
Abstract
The high efficacy of therapeutic cancer vaccines in preclinical studies has yet to be fully achieved in clinical trials. Tumor immune suppression is a critical factor that hampers the desired antitumor effect. Here, we analyzed the combined effect of a cancer vaccine and the receptor tyrosine kinase inhibitor sunitinib. Sunitinib was administered intraperitoneally, alone or in combination with intramuscular immunization using a viral vector based cancer vaccine composed of Semliki Forest virus replicon particles and encoding the oncoproteins E6 and E7 (SFVeE6,7) of human papilloma virus (HPV). We first demonstrated that treatment of tumor-bearing mice with sunitinib alone dose-dependently depleted myeloid-derived suppressor cells (MDSCs) in the tumor, spleen and in circulation. Concomitantly, the number of CD8+ T cells increased 2–fold and, on the basis of CD69 expression, their activation status was greatly enhanced. The intrinsic immunosuppressive activity of residual MDSCs after sunitinib treatment was not changed in a dose-dependent fashion. We next combined sunitinib treatment with SFVeE6,7 immunization. This combined treatment resulted in a 1.5- and 3-fold increase of E7-specific cytotoxic T lymphocytes (CTLs) present within the circulation and tumor, respectively, as compared to immunization only. The ratio of E7-specific CTLs to MDSCs in blood thereby increased 10- to 20-fold and in tumors up to 12.5-fold. As a result, the combined treatment strongly enhanced the antitumor effect of the cancer vaccine. This study demonstrates that sunitinib creates a favorable microenvironment depleted of MDSCs and acts synergistically with a cancer vaccine resulting in enhanced levels of active tumor-antigen specific CTLs, thus changing the balance in favor of antitumor immunity.
Abbreviations:
- ARG1, arginase-1
- CTL, cytotoxic T lymphocyte
- DC, dendritic cell
- HPV, human papilloma virus
- Flt3, Fms-like tyrosine kinase 3
- iNOS, nitric oxide synthase
- MDSC, myeloid-derived suppressor cell
- mRCC, metastatic renal cell carcinoma
- PBMC, peripheral blood mononuclear cell
- rSFV, recombinant Semliki forest virus
- Treg, regulatory T cell
- TGFβ, transforming growth factor β
- VEGF, vascular endothelial growth factor receptor.
Introduction
Immunotherapy against cancer aims at the induction and subsequent migration of tumor-antigen specific T cells into the tumor. This can be achieved by therapeutic immunizations generating tumor antigen-specific T cells in combination with treatments that enhance intratumoral infiltration of T cells.Citation1,2 However, intratumoral immunosuppressive cells may hamper immune-based tumor eradication, despite the presence of tumor antigen-specific T cells.Citation3
Myeloid-derived suppressor cells (MDSCs) are immunosuppressive cells of myeloid origin composed of immature macrophages, granulocytes and dendritic cells. Increased levels of MDSCs have been reported in spleen, bone marrow and circulation in tumor-bearing animals and cancer patients.Citation4 Intratumoral MDSC levels correlate with tumor progression, metastasis and resistance to cancer therapies.Citation5,6 In mice, MDSCs are identified by the simultaneous expression of the myeloid antigens CD11b and Gr-1 on their cell membrane. MDSCs can suppress T-cell functions via inducible nitric oxide synthase (iNOS) and arginase-1 (ARG1).Citation7,8 To overcome the immunosuppressive effects of MDSCs and potentiate antitumoral immune responses, depletion or functional inhibition of MDSCs in combination with immunization regimens could be an option. Citation9,10
Sunitinib malate is an oral, broad spectrum, receptor tyrosine kinase inhibitor currently used as a first-line therapy of metastatic renal cell carcinoma and for treatment of gastrointestinal stromal tumors resistant to conventional therapy.Citation11-13 The mechanism of action of sunitinib consists of blockade of several receptor tyrosine kinases localized on both tumor cells and MDSCs, such as vascular endothelial growth factor receptor (VEGFR), Flt3 Fms-like tyrosine kinase 3 (Flt3) and c-kit (CD117).Citation14 Previous studies have shown that blockade of these receptors skew MDSCs towards an immunosuppressive phenotypeCitation15 and reverse immune tolerance in a variety of murine tumor models.Citation16-18
We developed a cancer vaccine against human papillomavirus (HPV) induced cancer, based on a recombinant alphavirus, namely Semliki Forest virus (rSFV). The rSFV replicon particles encode a fusion protein of HPV type 16 E6 and E7 derived from HPV type 16 (termed SFVeE6,7). Immunization with SFVeE6,7 particles generates high levels of HPV-specific T cells and eradicates established HPV-transformed tumors. Using an HPV tumor model in mice, we studied the effect of sunitinib on intratumoral MDSC accumulation and activity of intratumoral antigen-specific T cells. This study aimed to determine if the efficacy of a cancer vaccine could be enhanced by depletion or functional inactivation of the immunosuppressive MDSC population.
