
Abstract
Therapeutic vaccinations against cancer are still largely ineffective. Major caveats are inefficient delivery of tumor antigens to dendritic cells (DCs) and excessive immune suppression by Foxp3+ regulatory T cells (Tregs), resulting in defective T cell priming and failure to induce tumor regression. To circumvent these problems we evaluated a novel combinatorial therapeutic strategy. We show that tumor antigen targeting to DC-SIGN in humanized hSIGN mice via glycans or specific antibodies induces superior T cell priming. Next, this targeted therapy was combined with transient Foxp3+ Treg depletion employing hSIGNxDEREG mice. While Treg depletion alone slightly delayed B16-OVA melanoma growth, only the combination therapy instigated long-term tumor regression in a substantial fraction of mice. This novel strategy resulted in optimal generation of antigen-specific activated CD8+ T cells which accumulated in regressing tumors. Notably, Treg depletion also allowed the local appearance of effector T cells specific for endogenous B16 antigens. This indicates that antitumor immune responses can be broadened by therapies aimed at controlling Tregs in tumor environments. Thus, transient inhibition of Treg-mediated immune suppression potentiates DC targeted antigen vaccination and tumor-specific immunity.
Introduction
The development of immunotherapies for the successful treatment of cancer is a major challenge. For an effective elimination of tumor cells, it is essential to generate a large pool of tumor-specific effector CD8+ T cells that recognize specific peptide-MHC complexes on the surface of tumor cells with sufficient avidity. To accomplish this, proper activation of naive tumor-specific CD8+ T lymphocytes by DCs is required. The presentation of tumor antigens on MHC class I molecules together with co-stimulatory and pro-inflammatory cytokines drives CD8+ T cell expansion and concurrent acquisition of effector functions such as cytolytic activity and production of cytokines (i.e., interferon (IFN)γ and tumor necrosis factor (TNF).Citation1 However, tumor-infiltrating lymphocytes may be rendered incapable of rejecting tumor cells and/or remain insufficient in number due to anti-inflammatory properties of tumor cells.Citation2,3 Additionally, proper maturation of antigen-sampling DCs is prevented.Citation4 As a consequence, the DCs express low amounts of co-stimulatory molecules and secrete immunosuppressive cytokines. In the tumor-draining lymph node (TDLN), interaction of naive tumor-specific T cells with these DCs results in poor effector T cell generation. Instead, T cells differentiate into so-called induced Treg.Citation2,3,5 In fact, an expansion of Tregs has been noted in many murine and human malignancies, resulting in a profound blockade of tumor-specific immunity at multiple layers.Citation6
In addition to iTreg, also natural Treg (nTreg), which develop in the thymus, contribute to tumor-specific immune tolerance. By expressing high levels of CCR4, nTreg are recruited to CCL22 rich tumor microenvironments.Citation7-9 Here, nTreg actively expand and suppress other immune cells in a cell-contact dependent manner.Citation3,8 Thus, it is clear that various subpopulations of Tregs endowed with various suppressive functions co-exist in cancer patients. Together, these events enable tumors to escape the immune system and result in uncontrolled growth and expansion of the tumor cells.
The identification of the immunodominant epitopes of several tumor antigens facilitated the use of protein or peptide antigens as vaccines to boost tumor-immunity.Citation10 However, these types of vaccines require high amounts of antigens to be effective as they will also be internalized and/or presented by other cells than DCs.Citation11-15 Additionally, the efficacy of these vaccines is often limited in a therapeutic setting. To enhance cross-presentation of tumor antigens and to achieve a better priming of T cells, current vaccination strategies focus at the in vivo delivery of tumor-antigens as proteins or peptides specifically to DCs. Hereto, antigens can be tagged with antibodies or ligands specific for a DC-expressed receptor.Citation16 A particularly promising target in this respect is the endocytic C-type Lectin Receptor (CLR) DC-SIGN, which is expressed on human immature DCs, providing the opportunity to specifically target DCs and additionally mediate fast and efficient uptake of antigens. Antigens taken up via DC-SIGN end up as epitopes in MHC class II and I molecules enhancing antigen-specific CD4+ and CD8+ T cell responses.Citation17-19 As no functional homolog of DC-SIGN exists in mice,Citation20 we generated humanized mice expressing human DC-SIGN (hSIGN) on conventional DCs.Citation21 Importantly, delivery of antigens via anti-DC-SIGN monoclonal antibodies (aDC-SIGN) enhances T cell responses in vitro and in vivo.Citation18,19,22,23
An advantage of targeting antigens to CLRs is the possibility to modify antigens with the natural ligands of CLR, i.e., carbohydrates, which are small compared to antibodies. Glycans are naturally expressed throughout the body and therefore poorly immunogenic. The natural ligands of DC-SIGN comprise high-mannose oligosaccharides and fucose-containing Lewis-type determinants, such as LewisB (LeB) and LewisX (LeX).Citation24 We and others have shown that glycan-modified protein and peptide antigens, either soluble or engineered into liposomes and dendrimers, are efficiently internalized and shuttled into the MHC-II and MHC-I presentation route, leading to enhanced CD4+ and CD8+ T cell activation.Citation25-29 In addition, we showed that in combination with an adjuvant, DC-SIGN triggering modulates cytokine responsesCitation26 and elicits strong CD4+ and CD8+ effector T cell responses in murine and human models.Citation23,26 However, whether targeting antigens to CLRs such as DC-SIGN using glycans yields superior tumor immunity than using specific antibodies is not known at present.
The cytolytic function of CD8+ T cells in the tumor micromilieu can be restored by suppressing Treg numbers and/or function.Citation2,30 Due to the initial identification of Tregs by high CD25 expression,Citation31 administration of anti-CD25 antibodies (PC61 in mice) is commonly used to deplete Tregs. However, CD25 is also expressed by effector T cells and other immune cells, herewith reducing the efficacy of this strategy. This is illustrated by the finding that removal of Tregs only before tumor inoculation facilitated tumor rejection. By contrast, injection of PC61 simultaneously or after tumor inoculation was not effective, as tumor-resident Tregs were not depleted and/or also clonal expansion of tumor-specific effector T cells was inhibited.Citation32,33 Moreover, CD25− Treg escape this strategy. Several clinical trials aiming to deplete CD25+ Treg in cancer patients using recombinant immunotoxins such as dinileukin diftitox (Ontak) and LMB-2Citation34,35 have shown marginal results. A recent study even reported induction of tolerance following Ontak therapy.Citation36 Similar to the observations in the murine studies, results in patients may have been confounded by the co-depletion of tumor-reactive effector T cells.
