stimulation with peptide-pulsed mature dendritic cells (mDC), were tested against autologous DC.LacZ or DC.MAGE-A6 using the IFNγ ELISPOT assay. Data are representative of 3 independent experiments. Mean ± SEM values are presented; statistical analyses were performed by single-tailed Student's T test; p < 0.05.
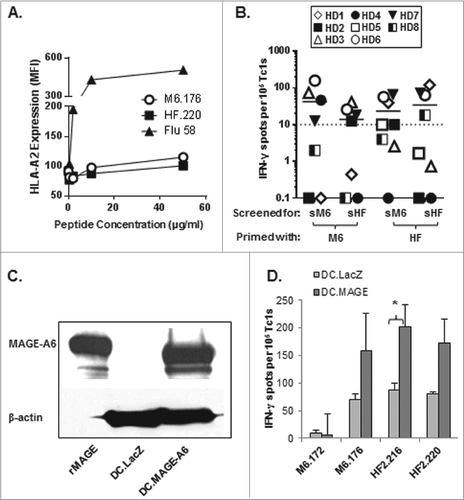
To further confirm the immunogenicity of the short epitopes, MAGE-A6176–185 and HF-2220–229 peptides were compared to their long counterparts for their ability to sensitize CD8+ T cells against naturally-processed MAGE-A6 using the IFNγ ELISPOT assay. CD8+ T cells were tested against autologous DCs transduced with AdV.LacZ (adenovirus expressing irrelevant LacZ antigen) or AdV.MAGE-A6 (). We found that DCs infected with AdV.MAGE-A6, but not AdV.LacZ, were recognized by the peptide-primed CD8+ T cells. When taken together, these data suggest that both the MAGE-A6176–185 and HF-2220–229 peptides effectively stimulate CD8+ T cells reactive against HLA-A2+ MAGE-A6+ tumor cells in vitro.
Long peptides, particularly HF-2216–229, induce cross-reactive CD8+ T-cell recall responses in vitro
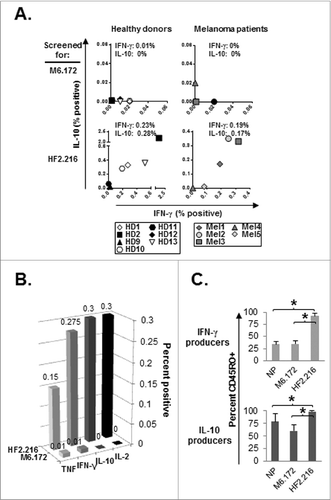
We observed that 7/8 healthy donors tested ( and Table SI) were able to mount Tc1 responses against the MAGE-A6172–187 and/or HF-2216–229 peptides after only a single round of IVS employing either of these 2 homologues. We hypothesized that these responses were mediated by cross-reactive memory CD8+ T cells that had been naturally primed in vivo against the environmental bacteria Mycoplasma penetrans, which encodes the HF-2216–229 peptide. To evaluate this, we examined MAGE-A6172–187 and HF-2216–229 memory CD8+ T-cell responses among T cells obtained from healthy donors as described in Materials and Methods (Fig. S3). When used as stimuli, the MAGE-A6172–187 and HF-2216–229 peptides induced unique patterns of “recall” cytokine secretion (). The first major difference was in the frequency and magnitude of Tc1 responses to the 2 peptides. Peptide-specific cytokine responses were more frequent and stronger when induced by HF-2216–229 than by MAGE-A6172–187. Tc1 memory response to HF-2216–229 was detectable in 6/6 healthy donors evaluated, while the MAGE-A6172–187 recall response was identified in 4/7 healthy donors. On average, HF-2216–229 peptide induced approximately 20-fold greater frequency of responders when compared to the MAGE-A6172–187 peptide. The second difference involved the quality of the T-cell responses evoked by the individual IVS procedures. The MAGE-A6172–187 peptide induced primarily Type-1 (IFNγ) responses, whereas the HF-2216–229 peptide promoted mixed Type-1/regulatory recall responses based on coordinate IFNγ/IL-10 production. This distinctive cytokine-secretion pattern of HF-2216–229-specific CD8+ T cells extended beyond IFNγ/IL-10 production patterns. Cytofluorimetric analyses further showed that, unlike MAGE-A6172–187-based stimulation, activation with the HF-2216–229 peptide induced high frequencies of IL-2-producing CD8+ T cells, which are indicative of memory responses ().Citation30 To confirm this, we evaluated CD45RO expression on IFNγ and IL-10-producing CD8+ T-cell responders (Fig. S3 and 3C). HF-2216–229-specific cells, but not MAGE-A6172–187-specific, were almost entirely CD45RO+, suggesting that these responders were primarily recruited from the memory pool of the CD8+ T cells.
Figure 3. CD8+ T cell memory responses to MAGE-A6172–187 and HF-2216–229 long peptides. (A) Healthy donor (HD) or melanoma (Mel) patient-derived CD8+ T-cell memory responses were evaluated against MAGE-A6172–187 (M6.172) or HF-2216–229 (HF2.216) peptides. CD8+ T cells purified from fresh healthy donor or melanoma patient PBMCs were co-cultured with autologous monocytes and 10μg/mL peptides for 1.5 h, followed by 10ug/mL brefeldin A for an additional 3.5 h. CD8+CD3+cytokine+ populations were evaluated by immunofluorescence staining and flow cytometry. Non-specific cytokine responses were subtracted from the presented peptide-specific responder percentages. Median percentages of peptide-specific cytokine responders are reported in interferon γ (IFNγ) vs. IL-10 dot-plots. Note: for HD12, insufficient numbers of cells were obtained to allow for testing of HF-2216–229-specific memory responses. ((B) and C) Overall CD8+ T-cell cytokine responses against MAGE-A6172–187 or HF-2216–229 peptide, and CD45RO staining on cytokine-positive cells from healthy donors were also measured. CD8+ T cells isolated from 7 healthy donor PBMCs were tested for production of multiple cytokines by intracellular staining and CD45RO expression levels via fluorescence cytometry. (B) Median percentages of peptide-specific cytokine-positive responders are shown for TNF, IFN-γ, IL-10, and IL-2. (C) Percentage of CD45RO+ memory cells among IFNγ+ or IL-10+ CD8+ T-cell responders. Median percentage values are shown;statistical analyses were performed by single-tailed Student's T test; *p < 0.05.