Results
Effect of sunitinib on intratumoral and intrasplenic levels of MDSCs and Tregs
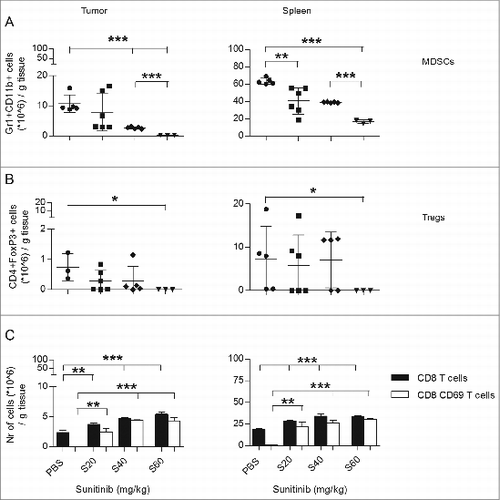
We first determined the effect of sunitinib on MDSC accumulation in tumors and spleens of TC-1 tumor-bearing mice. Starting day 15 after tumor inoculation, mice received sunitinib daily for a period of 9 days. Subsequently, the levels of MDSCs in tumors and spleens were assessed by immunostaining and fluorescence cytometry (Fig. S1). The absolute numbers of both intratumoral and intrasplenic MDSCs decreased dose-dependently (, Fig. S2B). As MDSCs have been shown to induce expansion of regulatory T cells (Tregs,Citation19 we also determined the effect of sunitinib on Treg levels. Sunitinib slightly, but not significantly, decreased numbers of intratumoral and intrasplenic Tregs. Only the highest dose of sunitinib resulted in a significant decrease of Tregs (). However, as this highest dose of sunitinib resulted in weight loss of the mice, this dosage was not used in further experiments.
Figure 1. The effect of sunitinib treatment on intratumoral MDSC, Tregs, CD8+ T cells and activation status of the CD8 T cell population. C57Bl6 mice were subcutaneously inoculated with TC-1 tumor cells (n = 3–6 mice/group). On day 15 after tumor inoculation, sunitinib treatment was started, daily, i.p., for a period of 9 consecutive days. Three increasing dosages of sunitinib were used: 20, 40 and 60 mg/kg body weight. Mice were sacrificed and tumors and spleens were harvested. The levels of (A) MDSCs (CD11b+Gr1+), (B) Tregs (CD4+FoxP3+), (C) CD8+ T cells (CD8+; black bars) and the activation status of CD8+ T cells (CD69+; white bars) were analyzed by immunostaining and multicolor fluorescence cytometry. Experiments were repeated twice. Shown here are averages and SD for each experimental group (*p < 0.05; **p < 0.01; ***p < 0.001).
Effect of sunitinib on intratumoral and intrasplenic levels of CD8+ T cells
We next, set out to determine the effect of sunitinib on the intratumoral and intrasplenic levels of CD8+ T cells, as well as the fraction of CD8+ T cells positive for CD69, the earliest inducible cell surface glycoprotein acquired during lymphocyte activation (Fig. S3).Citation20 The absolute numbers of both intratumoral and intrasplenic CD8+ T cells increased dose-dependently (, black bars, and Fig. S2C). More than 95% of CD8+ T cells isolated from the primary tumors and spleens of the untreated group were negative for the activation marker CD69. Notably, more than 50% of CD8+ T cells were positive for CD69 after sunitinib treatment, irrespective of the dosage employed (, white bars). These results indicate that sunitinib treatment not only increases the numbers but also the expression of the activation marker CD69 on the surface of intratumoral and intrasplenic CD8+ T cells (Fig. S3).