Bacterial artifical chromosome (BAC)-transgenic DEREG mice express the human diphtheria toxin receptor (DTR) under the control of the FoxP3 locus and thereby allow the selective depletion of Tregs without affecting other T cell populations.Citation37 Indeed, using these mice we could establish that specific and transient removal of Treg was sufficient to induce partial regression of established B16-OVA melanoma, demonstrating the potent inhibitory effects of Tregs on antitumor immunity.Citation30 Moreover, tumor growth could be further delayed when mice received a combination treatment consisting of Treg depletion and vaccination with tumor antigen mixed with TLR9 agonist. However, whether antigen targeting to DCs combined with Treg depletion has superior activity has not been investigated.
Although promising results have been obtained by depleting Treg or by targeting antigens to DC specifically, long-term tumor regression is difficult to achieve. We therefore examined whether a novel combinatorial immunotherapy consisting of a DC-SIGN targeting vaccine and specific, transient, Treg depletion has superior efficacy in combating established melanoma compared to monotherapies. For this purpose we crossed humanized hSIGN mice with DEREG mice. Moreover, we also directly compared glycan- with antibody-based DC-SIGN targeting vaccines for their efficacy. Hereto, the model tumorantigen OVA was modified with either LeB or anti-DC-SIGN mAb.
We show that the combination of Treg depletion with a DC-SIGN-targeting vaccine resulted in superior expansion of OVA-specific CD8+ T cells, which infiltrated B16-OVA melanoma tumors, resulting in long-term tumor control.
Results
Improved antigen-specific T cell responses by targeting antigen to DC-SIGN
To compare T cell activation induced by DCs after antigen targeting to DC-SIGN, we modified ovalbumin (OVA) either with LeB glycans (OVA-LeB) or DC-SIGN-specific monoclonal antibodies (OVA-aDC-SIGN). Correct modification of LeB- and aDC-SIGN-coupled OVA was determined by ELISA using specific antibodies to LeB as well as DC-SIGN-Fc chimeric molecules (). Our data show that only OVA-LeB was specifically recognized by anti-LeB. Anti-LeB antibodies did not bind aDC-SIGN- or isotype Ab-modified and native OVA. DC-SIGN-Fc binding was detected to OVA-LeB and OVA-aDC-SIGN and was abrogated by the addition of the Ca2+ chelator EDTA, indicating functional modification. Clearly, DC-SIGN-Fc did not recognize isotype Ab-modified and native OVA. The equal recognition of all antigen preps by anti-OVA antibodies indicates that OVA is not denatured during the chemical conjugation process.
Figure 1. Enhanced proliferation of T cells by antigen targeting to DC-SIGN. (A) Glycan and antibody-modified OVA efficiently bind human DC-SIGN. LeB-, aDC-SIGN-, isotype Ab-modified and native OVA were analyzed by ELISA using anti-OVA or anti-LeB antibodies or binding to DC-SIGN using DC-SIGN-Fc chimeric molecules. Depicted results are representative of four independent experiments. (B–E) hSIGN and WT BMDCs were pulsed with indicated concentrations of OVA-aDC-SIGN, OVA-isotype (B+C), OVA-LeB or native OVA (D+E) and subsequently co-cultured with purified CD4+ OT-II T cells or CD8+ OT-I T cells. Expansion of OVA-specific T cells was determined using 3H-thymidine incorporation. Data are shown as means +/˗ SD of triplicate cultures. * P < 0.05. Results shown are representative of three independent experiments.
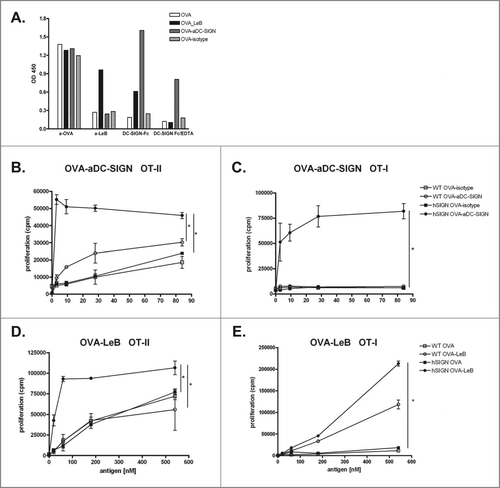
BMDCs from hSIGN and WT mice were loaded with equimolar amounts of OVA-aDC-SIGN or OVA conjugated with isotype control Abs (OVA-isotype) and subsequently co-cultured with OVA-specific CD4+ or CD8+ T cells. Internalized OVA-aDC-SIGN is shuttled into the MHC class II presentation route as evident from vigorous proliferation of OVA-specific CD4+ T cells (). Moreover, the response induced by DC-SIGN mediated targeting was much more efficient than that induced by control OVA-isotype, as the same degree of CD4+ T cell proliferation could be induced with >80-fold less OVA. OVA-aDC-SIGN also efficiently entered a cross-presentation route resulting in presentation on MHC class I molecules and activation of OVA-specific CD8+ T cells (). The enhanced presentation of OVA antigens in MHC-II and I was specifically induced upon DC-SIGN-mediated uptake, as neither OVA-isotype nor WT DCs evoked such strong OT-II and OT-I T cell proliferation. Similarly, and as reported earlier,Citation28 glycan-modified OVA internalized by DC-SIGN is shuttled into both MHC class II and I presentation routes as revealed from increased proliferation of OVA-specific CD4+ and CD8+ T cells (). Yet, while targeting DC-SIGN with OVA-LeB induces comparable activation of CD4+ T cells as OVA-aDC-SIGN, we found that cross-presentation of OVA is much more enhanced using OVA-aDC-SIGN than OVA-LeB. Moreover, we found that approximately 10- to 50-fold lower amounts of OVA were sufficient when conjugated to aDC-SIGN to evoke similar CD8+ T cell responses as OVA-LeB (i.e., 3 nM vs. 183 nM, respectively). Thus, both DC-SIGN targeting formulations increased specific activation of CD4+ and CD8+ T cells by enhancing antigen presentation, albeit with some differences in cross-presentation.