MAGE-A6172–187- or HF-2216–229-specific memory CD8+ T cells in advanced-stage melanoma patients
Tumors inhibit antitumor T-cell immunity using a broad range of immunosuppressive mechanisms.Citation31 As such, we assessed whether the magnitude and balance of MAGE-A6172–187- and HF-2216–229–specific CD8+ T-cell memory responses in healthy individuals and advanced-stage melanoma patients varied (). MAGE-A6172–187-reactive memory responses were rare (2/5 patients) and lacked Type-1 functional skewing as we had observed in healthy normal HLA-A2+ donors (; Mel4 produced IL-10, while Mel5 produced IFNγ). Similar to healthy donors, HF-2216–229–based stimulation of melanoma patient-derived CD8+ T-cells induced mixed IFNγ/IL-10 production from 4/5 patients tested. These responses were on average 17-fold greater as compared to those elicited by MAGE-A6172–187 stimulation of CD8+ T lymphocytes from melanoma patients, based on the frequency of cytokine producing CD8+ T cells.
Table 2. HLA-A2+ healthy donor and melanoma patient demographic and clinical characteristics.
A further investigation of the functional capacity of Tc1 after MAGE-A6172–187 and HF-2216–229 peptide-based IVS of T lymphocytes from advanced-stage melanoma patients revealed that MAGE-A6172–187- and HF-2216–229-primed CD8+ T cells less effectively recognized MAGE-A6+ tumor cells than primed CD8+ T cells from health donors (, S2 and 4). Similar to healthy donor-derived T lymphocytes, the quality and magnitude of CD8+ T-cell responses varied with respect to the peptide used for IVS, the individual donor evaluated, and the tumor cell target tested. With exception of patient Mel6, the antitumor responses induced in melanoma patients were mostly IFNγ-skewed. The melanoma cell line SLM2 was poorly recognized in general. HF-2216–229 peptide-stimulated T cells from patient Mel4 responded by selectively producing IFNγ, in contrast to either MAGE-A6172–187- or HF-2216–229-stimulated CTLs from patient Mel6 that secreted granzyme B or mixed IFNγ/granzyme B, respectively. When evaluating T-cell responses against the cell line Mel526, the MAGE-A6172–187 peptide was found to induce antitumor immunologic response among T cells from patients Mel5 and Mel6, whereas the HF-2216–229 peptide stimulated responses in 4/5 donors tested (Mel3–6). Both patient Mel5 and Mel6 T cells induced by either peptide produced granzyme B, contrasting with T cells from patients Mel3 and Mel4 that responded to HF-2216–229 stimulation by exclusively producing IFNγ. Conversely, 4/5 patients stimulated with MAGE-A6172–187 (Mel3–6) produced IFNγ in response to the EBV-transformed B-lymphoblastoid cell line WS-LCL; however, only HF-2216–229-primed Mel5 and Mel6 CD8+ T cells produced IFNγ when restimulated with WS-LCL. Patient Mel6-derived CTLs stimulated with either peptide also secreted granzyme B in response to WS-LCL challenge. Altered reactivity of MAGE-A6172–187–stimulated T cells against WS-LCL could be attributed to its expression of high levels of HLA-A2 and/or co-stimulatory molecules, as well as its increased secretion of multiple pro-inflammatory factors in comparison to the melanoma cell lines (Fig. S1B and S1C). These data show that MAGE-A6172–187 and HF-2216–229 have unique immunogenic properties that may complement one another with regard to activation of a broad antitumor CD8+ T-cell repertoire.
HF-2216–229-stimulated CD8+ T cells exhibit a higher functional avidity for the MAGE-A6172–187 peptide than T cells primed against the natural MAGE-A6 peptide itself
To determine whether the differences observed in the recognition of peptide-loaded APCs and tumor targets by CTLs primed with MAGE-A6172–187 and HF-2216–229 peptides could be attributed to differences in responder T-cell functional avidity, we compared the abilities of peptide-primed CD8+ T cells generated from donor HD3 to recognize titrated doses of the MAGE-A6172–187 peptide pulsed onto autologous immature dendritic cells (iDC) in IFNγ ELISPOT assays. CD8+ T cells generated using the IVS protocol and primed with the MAGE-A6 peptide recognized APCs only when pulsed with a relatively high concentration of MAGE-A6172–187 peptide (with maximal response at a peptide dose of 2 μmol/L), whereas HF-2216–229-stimulated CD8+ T cells optimally recognized target cells pulsed with 16 nmol/L MAGE-A6172–187 peptide (i.e., a 125-fold lower concentration of the natural MAGE-A6 peptide; ). A similar pattern of functional avidity for the MAGE-A6172–187 peptide was also observed in HD4 (data not shown). These data suggest that HF-2216–229 peptide-stimulated T cells exhibit higher functional avidity for the MAGE-A6172–187 peptide epitope versus T cells activated in IVS using the MAGE-A6 peptide itself.
Figure 4. Functionality of MAGE-A6172–187 or HF-2216–229-stimulated CD8+ T-cell responders from advanced-stage melanoma patients. MAGE-A6172–187 or HF-2216–229 peptide-stimulated CD8+ T cells from melanoma patients were examined for their ability to recognize HLA-A2+, MAGE-A6+ tumor cell lines in vitro as described in . Spot counts greater than 10 over control were considered to be antigen-specific (dotted lines represent the cut-off).
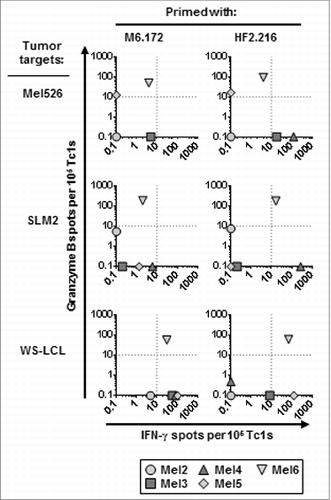
Figure 5. HF-2216–229 peptide stimulation induces high avidity, polyclonal CD8+ T cells capable of recognizing naturally-processed MAGE-A6 in an HLA-A2–restricted manner. In (A), to test the functional avidity of MAGE-A6172–187 (M6) and HF-2216–229 (HF2) peptide-primed CTLs, lymphocytes were evaluated for their ability to recognize titrated doses of the MAGE-A6172–187 peptide pulsed autologous HLA-A2+ iDC via interferon γ (IFNγ) ELISPOT assay. MAGE-A6172–187, MW = 1,772 g/mol (e.g.,, 10 μmol/L = 17.8 μg/mL); HF-2216–229, MW = 2,115 g/mol (e.g., 10 μmol/L = 21.2 μg/mL). Data from HD3 are shown, and are reflective of 2 independent experiments. NP (not pulsed with peptide) mature dendritic cells (mDC) were used for control in vitro stimulation. (B) MAGE-A6172–187–specific Vβ3+ and HF-2216–229-specific Vβ8+ T-cell clones from donors HD3 and HD12, respectively, were evaluated for their ability to recognize unpulsed (iDC), MAGE-A6172–187 (iDC/M6.172)– or HF-2216–229 (iDC/HF2.216)–pulsed autologous iDC, as well as the Mel526 and SLM2 melanoma cell lines in IFNγ and granzyme B ELISPOT assays. Analysis of 104 T effector cells and 104 control or peptide-pulsed antigen presenting cells (APCs) co-incubated in each assay well. The data presented are adjusted to represent the frequency of responders per 105 CD8+ T cells. BB7.2 anti-HLA-A2 antibody (10μg/mL) was added to replicate wells to confirm the HLA-A2-restriction of T-cell responses. Mean ± SEM values are presented. Statistical analyses were performed relative to BB7.2 blocking control by single-tailed Student's T test; *p < 0.05.