Combined effect of immunization and sunitinib on intratumoral and intrasplenic levels of total and E7-specific CD8+ T cells, MDSCs and Tregs
We next investigated the combined effect of sunitinib and viral vector immunization on intratumoral and intrasplenic levels of different immune cell populations. Mice were i.m. injected once on day 14 after tumor inoculation with 5×106 SFVeE6,7 particles. Sunitinib treatment was started the following day, with 20 or 40 mg/kg body weight i.p., and continued for a period of 9 consecutive days. On day 24 tumors and spleens were harvested and analyzed. The priming SFVeE6,7 immunization did not influence the depletion of intratumoral and intrasplenic MDSCs induced by sunitinib ( versus ). Also the number of Tregs was not altered by SFVeE6,7 priming ( versus ).
Figure 2. Therapeutic immunization and sunitinib enhance intratumoral and intrasplenic levels of total CD8+ T cells, but not MDSC and Tregs. Mice were s.c. inoculated with TC-1 tumor cells (n = 3 mice/group). On day 14 after tumor inoculation mice were vaccinated once, i.m., with 5 × 106 SFVeE6,7 particles. One day later, sunitinib treatment of 20 and 40 mg/kg body weight was started, i.p., for a period of 9 consecutive days. The levels of (A) MDSCs (Gr1+CD11b+), (B) Tregs (CD4+FoxP3+), (C) activated total CD8+ T cells (CD8+CD69+), and (D) activated E7-antigen specific CD8+ T cells (CD8+E7+CD69+) in tumors and spleens were analyzed by immunostaining and fluorescence cytometry on day 24. Experiments were repeated twice. Shown here are averages and SD for each experimental group (*p < 0.05).
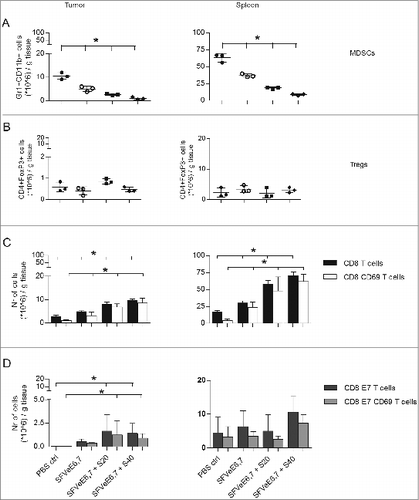
Notably, the combination of immunization and sunitinib led to an increase in intratumoral and intrasplenic CD8+ T cells, as compared to SFVeE6,7 immunization or sunitinib treatment alone ( versus ), coincident with an increase in the number of antigen-specific CD8+ T cells ( and ).
Figure 3. MDSCs isolated from tumors of TC-1 tumor-bearing mice suppress the anti-tumoral activity of CD8+ T cells. TC1 tumor-bearing mice (n = 5–7 mice/group) were divided in two subgroups, the first subgroup receiving PBS and the second receiving 40 mg/kg body weight sunitinib, i.p., for a period of nine consecutive days. At the end of the experiment, intratumoral MDSCs were isolated and positively sorted (CD11b+Gr1+). A separate group of mice received 2 SFVeE6,7 immunizations, in a dosage of 5 × 106 particles, on days 0 and 14. This group of mice was then sacrificed 10 days later and splenocytes were isolated and co-cultured with different ratios of MDSCs (isolated from the first two subgroups and positively sorted) for a period of 7 days. (A) A 51Chromium release assay was performed to determine the suppressive effect of MDSCs. The target cells used in this assay were C3 tumor cells expressing the full HPV16 genome. (B) On day 4 of co-culture, a portion of the cells were labeled with carboxyfluorescein succinimidyl ester (CFSE). On day 7 of co-culture, the E7-antigen specific CD8+ T-cell proliferation was determined by multicolor fluorescence cytometry. Experiments were repeated twice. Shown here are averages and SD for each experimental group (*p < 0.05; ***p < 0.001).