We next assessed whether these differences are also reflected in the generation of endogenous effector CD4+ and CD8+ T cells in vivo. hSIGN mice were injected subcutaneously (s.c.) with OVA-LeB or OVA-aDC-SIGN mixed with agonistic aCD40 antibodies. The frequency of antigen-specific effector CD8+ T cells in the spleen was determined by their ability to produce IFNγ and TNF after a 4h ex-vivo re-stimulation. Compared to native OVA/anti-CD40, immunization with OVA-LeB and OVA-aDC-SIGN induced higher percentages of IFNγ- and TNF-double-producing CD8+ T cells (). Similarly, IFNγ single-producing CD8+ T cell responses were highest in mice immunized with DC-SIGN targeting formulations (). By contrast, antigen-specific TNF single-producers were not enhanced (Fig.2A and not shown). In summary, these data clearly show that by targeting antigen to DC-SIGN the overall CD8+ effector T cell response is shifted toward IFNγ/TNF-double-producers (). The polyfunctionality of the OVA-specific T cells expanded under DC-SIGN-targeting conditions is also suggested by the increased cytokine production on a per cell basis ().
Figure 2. Immunization with OVA-LeB and OVA-aDC-SIGN increases T cell priming in vivo. hSIGN mice were immunized s.c. with either OVA-LeB, OVA-aDC-SIGN or native OVA mixed with anti-CD40 using a prime-boost protocol. Spleens were examined for the frequency of OVA-specific CD8+ and CD4+ effector T cells 1 week after boosting. (A–C) OVA-aDC-SIGN and OVA-LeB augmented generation of poly-functional effector CD8+ T cells, as revealed by higher frequencies of IFNγ- and TNF-double and IFNγ-single producing CD8+ T cells after o/n re-stimulation with OVA257-264. (D) Targeting antigen to DC-SIGN also enhanced induction of CD4+ effector T cells as shown by intra-cellular IFNγ-staining after 2 d restimulation with OVA262-276. Dashed lines represent frequencies of cytokine-secreting T cells in naive mice. n = 5 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001. Results shown are representative of two independent experiments.
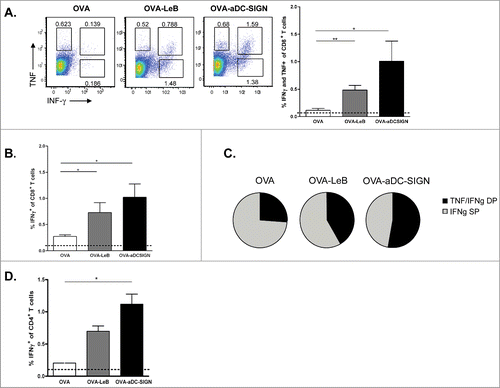
Next we analyzed the induction of CD4+ T effector cells as they are crucial for the generation of optimal effector and memory CD8+ T cell responses as well as promote accumulation of antigen-specific CD8+ T cells in the tumor.Citation38,39 Analysis of OVA-specific CD4+ T cell responses revealed that in comparison to untargeted OVA, immunization with both DC-SIGN targeting vaccines augmented the frequencies of OVA-specific IFNγ producing T cells (). These data show that targeting antigens to DC-SIGN on DCs using glycans or specific antibodies not only induced clonal expansion of antigen-specific CD4+ and CD8+ T-cells but also promotes their differentiation into cytokine-producing effector T cells.
Superior tumor control by combining Treg depletion with DC-SIGN targeted vaccination
These observations prompted us to examine the therapeutic efficacy of the DC-SIGN targeting vaccines in hSIGN mice bearing the aggressive B16-OVA melanoma. Vaccination was started on day 3 after tumor inoculation, followed by two boost vaccinations on days 10 and 17. However, we observed that three subsequent s.c. doses of either OVA-LeB or OVA-aDC-SIGN only slightly delayed B16-OVA tumor growth (). Tumor growth was even less affected when mice were immunized with non-targeted OVA. The marginal effect of the DC-SIGN targeting vaccines on tumor growth may result from the presence of tumor-associated Treg that are known to compromise the effector function of tumor-specific effector T cells.Citation2,30 To address this possibility, we crossed hSIGN mice with bacterial artificial chromosome (BAC)-transgenic DEREG mice in which FoxP3+ Treg can be depleted in a specific and controlled fashion by i.p. injection of DT. Indeed, depletion of Tregs in B16-OVA melanoma-bearing mice on day 7 and 8 combined with vaccination with non-targeted tumor antigen OVA delayed tumor growth (). Combining Treg depletion with adjuvant administration also delayed tumor growth. However, only when Treg depletion was combined with a DC-SIGN-targeting vaccine, a significant delay in tumor growth was observed (i.e., 10–70 days). Furthermore, the majority of mice survived long-term with small controlled tumors (). Importantly, comparable responses were induced by the antibody- and the glycan-based vaccine.
Figure 3. DC-SIGN-targeted vaccination combined with Treg depletion is a superior novel tumor therapy. (A) WT and hSIGN mice or B, DEREG and hSIGNxDEREG mice were challenged s.c. with B16-OVA tumor cells. Mice were vaccinated s.c. on days 3, 10 and 17 with either OVA-LeB, OVA-aDC-SIGN or native OVA mixed with anti-CD40. Control mice received PBS or anti-CD40. B, Mice were depleted of Tregs by DT injection on days 7 and 8 after tumor inoculation. (A–B) Individual tumor growth; (C) Mean tumor size in mice that survived long-term compared to that in untreated mice. (P < 0.05, OVA-LeB vs. untreated and OVA; OVA-aDC-SIGN vs. untreated; P < 0.01 OVA-aDC-SIGN vs. OVA). Data represent cumulative results from three independent experiments with 16-21 mice/group.