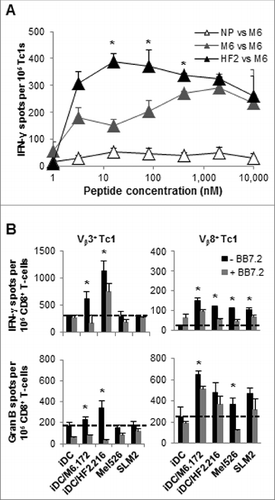
CD8+ T cells induced by MAGE-A6172–187 and HF-2216–229 peptide-based IVS display diverse repertoires
We next surveyed the clonality and antigen specificity of MAGE-A6172–187 vs. HF-2216–229 peptide-primed Tc1 in donors with previously measurable responses to these peptides. Highly-enriched populations of MAGE-A6172–187 and HF-2216–229-specific CD8+ T cells were isolated via flow cytometry from HD3 and HD12, respectively, and tested for their ability to recognize MAGE-A6172–187 and HF-2216–229 peptide–pulsed autologous iDC using the IFNγ cytokine secretion assay. Simultaneously, these cells were evaluated for TCR Vβ gene product expression by FACS. Multiple MAGE-A6172–187 (data not shown) and HF-2216–229–specific CD8+ T-cell clones (Fig. S4) cross-reacted against both peptides. We sorted and expanded Vβ3+ and Vβ8+ CD8+ T-cell clones from MAGE-A6172–187 and HF-2216–229-primed CTL populations, respectively, and consequently evaluated their abilities to recognize the 2 synthetic peptides as well as HLA-A2+ MAGE-A6+ melanoma lines in IFNγ and granzyme B ELISPOT assays. As illustrated in , Vβ3+ clone recognized both the priming MAGE-A6172–187 and, particularly, the homologous HF-2216–229 peptide, with IFNγ secretion being the most dominant response. These T cells did not recognize either the Mel526 or SLM2 melanoma cell lines, suggesting that this CTL population responded against a MAGE-A6172–187-derived epitope that was not efficiently presented by relevant melanoma target cells. In comparison, HF-2216–229-primed Vβ8+ clone effectively recognized both synthetic peptides, as well as both melanoma cell lines based on IFNγ and, particularly, granzyme B secretion (). Based on the MAGE-A6172–187 and HF-2216–229 GenBank homology searches, multiple MAGE-A proteins (MAGE-A2, MAGE-A3 and MAGE-A12, in particular) share the homologous core sequence against which potential cross-reactive T cell responses could also be induced using the evaluated peptides. Using RT-PCR we screened for HLA-A-matched control cell lines that do not express MAGE-A2, -A3, -A6 or -A12 as negative controls unsuccessfully (data not shown). Consequently, we used HLA-A2-specific blocking monoclonal antibody (BB7.2), which reduced the levels of T-cell secreted effector molecules in response to relevant target stimulation, supporting the HLA-A2–restricted nature of target cell recognition by these Vβ3+ and Vβ8+ Tc1 effector cell populations. These data suggest that the MAGE-A6172–187 and HF-2216–229-cross-reacting CTL clonal repertoire is large and capable of recognizing multiple epitopes derived from these homologous peptides. A closer evaluation of the HF-2216–229–primed Tc1 population confirmed this assumption as multiple CTL clones cross-reacted against both peptides, however only 5 T-cell subpopulations (Vβ7.1+, Vβ12+, Vβ13.6+, Vβ16+ and the aforementioned Vβ8+) contained responder cells that could also react detectably against naturally-processed and HLA-A2-presented MAGE-A6-derived peptides (i.e., presented by DC.MAGE-A6; Fig. S4D).
Discussion
The MAGE-A family is composed of 15 proteins Citation32 that are cumulatively expressed by more than half of all human cancers.Citation16 Among healthy adult tissues, MAGE-A proteins are expressed only in spermatogonia and spermatocytes, and, MAGE-A3 and MAGE-A4 are expressed in placenta.Citation13,14 As both testis and placenta represent immunologically-privileged organs due to their lack or deficiency in MHC Class I expression, any potential vaccination strategies targeting MAGE-A proteins would be expected to have only limited adverse (autoimmune) effects in patients. Indeed, only minimal toxicity has been observed after MAGE-A3-based vaccine in a completed Phase III clinical trial in the setting of melanoma.Citation33 However, MAGE-A and other TAAs in the cancer-testes family can be expressed by medullary thymic epithelial cells, which present self-antigens during the process of T-cell selection in the thymus.Citation23 Most antigen-specific T lymphocytes are believed to undergo thymic and peripheral clonal selection or to acquire a regulatory phenotype in accordance with operational tolerance to “self” antigens. As such, only low-avidity and/or regulatory T lymphocytes specific for these antigens are expected to remain in circulation under non-pathologic conditions, which may limit the protective capacity of the MAGE-A6-specific T-cell repertoire in cancer patients.