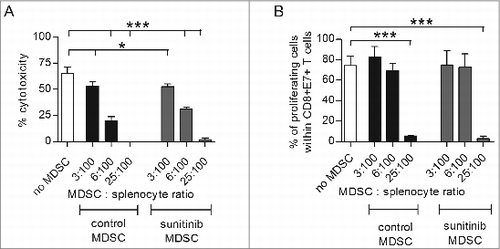
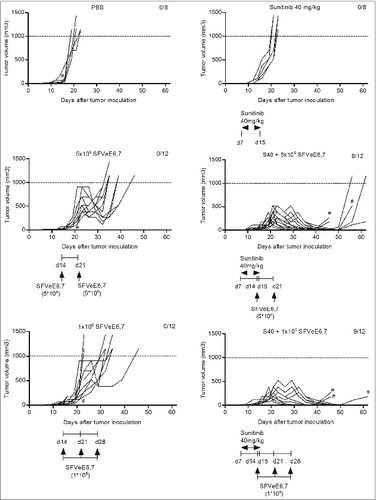
Figure 4. Combination of sunitinib and therapeutic vaccination abrogates tumor growth. Mice (n = 4–6/group) were s.c. inoculated with TC-1 tumor cells. On day 7 after tumor inoculation, sunitinib treatment was started, daily, i.p., for a period of 9 consecutive days. Mice where then immunized with SFVeE6,7 i.m., either on days 14 and 21 after tumor inoculation with a dosage of 5 × 106 particles, or on days 14, 21, and 28 with a dosage of 1 × 106 particles, respectively. Tumor measurements were performed periodically; when tumor size exceeded 1000 mm3 or when tumors protruded through the skin (#) mice were sacrificed due to ethical reasons. Experiments were repeated at least twice. Shown here are the tumor growth curves for each experimental group, per mouse.
Remarkably, with a sunitinib dose of 40 mg/kg body weight, the ratio of E7-specific CD8+ T cells to MDSCs increased 12.5-fold, from 0.1:1 upon immunization only, to 1.25:1 after combined sunitinib and immunization treatment.
Intrinsic immunosuppressive activity of MDSCs after sunitinib treatment
Next, MDSCs were isolated and their intrinsic suppressive activity was determined ex-vivo. MDSCs isolated from TC-1 tumors of PBS- or sunitinib-treated mice were co-cultured with splenocytes isolated from SFVeE6,7 immunized mice. Cytolytic activity and proliferation of CD8+ T cells were measured after 7 days of co-culture.
Cytolytic activity of CD8+ T cells, in the absence or presence of MDSCs, was measured in a chromium51 release assay, in which C3 tumor cells were used as target cells and the effector to target cell ratio was 100:1. The percentage of cytotoxicity decreased from ∼70-75% in the asbsence of exogenous MDSCs to ∼55% and ∼25-30% at MDSC to splenocyte ratios of 3:100, and 6:100, respectively. The addition of 25 MDSCs per 100 splenocytes completely blocked cytolysis (). The inhibitory effects between MDSCs isolated from tumors of the PBS or sunitinib treated groups did not differ.
Proliferation of E7-antigen specific T cells in the absence or presence of MDSCs at the above-mentioned ratios were measured by carboxyfluorescein succinimidyl ester (CFSE)-labeling of cells on day 4 and flow cytometric analysis on day 7 of co-culture. In the absence of additional MDSCs, the percentage of proliferation was ∼80%, similar to that when MDSCs were added at an MDSC to splenocyte ratio of 3:100 and 6:100, respectively. However, at an MDSC to splenocyte ratio of 25:100 the percentage of proliferation decreased to 1–5% ( and Fig. S5). This inhibitory effect on proliferation was also not influenced by sunitinib treatment.
We next analyzed changes in mRNA levels of different enzymes and factors that have been previously implicated as being responsible for MDSC-mediated immune suppression, such as arginase-1 (ARG1), inducible nitric oxide synthase (iNOS) and transforming growth factor β (TGFβ).Citation8,21 Thus, we analyzed changes in mRNA levels of these factors in MDSCs isolated from tumors of PBS- or 40 mg/kg body weight sunitinib-treated mice. The fold expression of these factors in MDSCs isolated from tumors of sunitinib-treated mice was similar to those in MDSCs isolated from tumors of PBS-treated mice (Table S2).