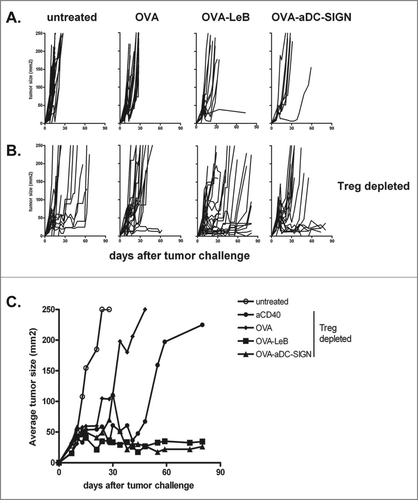
Thus, the circumvention of Treg-mediated suppression enhances the efficacy of vaccines targeting hSIGN.
Combination therapy promotes maximal tumor infiltration by OVA-specific lytic CD8+ T cells
Having shown a strong tumor regression in mice that received the combination therapy we next performed detailed analysis of the underlying cellular interactions. We have previously shown that the therapeutic effect of Treg depletion on B16-OVA melanoma regression relied on CD8+ T cells.Citation30,40 In line with previous data, depletion of Tregs led to the increased infiltration of tumors with CD8+ T cells but not CD4+ T cells (). Virtually all tumor-infiltrating CD8+ T cells were fully activated after Treg depletion as revealed by a CD62LlowCD44high phenotype (). Moreover, frequencies of activated CD8+ T cells in spleen and TDLN were enhanced after Treg depletion. Notably, CD4+ T cell activation was similarly enhanced in all tissues tested (). The activated CD8+ T cells found after Treg depletion were not OVA-specific as shown by the lack of staining with H2-Kb/SIINFEKL-pentamers (, aCD40 treated group), underlining that B16-associated OVA is poorly immunogenic, even after Treg depletion. When Treg depletion was combined with DC-SIGN-targeted vaccination, the frequencies of tumor-infiltrating CD8+ and CD4+ T cells did not further increase (). However, the combination therapy generated a large population of OVA-specific CD8+ T cells in the TDLNs and spleens of tumor-bearing mice (). Strikingly, these activated tumor antigen-specific CD8+ T cells accumulated in the tumor tissue (40–50% of the intra-tumoral CD8+ T cells; ), illustrating that they could effectively infiltrate melanomas. Both DC-SIGN-targeting vaccines showed similar potency of inducing antigen-specific CD8+ T cells in the tumor, TDLN and systemically.
Figure 4. Combination therapy maximizes tumor infiltration by OVA-specific effector CTLs. DEREG and hSIGNxDEREG mice were challenged s.c. with B16-OVA tumor cells. hSIGNxDEREG mice were vaccinated with either OVA-LeB, OVA-aDC-SIGN or native OVA mixed with anti-CD40 on days 3 and 10. Control mice received PBS. All mice were injected with DT on days 7 and 8 after tumor inoculation. On day 14 after tumor inoculation, mice were sacrificed and TILs, TDLNs and spleens were analyzed by flow cytometry to determine the frequency of (A) CD4+ and CD8+ T cells; (B) activated T cells defined by CD62LloCD44hi phenotype and (C) H2-Kb/SIINFEKL-pentamer-positive CD8+ T cells. (D) IFNγ production by activated CD8+ T cells in TDLN and spleen was determined by intracellular staining after OVA-specific restimulation ex vivo. Representative flow cytometric analyses are shown (right). Each dot represents one mouse. n = 5 mice/group. (left). (E) Additionally, frequencies (top panels) as well as absolute cell numbers (lower panels) of IFNγ production by activated CD8+ T cells in TDLN was determined by intracellular staining after TRP-2 and gp100-specific restimulation ex vivo. Each dot represents one mouse. n = 5 mice/group. *P < 0.05, **P < 0.01, *** P < 0.001. Graphs shown are representative of two independent experiments.
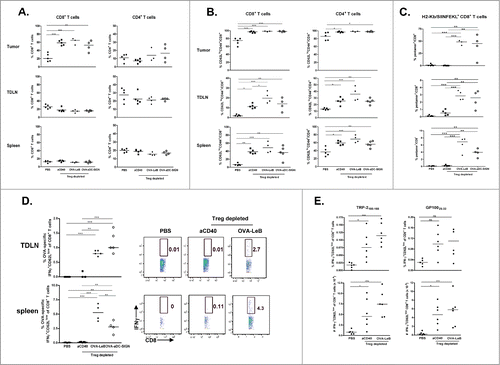
Furthermore, we demonstrate that these CD8+ T cells are functional effector cells in tumor-bearing mice, given that they vigorously produced IFNγ upon restimulation with SIINFEKL peptide () and induce melanoma regression ( andCitation30).
Even though none of the CD8+ T cells that infiltrated the tumor mass in non-vaccinated Treg depleted mice were specific for the OVA tumor antigen (), it is very likely that these CD8+ T cells possess specificity to endogenous melanoma antigens as delayed tumor growth was observed in these mice and it has been shown that mainly tumor antigen-specific T cells infiltrate tumors.Citation39 Indeed, we detected CD8+ T-cells with reactivity against gp100 and TRP2 in the TDLN () but not the spleens of Treg depleted mice (not shown).
Together, we show for the first time that a novel combination therapy consisting of transient Treg depletion and a DC-SIGN-targeting vaccine results in superior generation of a broad repertoire of tumor-specific effector CD8+ T cells and tumor control.
Discussion
Here we show that the specific combination of temporary Treg depletion and vaccination with DC-SIGN targeting formulations induces large numbers of effector T cells that infiltrate the tumor area and causes long-term tumor regression. Thus, this novel strategy is powerful enough to overcome tumor-induced immune suppression, a major obstacle for current immunotherapies. Although optimal priming is accomplished by targeting antigens to DC-SIGN, our data clearly demonstrate that depletion of tumor-associated Treg is needed to render this type of immunotherapy effective.