One approach to activate previously silent T lymphocytes against “self” tumor antigens is through the use of immunogenic microbial “mimic” epitopes that share a high degree of sequence homology with TAA-derived peptides. Immunization with an epitope mimic may prime a pool of precursor or memory lymphocytes exhibiting high (cross-reactive) functional avidity against TAA-derived epitopes. Our group has previously described 2 pan-HLA-DR-presented T-helper epitopes, MAGE-A6172–187 and its highly-immunogenic homolog HF-2216–229 derived from the Mycoplasma penetrans HF-2 permease protein. Mycoplasma penetrans HF-2 is a common, opportunistic microbe that is clinically observed in cases of human immunodeficiency virus (HIV-1) infection. It has also been suggested to represent a primary cause of non-HIV–related urethritis and respiratory disease, particularly among children.Citation34 We had previously noted that both MAGE-A6172–187 and HF-2216–229 peptides were capable of stimulating MAGE-A6-specific CD4+ T-cell responses in the majority of healthy donors and melanoma patients tested, with HF-2216–229-reactive CD4+ T-cells being derived from a memory pool of donor T cells and demonstrating the ability to recognize both MAGE-A6172–187-pulsed target cells as well as HLA-DR-matched, MAGE-A6+ tumor cell lines. Such immunologic molecular mimicry had also previously been reported for a peptide derived from the herpes simplex virus type 1 glycoprotein C that elicited CD8+ T cells in vitro that cross react against the HLA-A2–presented, melanoma-associated MART-127–35 peptide epitope.Citation35 These data supported the strategic use of homologous, pathogen-derived peptides to break operational tolerance mechanisms and promote effective immunity against “self” TAA.Citation35
The ability to optimally induce robust antitumor immunity is believed to require the coordinated activation of both Type-1 CD4+ helper T cells and CD8+ CTLs, and can be achieved using long synthetic peptides containing both CD4+ and CD8+ T-cell epitopes.Citation36 With this in mind, we investigated whether the MAGE-A6172–187 and HF-2216–229 peptides could also induce cross-reactive CD8+ T cells capable of recognizing MAGE-A6+ tumor cells in vitro. Indeed, we observed this to be the case for the majority of HLA-A2+ healthy donors tested. Notably, donors that were unresponsive to a given peptide could be sensitized by the other peptide leading to cross CD8+ T-cell reactivity against either peptide and to MAGE-A6+ tumor cells. Both MAGE and mycoplasma-specific responses were rapidly induced by a single round of IVS, however only the Mycoplasma penetrans HF-2216–229 peptide induced elevated frequencies of CD45RO+ memory Tc1 exhibiting a high degree of functional avidity, as manifest by IL-2 production in response to antigen-specific stimulation. While we have not provided evidence for the ability of our peptide-stimulated Tc1 cells to also recognize Mycoplasma penetrans-infected cells (that process and present endogenous HF-2-derived epitopes) our data suggest that the majority of healthy individuals have experienced this environmental pathogen Citation36 and have circulating levels of antigen-specific memory CD4+ and CD8+ T-cells. In line with these observations, a previous study has shown that short-term co-cultures of PBMCs with this pathogen lead to the rapid activation of CD8+ and CD4+ T cells,Citation37 suggesting that Mycoplasma penetrans infection readily promotes adaptive T-cell mediated immunity.
With regard to the quality and frequency of peptide-specific and antitumor CD8+ T-cell responses, there was a clear dichotomy of induced granzyme B and IFNγ secretion. This could simply reflect the functional capacity of individual Tc1 populations in these cultures, or alternatively, the differential register of functional avidity for such T effector cells based on aggregate signals provided by target cell HLA-A2/MAGE-A6 peptide complexes, co-stimulatory and co-inhibitory molecules.Citation38-40 Our data suggest the existence of Tc1 populations of various MAGE-A6172–187- and HF-2216–229–derived epitope specificities in these cultures that may account for these functional differences. We showed that the “subunit” MAGE-A6176–185 and HF-2220–229 peptides are cross-reactive, HLA-A2-presented epitopes that are contained within the MAGE-A6172–187 and HF-2216–229 peptides, respectively. We observed variability in responder frequencies to the short and matching long peptides, as well as to tumor cell targets that intrinsically express the MAGE-A6 gene product. This could be attributed to differential downstream processing of short and long peptides by APCs,Citation41 and also suggest that there may be multiple HLA-A2-restricted homologous epitopes encoded within the long peptides. Additional neural network algorithm screenings of the MAGE-A6172–187 and HF-2216–229 (long) peptides suggest that they may also share homologous epitopes presented by A*2403, B*1402, B*3901, and B*4001 molecules (data not shown). Hence, although it remains to be formally proven, these data suggest that the MAGE-A6172–187 and HF-2216–229 may elicit MAGE-A6-reactive CD8+ T-cell responses in a substantial proportion of cancer patients harboring MAGE-A6+ lesions. When taken together with our previously published CD4+ T-cell data,Citation29 we believe that these peptides have significant clinical potential due to their ability to coordinately stimulate Type-1 CD4+ and CD8+ T-cell responses against the MAGE-A6 TAA.
We observed that HLA-A2+ melanoma patients were comparatively deficient in their ability to mount memory responses against the MAGE-A6172–187 peptide when compared with healthy HLA-A2+ donors, and that the quality of these responses tended toward monofunctionality, which may be impacted by disease status. For instance, patient Mel4, who had distant metastases (lung/nodal) after surgery, produced mainly IL-10 in response to MAGE-A6172–187. Patient Mel5 produced IFNγ in response to specific stimulation and was the only patient with no evidence of disease at the time of analysis. Granzyme B production by melanoma patient CD8+ T cells in response to HLA-A2+ MAGE-A6+ tumor cell line targets was also suppressed in the majority of patients tested when compared to healthy donors. Of the patients evaluated in this study, only peptide-activated CD8+ T-cells from patients Mel5and Mel6 secreted granzyme B in response to MAGE-A6+ tumor cells, while Mel4 produced no detectable granzyme B after co-culture with these target cell lines. When taken together, these data suggest that the circulating anti-MAGE-A6 CD8+ T-cell repertoire in melanoma patients may be relegated to a state of low functional avidity or to hyporesponsiveness.
In comparison, melanoma patients retain protective anti-Mycoplasma penetrans memory Tc1 responses that are qualitatively and quantitatively similar to those of healthy individuals. HF-2216–229 peptide induced mixed IFNγ/IL-10 responses in all tested healthy individuals and the majority of melanoma patients evaluated (i.e., the only non-responder patient was Mel4). Due to the importance of Th1/Tc1-mediated immunity in effective protection against tumors,Citation7,9 it is likely that a future HF-2216–229 immunization strategies against MAGE-A6+ tumors should include a Type-1 skewing modality. Type-1 polarizing mature dendritic cells (mDC), which we have previously shown to be potent mediators of both adaptive and innate Type-1 immunities,Citation42-44 could be a potent immunization platform for enhancing cross-reactive immunity and treating patients harboring MAGE-A6+ malignancies. Given the link between clinical Mycoplasma infections and pulmonary disease,Citation34 and taken together with circumstantial reports of spontaneous cancer regression in patients (particularly of hematologic malignancies) recovering from pneumonia,Citation45,46 it is tempting to consider the possible involvement of exploiting cross-reactive immunity in these complex biological processes.