Taken together, these results suggest that sunitinib treatment induced a decrease in circulating as well as intratumoral MDSCs, while no change in intrinsic activity of intratumoral MDSCs was observed upon sunitinib treatment.
In vivo antitumor response of combinatorial treatment and effect on blood immune cells
We have previously shown that optimal immunization with an SFVeE6,7 dosage of 5×106 particles administered i.m. on days 7, 14, 21 after TC-1 tumor inoculation leads to complete tumor regression in all mice.Citation22 In order to evaluate the additive effect of sunitinib we immunized mice with two different suboptimal immunization schedules and dosages. For the first suboptimal regime, mice were immunized with the standard dose of 5 × 106 SFVeE6,7 particles, administered i.m. starting at a late time point after tumor inoculation and only twice, i.e., on days 14 and 21 after tumor inoculation. The second suboptimal regimen involved immunization with a suboptimal dose of vaccine, i.e. 1×106 SFVeE6,7 particles, administered i.m. also starting at a late time-point, i.e., 14, 21 and 28 after tumor inoculation. These stringent suboptimal SFVeE6,7 regimens, used in combination with sunitinib, would allow us to monitor temporal changes in levels of circulating immune cells as well as tumor growth. Sunitinib treatment (40 mg/kg body weight, i.p.) was started on day 7 after tumor inoculation. This created an intratumoral immune environment deprived of MDSCs and enriched in CD69+ E7-specific CD8+ T cells (Fig. S6A and B).
Sunitinib alone neither delayed nor promoted tumor growth as compared to the untreated group () and mice had to be sacrificed at the same time point. This result indicates that, despite the fact that concentrations of sunitinib higher than 1 μM induce TC-1 cell death in vitro, 40 mg/kg body weight as employed in vivo does not seem to directly affect tumor growth. The 2 suboptimal SFVeE6,7 immunization regimens alone still resulted in a delay in tumor growth as compared to the PBS or sunitinib treated groups (). In contrast, in the group of mice receiving sunitinib followed by the first suboptimal immunization regime, (i.e., only receiving 2 immunizations, starting at a late time point with a standard dose of SFVeE6,7) 66% survived up to day 62 post inoculation (). Notably, the combination of a low dose immunization with sunitinib increased survival up to 75% (). It should be noted that, for ethical reasons, mice had to be sacrificed when tumors protruded through the skin, irrespective of tumor size. This explains why, despite the fact that most of the tumors regress in the combined treatment groups, not all the mice survived up to day 60.
Upon in vitro incubation of TC1 cells with increasing concentrations of sunitinib a >20% decrease in viability was observed at concentrations higher than 2.5 μM of sunitinib (Fig. S7). Similar effects have been previously observed by others.Citation23-25 Although a direct comparison with the in vivo treatment is difficult, this concentration is apparently not achieved in vivo, as the in vivo treatment with sunitinib at a dose of 40 mg/kg did not arrest tumor growth or delay tumor development.
Within these groups of mice we also analyzed the changes in immune cells in the blood during the treatment period. Seven days after tumor inoculation, circulating MDSC levels were maximally 1% of the total peripheral blood mononuclear cell (PBMC) population. However, at days 14, 21 or 28 after tumor inoculation, circulating MDSC levels increased to almost 20% of the total PBMCs in the groups that did not receive sunitinib. In the sunitinib treated groups MDSCs increased from 1% to maximally 5% of the total PBMCs ().