Deletion of Treg significantly delayed growth of established B16 melanomas and permitted the emergence of activated CD4+ and CD8+ T cells in the spleen and TDLNs (). Additionally, increased frequencies of activated and functional CD8+ T cells emerged within the tumor, which is in line with earlier observations by us and others (, Citation30,40). As expected none of these CD8+ T cells were specific for the OVA tumor antigen (), yet they possessed specificity to other melanoma antigens such as TRP-2 and gp100 (), as delayed tumor growth was observed in these mice and it has been shown that mainly tumor antigen-specific T cells infiltrate tumors.Citation39 Intriguingly, monotherapy consisting of Treg depletion delayed tumor growth but did not result in long-term tumor control (). Our observation that Treg depletion deterred tumor growth resembles those of others.Citation40,41 Yet, in these studies the efficacy of Treg ablation monotherapy was higher than in ours. This might be related to different mouse models used, which showed a more complete deletion of FoxP3+ Tregs. However, this near complete Treg ablation might predispose to autoimmune symptoms,Citation42,43 which was not observed in our study. Additionally, discrepancy could be related to differences in aggressiveness of the B16-OVA tumors used. In contrast to implanting in vitro cultured B16-OVA tumor cells,Citation40 in our study tumor cells were passaged in vivo before implanting them in the experimental mice, herewith selecting for the most aggressive clones expressing low levels of OVA. Combining Treg depletion with a non-targeted vaccine resulted in a stronger delay of the tumor growth than Treg depletion alone, corroborating our previous findings.Citation30 This effect might be due to the action and presence of OVA-specific CD8+ T cells in addition to activated CD8+ T cells specific for other tumor antigens. Nevertheless, this strategy could not install long-term tumor control in the majority of mice. Only when Treg depletion is combined with DC-SIGN targeting vaccines, tumor control is achieved. Our data suggest that Tregs mainly control the effector function of tumor antigen-specific CTLs and/or their mobilization into the tumor. The increased intra-tumoral presence of activated CD8+ T cells may be facilitated by alterations in the tumor vasculature, making the tumor more permissive for T cell infiltration. Indeed, expression of intercellular adhesion molecule (ICAM) and vascular adhesion molecule (VCAM), involved in adhesion and transmigration of T cells, are increased on tumor endothelial cells in the absence of Treg and strongly correlate with infiltration and tumor rejection.Citation40 Whether these alterations in adhesive molecules result from direct influence of Treg on the endothelial cells or from the action of inflammatory factors that become available in the absence of Treg is unclear.Citation32,44 However, one can speculate that Tregs also suppress the expansion or survival of CTLs given the strong increase of OVA-specific as well as gp100- and TRP-2-specific CTLs following combination therapy. Importantly, and in line with previous studies,Citation30,45 none of the mice that were temporarily depleted of Treg developed autoimmunity, as we did not observed vitiligo or uveitis, common bystander effects of systemic cutaneous melanoma reactivity. Furthermore no generalized autoimmunity was observed in any of the groups.
We found that the DC-SIGN targeting vaccines not only promote effector CD8+ T cell induction but also evoke generation of effector CD4+ T cells. Even though we used agonistic CD40 antibodies as an adjuvant, which licenses DCs and thus bypasses CD4+ T cell help for priming CD8+ T cells,Citation46 CD4+ T cell help is needed for optimal generation and maintenance of CD8+ T cells.Citation47 In fact, memory CD8+ T cells may be formed in mice vaccinated with the DC-SIGN targeting formulations, as only in those mice long-term tumor protection was observed when the vaccination was combined with Treg depletion. Furthermore, the activated tumor-specific CD4+ T cells may play an important role at later stages in the antitumor response as their help is required for tumor-specific CD8+ T cells to accumulate, survive, and function within the tumor.Citation39
Until now, the majority of vaccination studies with non-targeted peptide tumor antigens showed only marginal success at promoting long term survival.Citation48 Presumably, these studies may benefit from being combined with Treg depleting agents. So far, clinical studies focused on CD25 to deplete Tregs using either IL-2-Diphteria Toxin fusion protein (dinileukin diftitox or ONTAK) and LMB-2 in cancer patients.Citation34,35 However, even though tumor-specific T cells were elicited, no clinical responses have been reported. One disadvantage of using CD25-directed Treg depletion is that besides Treg multiple immune cells are eliminated that are crucial players in the antitumor response, such as activated T and NK cells and even DCs. Furthermore, limited efficacy may also be related to the fact that intra-tumor CD25+ Tregs as well as CD25− Tregs are not affected by this treatment. Thus, more selective agents are needed to reduce the number and/or suppressive function of Tregs in humans.
Current strategies are directed on the use of blocking antibodies against T cell inhibitory receptors such as programmed death-1 receptor (PD-1) or CTLA4.Citation49 These antibodies may act as a double edged-sword as they do not only boost effector T cell responses but also inhibit Treg suppressive function.Citation49 Even in advanced stages, improved clinical outcome has been observed in a limited set of patients. A phase three study in metastatic melanoma patients who had undergone previous treatments showed that the clinical grade anti-CTLA-4 antibody ipilimumab either alone or with gp100 peptide vaccination, improved overall survival as compared with gp100 vaccination alone.Citation50 Anti-CTLA-4 has been shown to elicit higher avidity tumor-specific T cells when used with vaccines.Citation51 However, this agent is not tumor-specific and generates a full-blown (systemic) immune activation causing severe side effects including significant autoimmunity such as colitis, rash, and endocrinopathy.Citation50 Also blockade of PD-1, an inhibitory receptor expressed by T cells, can overcome immune resistance and enhance T-cell responses.Citation52 Moreover, patients may benefit from combinations of these two agents as it was shown in the murine melanoma model that treatment comprising of GVAX with a combination of anti-PD-1 and CTLA-4 antibodies expanded the number of tumor-infiltrating T cells while reducing tumor-associated suppressive cells like Tregs.Citation53
In the present study, we found that specific targeting of DC-expressed DC-SIGN using glycan- and antibody-modified antigens induced comparable T cell responses in vivo. Future studies will reveal whether or not these DC-SIGN targeting modalities generate different effector/memory T cell populations.
Together, our study clearly shows that DC-specific vaccination strategies significantly benefit from transient elimination of immunosuppressive Tregs. We showed that transient depletion of Treg allows the local appearance of melanoma-antigen-specific CD8+ T-cells. Thus, antitumor immune responses can be broadened by therapies aimed at controlling Tregs in tumor environments. This novel combination therapy may serve as prototype for the development of effective next generation immunotherapies in cancer patients.
Methods
Mice
hSIGN and DEREG transgenic mice were described previously.Citation21,37 hSIGN, hSIGNxDEREG mice, OT-I and OT-II tg were bred at the animal facilities of Twincore (Hannover, Germany) or VU University (Amsterdam, Netherlands) under specific pathogen-free conditions and used at 8–16 weeks of age. All experiments were performed according to institutional and national guidelines.