In summary, MAGE-A6172–187 and HF-2216–229 share structural homology and complex immunologic cross-reactivity. These MAGE-A6/mycoplasma epitopes may prove useful in the development of novel cancer vaccines or for use in immunomonitoring strategies for patients harboring MAGE-A6+ tumor lesions. Based on neural network predictions, the current long peptides contain epitopes that may be pan-presented by an extended range of HLA Class I and II allotypes, a property which may make them broadly-applicable in clinical vaccines.
Materials and Methods
Cell lines
Cell lines included TAP-deficient T2 cells (ATCC),Citation47 HLA-A2+ MAGE-6+ melanoma cell lines Mel526 Citation48,49 and SLM2,Citation50 and the Epstein-Barr Virus (EBV)-transformed B-lymphoblastoid cell line (WS-LCL; HLA-A2+ MAGE-6+).Citation51 Cell lines were cultured in RPMI 1640 medium (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 1% HEPES, and 1% L-glutamate (Invitrogen). Cultures were maintained in a humidified 37°C incubator under 5% CO2 tension. Cell lines were negative for Mycoplasma contamination as shown by GEN-PROBE Mycoplasma Tissue Culture Non-Isotopic Rapid Detection System (Gen-Probe, Inc.; San Diego, CA).
Peptides
The MAGE-A6172–187 and Mycoplasma penetrans HF-2 permease HF-2216–229 peptides were previously described.Citation29 Homologous MHC Class I-binding epitopes within these peptides (MAGE-A6176–185 and HF-2220–229; ) were identified using the SYFPEITHI epitope prediction algorithm (http://www.syfpeithi.de).Citation52 These peptides, and the flu matrix FluM158–66, were synthesized by 9-fluorenylmethoxycarbonyl chemistry by the University of Pittsburgh Cancer Institute's (UPCI) Peptide Synthesis Facility. Peptides were >95 % pure based on high-performance liquid chromatography (HPLC) and tandem mass spectrometry analyses performed by the UPCI Protein Sequencing Facility.
Recombinant adenoviral vectors (AdV)
AdV vectors encoding E.coli ß-galactosidase gene (AdV.LacZ) and full-length MAGE-A6 (AdV.MAGE-A6) were obtained from the University of Pittsburgh Vector Core Facility. Both AdV are E1/E3-deleted and replication-defective. Multiplicity of infection (MOI) was calculated based on plaque forming units (pfu) per mL.
Isolation of patient and healthy donor peripheral blood mononuclear cells
Peripheral blood was obtained from 13 HLA-A2+ normal donors and 6 HLA-A2+ melanoma patients with informed consent (), under Institutional Review Board-approved protocols (UPCI #04–001 and 96–099). Peripheral blood mononuclear cells (PBMCs) were separated from whole blood using Ficoll-Hypaque gradient centrifugation (Cellgro; Mediatech, Inc., Herndon, VA) as previously described.Citation29
Monocyte purification and DC generation
CD14+ monocytes were isolated by positive selection from PBMC using magnetic-activated cell sorting (MACS; Miltenyi Biotech, Auburn, CA). A portion of monocytes was used as APCs for antigen-specific memory response tests or for ELISPOT assays. iDC were generated from the remaining monocytes as previously described.Citation43 On day 5 of culture, iDC were pulsed with test peptides for 2 h at 37°C, after which 1000 U/mL IFNγ and 250 ng/mL lipopolysaccharide (LPS;Sigma-Aldrich) were added directly to culture to mature DCs. After an overnight incubation at 37°C, peptide-pulsed mature DC (mDC) were harvested and used for in vitro stimulation (IVS) experiments.
CD8+ Tcell isolation from PBMCs and in vitro stimulation assay
CD8+ T cells were isolated from CD14-negative PBMCs by positive MACS selection (Miltenyi Biotech), and then either used for antigen-specific memory response tests or cryopreserved until needed to establish T cell co-cultures with autologous peptide-pulsed mDCs. Cells were co-cultured at a 5:1 T:mDC cell ratio in T-cell media [TCM; AIM-V supplemented with 5% human AB serum (Mediatech, Inc.., Manassas, VA)] for 11 d For functional avidity tests, CTLs underwent a second round of IVS at a 10:1 T:mDC cell ratio in TCM supplemented with 20 IU/mL rhIL-2 (eBioscience; San Diego, CA) and 10 ng/mL rhIL-7 (PeproTech; Rocky Hill, NJ) for 11 d.
Elispot
Following IVS culture, the frequencies of peptide-specific cytotoxic CD8+ T-cell responders were measured using anti-human human IFNγ (1-D1K and mAb7-B6–1-Biotin) or granzyme B (GB10 and GB-11-biotin) ELISPOT assays using antibody pairs from Mabtech, Inc.. (Cincinnati, OH) and MultiScreenTM HTS plates from Millipore (Bedford, MA), as previously described.Citation29 Autologous monocytes loaded with 10μg/mL of test peptides or AdV-transduced DCs or HLA-A2+ cell lines (Mel526, SLM2, WS-LCL) were used as antigen presenting cells. All tests were performed in triplicate wells, and mean values ± SEM presented.
Cytokine and chemokine measurements
Cell culture supernatants were assessed for cytokine and chemokine secretion by Luminex arrays using BioSource Cytokine 25-plex kit.
Cell staining with fluorochrome-conjugated antibodies and cytofluorimetric analysis
Single step staining of cell surface antigens was performed using the following fluorophore-conjugated antibodies: CD45RO-FITC, CD3-ECD, CD4-PE-Cy7, CD8-APC, CD80-PE, CD86-PE (Beckman Coulter; Brea, CA), HLA-A2 (BB7.2 mAb)-FITC, CD40-PE-Cy7 and CCR7-PE-Cy7 (BD PharMingen, San Diego, CA), as previously described.Citation44 Cell staining was analyzed using the CyAn flow cytometer (Beckman Coulter), and data analysis was performed with Summit v4.3 software (Beckman Coulter).
Reverse transcription polymerase chain reaction (PCR)
Expression of MAGE-A6 and β-actin mRNA was measured by RT-PCR, as previously described.Citation53
Western blot analysis
iDCs were transduced with Ad.LacZ or Ad.MAGE-A6 (MOI 250), as previously reported.Citation29 Following the 48 h incubation, DCs were collected and lysed using the freeze-thaw method, and analyzed along with recombinant MAGE-A6 control by Western blotting for MAGE-A6 and β-actin protein levels, as previously described.Citation29
Antigen-specific CD8+ T-cell memory responses
CD8+ T cells purified from fresh healthy donor or frozen melanoma patient PBMCs were co-cultured with autologous monocytes and 10μg/mL appropriate peptides for 1.5 h in TCM at 37°C. Cells co-cultured without peptides were used as a negative control. Afterwards, brefeldin A (Sigma) was added to cultures at 10μg/mL, and cells were incubated for an additional 3.5 h at 37°C. Following the incubation, cells were analyzed for CD3, CD8 and CD45RO expression, and intracellular IFNγ, IL-10, IL-2 or TNFα using PE conjugated antibodies (BD Pharmingen) per manufacturers’ protocols, and analyzed by flow cytometry.
T2-based HLA-A2 peptide-binding assay
T2 cells were loaded with titrated amounts of peptide (≤50 μg /mL) and incubated with 5μg/mL of β2-microglobulin (Sigma) and 5μg/mL BB7.2 (anti-HLA-A2) antibody in serum-free AIM-V media (GIBCO) for 18 h at 37°C. Afterwards, cells were stained with PE-conjugated polyclonal goat anti-mouse immunoglobulin F(ab’)2 (Dako, Carpinteria, CA; 1:20 working dilution) at 4°C for 30 min, and fixed with 2% w/v paraformaldehyde (Sigma) solution in PBS. Elevated HLA-A2 expression as measured by flow cytometry correlated with greater peptide binding.
Generation of MAGE-A6172–187 and HF-2216–229 specific CD8+ T-cell clones
CD8+ T cells isolated from donors HD3 and HD12 underwent a single round of IVS with MAGE-A6172–187 and HF-2216–229 peptides, respectively. Peptide–reactive IFNγ-secreting cells were enriched by FACS sorting using the IFNγ Cytokine Secretion Assay (Miltenyi Biotech). CD8+ T cells were then expanded and maintained by weekly stimulations using irradiated (5,000 rads) allogeneic PBMCs (106 cells/mL) as feeders in TCM supplemented with 200 units/mL rhIL-2 (eBioscience) and 1μg/mL phytohemagglutinin (Sigma). Subsequently these cells were evaluated for antigen recognition against a range of relevant or control target cells by fluorescence cytometry. The T-cell receptor (TCR) Vβ repertoire of the IFNγ responders was also evaluated by cytofluorimetric analysis using the IOTest Beta Mark kit (Beckman Coulter) in conjunction with the IFNγ Miltenyi Cytokine Secretion Assay and with Pacific Blue-conjugated CD3 (BD Pharmingen). Consequently, Vβ3+ (from MAGE-A6172–187-reactive CTLs) and Vβ8+ (from HF-2216–229–reactive CTLs) CD8+ T-cell clones were isolated by fluorescence-activated cell sorting, expanded in vitro for 11 days, and then analyzed for antigenic specificity.
Statistical analysis
Statistical analyses were performed by single-tailed Student's T test, with p < 0.05 considered statistically significant.
Authorship
LV and LHB were accountable for the conception and design of research and writing the manuscript; LV performed all in vitro experiments and data analysis; JS performed Western blot and RT-PCR analyses of tumor cell lines; JMK provided melanoma patient PBMC and contributed to writing the manuscript; and WJS provided the WS-LCL cell line and contributed to writing the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
954501_Supplementary_Materials.zip
Download Zip (432.5 KB)Funding
This study was supported by the UPCI Melanoma and Skin Cancer SPORE career development award (L.V.); NIH 1P50CA121973 (J.M.K.; Project 2, L.H.B.; Project 4, W.J.S.); and the Henry L. Hillman Foundation (L.H.B.). We thank the following UPCI shared resources (supported in part by NIH P30CA047904): Peptide Synthesis Facility; Protein Sequencing Facility; Flow Cytometry Facility (cell sorting); the Vector Core Facility (Ad.LacZ and Ad.MAGE-A6) and the Immunologic Monitoring Laboratory (Luminex and Mycoplasma tests).
References
- Gromet MA, Epstein WL, Blois MS. The regressing thin malignant melanoma: a distinctive lesion with metastatic potential. Cancer 1978; 42:2282-92; PMID:719608; http://dx.doi.org/10.1002/1097-0142(197811)42:5<2282::AID-CNCR2820420528>3.0.CO;2-V
- Tihan T, Filippa DA. Coexistence of renal cell carcinoma and malignant lymphoma. A casual relationship or coincidental occurrence. Cancer 1996; 77:2325-31; PMID:8635103; http://dx.doi.org/10.1002/(SICI)1097-0142(19960601)77:11<2325::AID-CNCR22>3.0.CO;2-Y
- Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med 1996; 183:725-9; PMID:8642276; http://dx.doi.org/10.1084/jem.183.3.725
- Clemente CG, Mihm MCJ, Bufalino R, Zurrida S, Collini P, Cascinelli N Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996; 77:1303-10; PMID:8608507; http://dx.doi.org/10.1002/(SICI)1097-0142(19960401)77:7<1303::AID-CNCR12>3.0.CO;2-5
- Old LJ, Chen YT. New paths in human cancer serology. J Exp Med 1998; 187:1163-7; PMID:9547328; http://dx.doi.org/10.1084/jem.187.8.1163
- Penn I. Malignant melanoma in organ allograft recipients. Transplantation 1996; 61:274-8; PMID:8600636; http://dx.doi.org/10.1097/00007890-199601270-00019
- Fallarino F, Grohmann U, Bianchi R, Vacca C, Fioretti MC, Puccetti P. Th1 and Th2 cell clones to a poorly immunogenic tumor antigen initiate CD8+ T cell-dependent tumor eradication in vivo. J Immunol 2000; 165:5495-501; PMID:11067902; http://dx.doi.org/10.4049/jimmunol.165.10.5495
- Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 2005; 434:88-93; PMID:15744305; http://dx.doi.org/10.1038/nature03337
- Surman D, Dudley M, Overwijk W, Restifo N Cutting edge: CD4+ T cell control of CD8+ T cell reactivity to a model tumor antigen. J Immunol 2000; 164:562-5; PMID:10623795; http://dx.doi.org/10.4049/jimmunol.164.2.562
- Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 2010; 70:8368-77; PMID:20940398; http://dx.doi.org/10.1158/0008-5472.CAN-10-1322
- Crotzer VL, Blum JS. Autophagy and adaptive immunity. Immunology 2010; 131:9-17; PMID:20586810; http://dx.doi.org/10.1111/j.1365-2567.2010.03309.x
- van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991; 254:1643-7; PMID:1840703; http://dx.doi.org/10.1126/science.1840703
- Jungbluth AA, Wilson A, Silva J, Iversen K, Frosina D, Zaidi B, Coplan K, Eastlake-Wade SK, Castelli SB, Spagnoli GC, et al. Expression of cancer-testis (CT) antigens in placenta. Cancer Immun 2007; 7:15-26; PMID:17715884
- Takahashi K, Shichijo S, Noguchi M, Hirohata M, Itoh K. Identification of MAGE-1 and MAGE-4 proteins in spermatogonia and primary spermatocytes of testis. Cancer Res 1995; 55:3478-82; PMID:7627949
- Furuta J, Umebayashi Y, Miyamoto K, Kikuchi K, Otsuka F, Sugimura T, Ushijima T. Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci 2004; 95:962-8; PMID:15596045; http://dx.doi.org/10.1111/j.1349-7006.2004.tb03184.x
- Jungbluth AA, Busam KJ, Kolb D, Iversen K, Coplan K, Chen Y-T, Spagnoli GC, Old LJ. Expression of MAGE-antigens in normal tissues and cancer. Int J Cancer 2000; 85:460-5; PMID:10699915; http://dx.doi.org/10.1002/(SICI)1097-0215(20000215)85:4<460::AID-IJC3>3.0.CO;2-N
- Gibbs P, Hutchins AM, Dorian KT, Vaughan HA, Davis ID, Silvapulle M, Cebon JS. MAGE-12 and MAGE-6 are frequently expressed in malignant melanoma. Melanoma Res 2000; 10:259-64; PMID:10890380; http://dx.doi.org/10.1097/00008390-200010030-00008]
- Tatsumi T, Kierstead LS, Ranieri E, Gesualdo L, Schena FP, Finke JH, Bukowski RM, Brusic V, Sidney J, Sette A, et al. MAGE-6 encodes HLA-DRβ1*0401-presented epitopes recognized by CD4+ T cells from patients with melanoma or renal cell carcinoma. Clinical Cancer Research 2003; 9:947-54; PMID:12631591
- Lin J, Lin L, Thomas DG, Greenson JK, Giordano TJ, Robinson GS, Barve RA, Weishaar FA, Taylor JMG, Orringer MB, et al. Melanoma-associated antigens in esophageal adenocarcinoma. Clinical Cancer Research 2004; 10:5708-16; PMID:15355897; http://dx.doi.org/10.1158/1078-0432.CCR-04-0468
- Liu B-B, Ye S-L, He P, Liu Y-K, Tang Z-Y. The tumour-associated antigen MAGE-1 is detectable in formalin-fixed paraffin sections of malignant melanoma. J Cancer Res Clin Oncol 1999; 125:685-9; PMID:10592101; http://dx.doi.org/10.1007/s004320050334
- Otte M, Zafrakas M, Riethdorf L, Pichlmeier U, Löning T, Jänicke F, Pantel K. MAGE-a gene expression pattern in primary breast cancer. Cancer Res 2001; 61:6682-7; PMID:11559535
- Rayman P, Wesa AK, Richmond AL, Das T, Biswas K, Raval G, Storkus WJ, Tannenbaum C, Novick A, Bukowski R, et al. Effect of renal cell carcinomas on the development of type 1 T-cell responses. Clin Cancer Res 2004; 10:6360S-6S; PMID:15448031; http://dx.doi.org/10.1158/1078-0432.CCR-050011
- Gotter J, Brors B, Hergenhahn M, Kyewski B. Medullary epithelial cells of the human thymus express a highly diverse selection of tissue-specific genes colocalized in chromosomal clusters. J Exp Med 2004; 199:155-66; PMID:14734521; http://dx.doi.org/10.1084/jem.20031677
- Cusick MF, Libbey JE, Fujinami RS. Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol 2012; 42:102-111; PMID:22095454; http://dx.doi.org/10.1007/s12016-011-8294-7
- Davies JM. Molecular mimicry: can epitope mimicry induce autoimmune disease? Immunol Cell Biol 1997; 75:113-26; PMID:9107563; http://dx.doi.org/10.1038/icb.1997.16
- Brocke S, Hausmann S, Steinman L, Wucherpfennig KW. Microbial peptides and superantigens in the pathogenesis of autoimmune diseases of the central nervous system. Semin Immunol 1998; 10:57-67; PMID:9529656; http://dx.doi.org/10.1006/smim.1997.0105
- Hudrisier D, Riond J, Burlet-Schiltz O, von Herrath MG, Lewicki H, Monsarrat B, Oldstone MBA, Gairin JE. Structural and functional identification of major histocompatibility complex class I-restricted self-peptides as naturally occurring molecular mimics of viral antigens. J Biol Chem 2001; 276:19396-403; PMID:11278441; http://dx.doi.org/10.1074/jbc.M008864200
- Moriyama H, Wen L, Abiru N, Liu E, Yu L, Miao D, Gianani R, Wong FS, Eisenbarth GS. Induction and acceleration of insulitis/diabetes in mice with a viral mimic (polyinosinic-polycytidylic acid) and an insulin self-peptide. Proc Natl Acad Sci 2002; 99:5539-44; PMID:11943868; http://dx.doi.org/10.1073/pnas.082120099
- Vujanovic L, Mandic M, Olson WC, Kirkwood JM, Storkus WJ. A mycoplasma peptide elicits heteroclitic CD4+ T cell responses against tumor antigen MAGE-A6. Clin Cancer Res 2007; 13:6796-806; PMID:18006782; http://dx.doi.org/10.1158/1078-0432.CCR-07-1909
- Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 2003; 4:225-34; PMID:12563257; http://dx.doi.org/10.1038/ni889
- Polak ME, Borthwick NJ, Gabriel FG, Johnson P, Higgins B, Hurren J, McCormick D, Jager MJ, Cree IA. Mechanisms of local immunosuppression in cutaneous melanoma. Br J Cancer 2007; 96:1879-87; PMID:17565341; http://dx.doi.org/10.1038/sj.bjc.6603763
- Katsura Y, Satta Y. Evolutionary history of the cancer immunity antigen MAGE gene family. PLoS ONE 2011; 6:e20365; PMID:21695252; http://dx.doi.org/10.1371/journal.pone.0020365