Generation of neo-glycoconjugates
For conjugation of LeB (Dextra labs, UK) to OVA, creating OVA-LeB, a bifunctional cross linker ((4-N-Maleimidophenyl) butyric acid hydrazide; MPBH; Pierce, Rockford, USA) was used as described previously.Citation28 In short, the hydrazide moiety of the linker is covalently linked to the reducing end of the carbohydrate via reductive amination. After 2 h incubation at 70°C, the mixtures were cooled down to RT. 1 mL ice-cold isopropanol (HPLC grade; Riedel de Haan, Seelze, Germany) was added and further incubated at ˗20°C for 1 h. The precipitated derivatized carbohydrates were pelleted and dissolved in 1 mM HCL. OVA (Calbiochem, Darmstadt, Germany) dissolved in PBS was added to derivatised carbohydrates of interest (10:1 molar equivalent carbohydrate:OVA) and conjugation was performed o/n at 4°C. Alternatively, anti-DC-SIGN antibodies (clone AZN-D1) were conjugated to OVA. Hereto, the AZN-D1 or an isotype control antibody were conjugated to OVA using the cross-linking agent sulfosuccinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate according to the manufacturer's protocol (Pierce). Glyco- or antibody-conjugates were separated from reaction-reductants using PD-10 desalting columns (Pierce, Rockford, USA). The concentration of OVA was determined using the bicinchoninic acid assay (Pierce). Presence of antibody or glycan on OVA was determined by ELISA, as described.Citation28,26
Antigen-presentation assays
T-cell proliferation assays were performed as previously described.Citation28 In short, DC were pulsed with indicated concentrations of OVA-LeB, OVA-aDC-SIGN, OVA-isotype ctrl or native OVA for 4 h before incubation with OVA-specific T-cells (OT-I or OT-II; 1:2 DC:T). [3H]-thymidine (1μCi/well; Amersham Biosciences, NJ, USA) was present during the last 16 h of a 72 h culture. [3H]-thymidine incorporation was measured using a Wallac microbeta counter (Perkin-Elmer, USA).
Vaccination and tumor therapy
Mice were injected s.c. either with 100 μg OVA-LeB or 10 μg OVA-aDC-SIGN mixed with 25 μg anti-CD40 Ab (1C10) on day 0 and day 14. Mice were sacrificed one week after boost and frequencies of OVA-specific cytokine-secreting T-cells were analyzed in the spleen by flow cytometry. B16-OVA melanoma cells were cultured as described.Citation30 For therapeutic vaccination experiments, mice were challenged with 2 × 105 tumor cells s.c. in the flank. Three days later, mice were vaccinated s.c. in the tailbase with 100 μg OVA, 100 μg OVA-LeB or 5 μg OVA-aDC-SIGN mixed with 25 μg anti-CD40 Ab (1C10) and were boosted 7 and 14 d later. Mice were injected with 1 μg DT i.p. on days 7 and 8 after tumor inoculation to deplete Tregs. As control, also WT and hSIGN mice were injected with DT. Tumor size was measured using calipers and expressed as length × width. Mice were sacrificed when tumors reached an area of 225 mm2.
Ex vivo isolation of TILs
To analyze T cell responses after immunotherapy, mice were sacrificed on day 14 after tumor inoculation. For TIL isolation, tumors were dissected, cut into small pieces and incubated in RPMI medium supplemented with 10% FCS, 50 U/mL penicillin, 50 μg/mL streptomycin, 1 mg/mL Collagenase D (Roche Diagnostics GmbH, Manheim, Germany) and 50 μg/mL DNase I (Roche) for 30 min at 37°C. Tumor fragments were passed through a cell strainer and digested for additional 30 min, followed by addition of 10 mM EDTA for the last 5 min. Red blood cells were lysed with ACK lysis buffer (0.15M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) and TILs were purified by Percoll gradient centrifugation.
T cell restimulation
For the detection of OVA-, TRP-2, gp100-specific IFNγ producing CD8+ T cells, lymphocytes were re-stimulated with 1.0 μg/mL OVA257-264, TRP-2180-188, gp10025-32, respectively in the presence of 5 μg/ml Brefeldin A (BD PharMingen) for 4–5 h. To address the presence of polyclonal IFNγ producing CD8+ and CD4+ T cells, cells were restimulated with 100 ng/ml phorbol 12-myristate 13-acetate (PMA), 10 μg/ml ionomycin and 5 μg/ml Brefeldin A. IFNγ production by OVA-specific CD4+ T cells was analyzed upon two day re-stimulation with OVA262-276.
Flow cytometry
Anti-CD4 (GK1.5), anti-CD8β (H35–17.2), anti-CD44 (IM7), anti-CD62L (MEL-14) and anti-IFNγ (XMG1.2) antibodies were purchased from eBioscience or BD PharMingen. OVA-specific CD8+ T cells were visualized by staining cells with H-2Kb/SIINFEKL pentamers (ProImmune, Oxford, UK). Anti-TNF antibody was purchased from BD PharMingen. Intracellular cytokine staining was performed after fixation with 2% paraformaldehyde and permeabilization with 0.5% Saponin.
Cells were acquired on LSRII (BD, Biosciences) or CyAn™ ADP (Beckman Coulter). Data analysis was performed with FlowJo software (Tree Star). Dead cells were excluded by PI, DAPI or ethidium bromide monoazide (Sigma).
Statistical analysis
Prism software (GraphPad 5.0) was used for statistical analysis. One way Anova, Student's t test or log-rank test (tumor experiments) was used. Statistical significance, assessed by calculating the P values, was defined as P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was financially supported by a grant from SenterNovem SII071030 (W.W.J.U., M.L.), NanoNextInitiative 03D03 (SE), FP7 Marie Curie ITN 213592 (M.P.), and EuroTransBio/PDVAC. C.T.M. was supported by the German National Academic Foundation.
References
- Seder RA, Darrah PA, Roederer M.T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol 2008; 8:247-58; PMID:18323851; http://dx.doi.org/10.1038/nri2274
- Menetrier-Caux C, Curiel T, Faget J, Manuel M, Caux C, Zou W. Targeting regulatory T cells. Target Oncol 2012; 7:15-28; PMID:22327882; http://dx.doi.org/10.1007/s11523-012-0208-y
- Nishikawa H, S Sakaguchi. Regulatory T cells in tumor immunity. Int J Cancer 2010; 127:759-67; PMID:20518016; http://dx.doi.org/10.1002/ijc.25429
- Chen DS, I Mellman. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013; 39:1-10; PMID:23890059; http://dx.doi.org/10.1016/j.immuni.2013.07.012
- Belkaid Y, G Oldenhove. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity 2008; 29: 362-71.; PMID:18799144; http://dx.doi.org/10.1016/j.immuni.2008.08.005
- Mayer CT, Berod L, Sparwasser T. Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front Immunol 2012; 3:183; PMID:22783257; http://dx.doi.org/10.3389/fimmu.2012.00183
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942-9; PMID:15322536; http://dx.doi.org/10.1038/nm1093
- Kryczek I, Liu R, Wang G, Wu K, Shu X, Szeliga W, Vatan L, Finlayson E, Huang E, Simeone D et al. FOXP3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Res 2009; 69:3995-4000; PMID:19383912; http://dx.doi.org/10.1158/0008-5472.CAN-08-3804
- Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, Betts GJ, Singh Y, Price DA, Godkin AJ et al. Analysis of the T-cell receptor repertoires of tumor-infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen-induced tumors. Cancer Res 2011; 71:736-46; PMID:21156649; http://dx.doi.org/10.1158/0008-5472.CAN-10-1797
- Haen SP, HG Rammensee. The repertoire of human tumor-associated epitopes - identification and selection of antigens and their application in clinical trials. Curr Opin Immunol 2013; 25:277-83; PMID:23619309; http://dx.doi.org/10.1016/j.coi.2013.03.007
- Sosman JA, Carrillo C, Urba WJ, Flaherty L, Atkins MB, Clark JI, Dutcher J, Margolin KA, Mier J, Gollob J et al. Three phase II cytokine working group trials of gp100 (210M) peptide plus high-dose interleukin-2 in patients with HLA-A2-positive advanced melanoma. J Clin Oncol 2008; 26:2292-8; PMID:18467720; http://dx.doi.org/10.1200/JCO.2007.13.3165
- Di Pucchio T, Pilla L, Capone I, Ferrantini M, Montefiore E, Urbani F, Patuzzo R, Pennacchioli E, Santinami M, Cova A, et al. Immunization of stage IV melanoma patients with Melan-A/MART-1 and gp100 peptides plus IFN-alpha results in the activation of specific CD8(+) T cells and monocyte/dendritic cell precursors. Cancer Res 2006; 66: 4943-51; PMID:16651452; http://dx.doi.org/10.1158/0008-5472.CAN-05-3396
- Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, Gailani F, Riley L, Conlon K, Pockaj B et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med 2011; 364:2119-27; PMID:21631324; http://dx.doi.org/10.1056/NEJMoa1012863
- Smith JW, Walker EB, Fox BA, Haley D, Wisner KP, Doran T, Fisher B, Justice L, Wood W, Vetto J et al. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J Clin Oncol 2003; 21:1562-73; PMID:12697882; http://dx.doi.org/10.1200/JCO.2003.09.020
- Walker EB, Haley D, Miller W, Floyd K, Wisner KP, Sanjuan N, Maecker H, Romero P, Hu HM, Alvord WG, et al. gp100(209-2M) peptide immunization of human lymphocyte antigen-A2+ stage I-III melanoma patients induces significant increase in antigen-specific effector and long-term memory CD8+ T cells. Clin Cancer Res 2004; 10:668-0; PMID:14760090
- Caminschi I, Maraskovsky E, Heath WR. Targeting dendritic cells in vivo for cancer therapy. Front Immunol 2012; 3:13; PMID:22566899; http://dx.doi.org/10.3389/fimmu.2012.00013
- Engering A, Geijtenbeek TB, van Vliet SJ, Wijers M, van Liempt E, Demaurex N, Lanzavecchia A, Fransen J, Figdor CG, Piguet V. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol 2002; 168:2118-26; PMID:11859097; http://dx.doi.org/10.4049/jimmunol.168.5.2118
- Kretz-Rommel A, Qin F, Dakappagari N, Torensma R, Faas S, Wu D, Bowdish KS. In vivo targeting of antigens to human dendritic cells through DC-SIGN elicits stimulatory immune responses and inhibits tumor growth in grafted mouse models. J Immunother 2007; 30:715-26; PMID:17893564; http://dx.doi.org/10.1097/CJI.0b013e318135472c
- Tacken PJ, de Vries IJ, Gijzen K, Joosten B, Wu D, Rother RP, Faas SJ, Punt CJ, Torensma R, Adema GJ et al. Effective induction of naive and recall T-cell responses by targeting antigen to human dendritic cells via a humanized anti-DC-SIGN antibody. Blood 2005; 106:1278-85; PMID:15878980; http://dx.doi.org/10.1182/blood-2005-01-0318
- Garcia-Vallejo JJ, Y van Kooyk. The physiological role of DC-SIGN: a tale of mice and men. Trends Immunol 2013; 34:482-6; PMID:23608151; http://dx.doi.org/10.1016/j.it.2013.03.001
- Schaefer M, Reiling N, Fessler C, Stephani J, Taniuchi I, Hatam F, Yildirim AO, Fehrenbach H, Walter K, Ruland J et al. Decreased pathology and prolonged survival of human DC-SIGN transgenic mice during mycobacterial infection. J Immunol 2008; 180:6836-45; PMID:18453604; http://dx.doi.org/10.4049/jimmunol.180.10.6836
- Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, Torensma R, Figdor CG. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release 2010; 144:118-26; PMID:20156497; http://dx.doi.org/10.1016/j.jconrel.2010.02.013
- Hesse C, Ginter W, Förg T, Mayer CT, Baru AM, Arnold-Schrauf C, Unger WW, Kalay H, van Kooyk Y, Berod L et al. In vivo targeting of human DC-SIGN drastically enhances CD8+ T-cell-mediated protective immunity. Eur J Immunol 2013; 43:2543-53; PMID:23784881; http://dx.doi.org/10.1002/eji.201343429
- Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol 2003; 170:1635-9; PMID:12574325; http://dx.doi.org/10.4049/jimmunol.170.4.1635
- Garcia-Vallejo JJ, Ambrosini M, Overbeek A, van Riel WE, Bloem K, Unger WW, Chiodo F, Bolscher JG, Nazmi K, Kalay H et al. Multivalent glycopeptide dendrimers for the targeted delivery of antigens to dendritic cells. Mol Immunol 2013; 53:387-97; PMID:23103377; http://dx.doi.org/10.1016/j.molimm.2012.09.012
- Unger WW, van Beelen AJ, Bruijns SC, Joshi M, Fehres CM, van Bloois L, Verstege MI, Ambrosini M, Kalay H, Nazmi K et al. Glycan-modified liposomes boost CD4+ and CD8+ T-cell responses by targeting DC-SIGN on dendritic cells. J Control Release 2012; 160:88-95; PMID:22366522; http://dx.doi.org/10.1016/j.jconrel.2012.02.007
- Aarnoudse CA, Bax M, Sánchez-Hernández M, García-Vallejo JJ, van Kooyk Y. Glycan modification of the tumor antigen gp100 targets DC-SIGN to enhance dendritic cell induced antigen presentation to T cells. Int J Cancer 2008; 122:839-46; PMID:17957800; http://dx.doi.org/10.1002/ijc.23101
- Singh SK, Stephani J, Schaefer M, Kalay H, García-Vallejo JJ, den Haan J, Saeland E, Sparwasser T, van Kooyk Y. Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol Immunol 2009; 47:164-74; PMID:19818504; http://dx.doi.org/10.1016/j.molimm.2009.09.026
- Yang L, Yang H, Rideout K, Cho T, Joo KI, Ziegler L, Elliot A, Walls A, Yu D, Baltimore D et al. Engineered lentivector targeting of dendritic cells for in vivo immunization. Nat Biotechnol 2008; 26:326-34; PMID:18297056; http://dx.doi.org/10.1038/nbt1390
- Klages K, Mayer CT, Lahl K, Loddenkemper C, Teng MW, Ngiow SF, Smyth MJ, Hamann A, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells improves effective therapeutic vaccination against established melanoma. Cancer Res 2010; 70:7788-99; PMID:20924102; http://dx.doi.org/10.1158/0008-5472.CAN-10-1736
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995; 155:1151-64; PMID:7636184
- Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med 2008; 205:2125-38; PMID:18725522; http://dx.doi.org/10.1084/jem.20080099
- Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor Rejection by in Vivo Administration of Anti-CD25 (Interleukin-2 Receptor +¦) Monoclonal Antibody. Cancer Res 1999; 59:3128-33; PMID:10397255
- Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest 2005; 115:3623-33; PMID:16308572; http://dx.doi.org/10.1172/JCI25947
- Morse MA, Hobeika AC, Osada T, Serra D, Niedzwiecki D, Lyerly HK, Clay TM. Depletion of human regulatory T cells specifically enhances antigen-specific immune responses to cancer vaccines. Blood 2008; 112:610-8; PMID:18519811; http://dx.doi.org/10.1182/blood-2008-01-135319
- Baur AS, Lutz MB, Schierer S, Beltrame L, Theiner G, Zinser E, Ostalecki C, Heidkamp G, Haendle I, Erdmann M et al. Denileukin diftitox (ONTAK) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting Treg. Blood 2013; 122:2185-94; PMID:23958949; http://dx.doi.org/10.1182/blood-2012-09-456988
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med 2007; 204:57-63; PMID:17200412; http://dx.doi.org/10.1084/jem.20061852
- Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003; 421:852-6; PMID:12594515; http://dx.doi.org/10.1038/nature01441
- Bos R, LA Sherman. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 2010; 70:8368-77; PMID:20940398; http://dx.doi.org/10.1158/0008-5472.CAN-10-1322
- Li X, Kostareli E, Suffner J, Garbi N, Hämmerling GJ. Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur J Immunol 2010; 40:3325-35; PMID:21072887; http://dx.doi.org/10.1002/eji.201041093
- Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 2013; 210: 2435-66; PMID:24127486; http://dx.doi.org/10.1084/jem.20130762
- Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 2007; 8:191-7; PMID:17136045; http://dx.doi.org/10.1038/ni1428
- Mayer CT, Ghorbani P, Kühl AA, Stüve P, Hegemann M, Berod L, Gershwin ME, Sparwasser T. Few Foxp3 regulatory T cells are sufficient to protect adult mice from lethal autoimmunity. Eur J Immunol 2014; 44:2990-3002 (epub ahead of print)PMID:25042334; http://dx.doi.org/10.1002/eji.201344315
- Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, Vardam TD, Weis EL, Passanese J, Wang WC et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest 2011; 121:3846-59; PMID:21926464; http://dx.doi.org/10.1172/JCI44952
- Teng MW, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ. Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res 2010; 70:7800-9; PMID:20924111; http://dx.doi.org/10.1158/0008-5472.CAN-10-1681
- Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998; 393:480-3; PMID:9624005; http://dx.doi.org/10.1038/31002
- Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med 1998; 187:693-702; PMID:9480979; http://dx.doi.org/10.1084/jem.187.5.693
- Haen SP, HG Rammensee. The repertoire of human tumor-associated epitopes - identification and selection of antigens and their application in clinical trials. Curr Opin Immunol 2013; 25:277-83; PMID:23619309; http://dx.doi.org/10.1016/j.coi.2013.03.007
- Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med 2009; 206:1717-25; PMID:19581407; http://dx.doi.org/10.1084/jem.20082492
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/10.1056/NEJMoa1003466
- Hodge JW, Chakraborty M, Kudo-Saito C, Garnett CT, Schlom J. Multiple costimulatory modalities enhance CTL avidity. J Immunol 2005; 174:5994-6004; PMID:15879092; http://dx.doi.org/10.4049/jimmunol.174.10.5994
- Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol 2012; 24:207-12; PMID:22236695; http://dx.doi.org/10.1016/j.coi.2011.12.009
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A 2010; 107:4275-80; PMID:20160101; http://dx.doi.org/10.1073/pnas.0915174107