- Trefzer U, Jouary T, Robert C, Santinami M, Levchenko E, Smithers B, Testori A, Peeters O, Kirkwood J, Dréno B. FC13 DERMA phase III trial of MAGE-A3 immunotherapy as adjuvant treatment in stage III melanoma: MAGE-A3 gene expression frequency and baseline demographics. Melanoma Res 2010; 20:e34-e35; http://dx.doi.org/10.1097/01.cmr.0000382815.90748.46
- Sasaki Y, Ishikawa J, Yamashita A, Oshima K, Kenri T, Furuya K, Yoshino C, Horino A, Shiba T, Sasaki T, et al. The complete genomic sequence of Mycoplasma penetrans, an intracellular bacterial pathogen in humans. Nucleic Acid Res 2002; 30:5293-300; PMID:12466555; http://dx.doi.org/10.1093/nar/gkf667
- Loftus DJ, Castelli C, Clay TM, Squarcina P, Marincola FM, Nishimura MI, Parmiani G, Appella E, Rivoltini L. Identification of epitope mimics recognized by CTL reactive to the melanoma/melanocyte-derived peptide MART-1(27-35). J Exp Med 1996; 184:647-57; PMID:8760818; http://dx.doi.org/10.1084/jem.184.2.647
- Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG, Meer DMAB-vd, Vloon APG, Essahsah F, Fathers LM, Offringa R, Drijfhout JW, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 2009; 361:1838-47; PMID:19890126; http://dx.doi.org/10.1056/NEJMoa0810097
- Sasaki Y, Blanchard A, Watson HL, Garcia S, Dulioust A, Montagnier L, Gougeon ML. In vitro influence of Mycoplasma penetrans on activation of peripheral T lymphocytes from healthy donors or human immunodeficiency virus-infected individuals. Infect Immun 1995; 63:4277-83; PMID:7591058
- Constant SL, Bottomly K. Induction of Th1 and Th2 CD4+ T cell responses: the alternative approaches. Annu Rev Immunol 1997; 15:297-322; PMID:9143690; http://dx.doi.org/10.1146/annurev.immunol.15.1.297
- Kumar V, Bhardwaj V, Soares L, Alexander J, Sette A, Sercarz E. Major histocompatibility complex binding affinity of an antigenic determinant is crucial for the differential secretion of interleukin 4/5 or interferon gamma by T cells. Proc Natl Acad Sci 1995; 92:9510-4; PMID:7568164; http://dx.doi.org/10.1073/pnas.92.21.9510
- Ma H, Kapp JA. Antigenic epitopes regulate the phenotype of CD8+ CTL primed by exogenous antigens. J Immunol 2000; 164:5698-703; PMID:10820246; http://dx.doi.org/10.4049/jimmunol.164.11.5698
- Rock KL, Farfán-Arribas DJ, Shen L. Proteases in MHC class I presentation and cross-presentation. J Immunol 2010; 184:9-15; PMID:20028659; http://dx.doi.org/10.4049/jimmunol.0903399
- Vujanovic L, Ballard W, Thorne SH, Vujanovic NL, Butterfield LH. Adenovirus-engineered human dendritic cells induce natural killer cell chemotaxis via CXCL8/IL-8 and CXCL10/IP-10. OncoImmunology 2012; 1:448-57; PMID:22754763; http://dx.doi.org/10.4161/onci.19788
- Vujanovic L, Szymkowski DE, Vujanovic NL, Butterfield LH Virally-infected and cytokine-matured human dendritic cells activate natural killer cells via cooperative activity of plasma membrane-bound TNF and IL-15. Blood 2010; 116:575-83; PMID:20430958; http://dx.doi.org/10.1182/blood-2009-08-240325
- Vujanovic L, Whiteside TL, Potter DM, Chu J, Ferrone S, Butterfield LH. Regulation of antigen presentation machinery in human dendritic cells by recombinant adenovirus. Cancer Immunol Immunother 2009; 58:121-33; PMID:18488218; http://dx.doi.org/10.1007/s00262-008-0533-2
- Bang S, Cheong J-W, Yang WI, Hahn JS. An unusual case of spontaneous remission of hodgkin's disease after a single cycle of COPP-ABV chemotherapy followed by infectious complications. Yonsei Med J 2005; 46:425-30; PMID:15988816; http://dx.doi.org/10.3349/ymj.2005.46.3.425
- Chen R-L, Chuang S-S. Transient spontaneous remission after tumor lysis syndrome triggered by a severe pulmonary infection in an adolescent boy with acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2009; 31:78-80; http://dx.doi.org/10.1097/MPH.0b013e31818ab30c
- Salter RD, Cresswell P. Impaired assembly and transport of HLA-A and -B antigens in a mutant TxB cell hybrid. EMBO J 1986; 5:943-949; PMID:3522223
- Schwartzentruber DJ, Topalian SL, Mancini M, Rosenberg SA. Specific release of granulocyte-macrophage colony-stimulating factor, tumor necrosis factor-alpha, and IFN-gamma by human tumor-infiltrating lymphocytes after autologous tumor stimulation. J Immunol 1991; 146:3674-81; PMID:1902860
- Tüting T, Wilson CC, Martin DM, Kasamon YL, Rowles J, Ma DI, Slingluff CL, Wagner SN, van der Bruggen P, Baar J, et al. Autologous human monocyte-derived dendritic cells genetically modified to express melanoma antigens elicit primary cytotoxic T cell responses in vitro: enhancement by cotransfection of genes encoding the Th1-biasing cytokines IL-12 and IFN-α. J Immunol 1998; 160:1139-47; PMID:9570527
- Tatsumi T, Kierstead LS, Ranieri E, Gesualdo L, Schena FP, Finke JH, Bukowski RM, Mueller-Berghaus J, Kirkwood JM, Kwok WW, et al. Disease-associated bias in T helper type 1 (Th1)/Th2 CD4+ T cell responses against MAGE-6 in HLA-DRB1*0401+ patients with renal cell carcinoma or melanoma. J Exp Med 2002; 196:619-28; PMID:12208877; http://dx.doi.org/10.1084/jem.20012142
- Herr W, Ranieri E, Olson W, Zarour H, Gesualdo L, Storkus WJ. Mature dendritic cells pulsed with freeze-thaw cell lysates define an effective in vitro vaccine designed to elicit EBV-specific CD4+ and CD8+ T lymphocyte responses. Blood 2000; 96:1857-64; PMID:10961887
- Rammensee H-G, Bachmann J, Emmerich NN, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenet 1999; 50:213-9; http://dx.doi.org/10.1007/s002510050595
- Blalock LT, Landsberg J, Messmer M, Shi J, Pardee AD, Haskell R, Vujanovic L, Kirkwood JM, Butterfield LH. Human dendritic cells adenovirally-engineered to express three defined tumor antigens promote broad adaptive and innate immunity. OncoImmunology 2012; 1:287-357; PMID:22737604; http://dx.doi.org/10.4161/onci.18628