Figure 5. Combination of therapeutic immunization and sunitinib stably decreases circulating MDSC levels, enhances levels of E7-specific CD8+ T cells and abrogates tumor growth. Mice (n = 4–6/group) were s.c. inoculated with TC-1 tumor cells. On day 7 after tumor inoculation, sunitinib treatment was started, daily, i.p., for a period of 9 consecutive days. Mice where then immunized with SFVeE6,7 i.m., either on days 14 and 21 after tumor inoculation with a dosage of 5 × 106 particles, or in days 14, 21 and 28 with a dosage of 1 × 106 particles, respectively. (A) The percentage of tumor-free survival was determined at the end of experiments. At specific time intervals, blood was drawn and used to identify the levels of (B) MDSCs (CD11b+Gr1+) and (C) E7-specific CD8+ T cells (E7+CD8+) by immnostaining and fluorescence cytometry. Statistical differences between groups that received sunitinib treatment, alone or in combination with immunization, and groups that received only SFVeE6,7 immunization or PBS are shown. Depicted here are averages and SD for each experimental group (*p < 0.05).
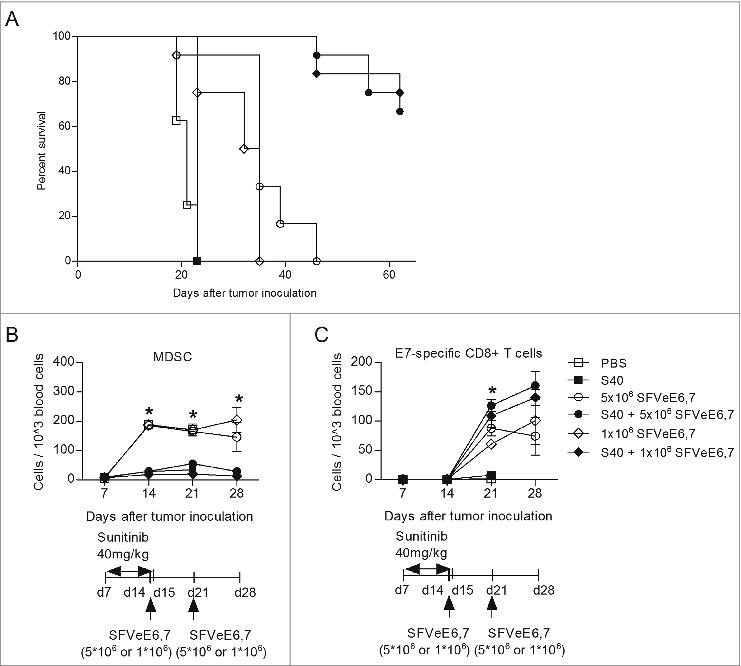
Levels of E7-specific CD8+ T cells were lower than 0.1% of the total PBMCs in all groups on days 7 and 14 after tumor inoculation. Notably, on day 21 post inoculation levels of E7-specific CD8+ T cells increased to ∼10-15% in all groups receiving SFVeE6,7. These levels were maintained constant up to day 28 (). Remarkably, the combination of immunization and sunitinib enhanced E7-specific CD8+ T cells:MDSC ratio 11-fold, specifically from 0.5:1 for the group receiving 2 dosages of 5 × 106 SFVeE6,7 to 5.55:1 for the group receiving both immunization and sunitinib. Similarly, a 21-fold increase in the E7-specific CD8+ T cells:MDSC ratio was observed between the group receiving 3 dosages of 1×106 SFVeE6,7 and the group receiving double treatment (). The proportion of total CD8+ T cells increased from 10% on days 7 and 14 post tumor inoculation to 17% of the total PBMCs in all groups receiving PBS or single treatment. In contrast, in mice that received SFVeE6,7 immunization combined with sunitinib, the levels of CD8+ T cells increased dramatically to 40% of total PBMCs (Fig. S8A).
Table 1. Ratios between total CD8+ T cells or HPV E7-specific CTLs and MDSCs in blood following sunitinib treatment alone or in combination with SFVeE6,7 immunizations at day 28 after tumor inoculation.
Circulating levels of neutrophils (CD11b+) and dendritic cells (CD11c+) were enhanced in all groups from ∼1% on day 7 post inoculation up to ∼10% and 10–20%, respectively, of total PBMCs on days 14, 21, 28 post inoculation (Fig. S8B and C).
Discussion
Tumor growth is often associated with the development and maintenance of an immunosuppressive tumor microenvironment. Accumulation of intratumoral MDSCs is one of the factors contributing to the phenomenon of tumor immune escape.Citation26,27 Recently, studies have reported that some chemotherapeutic drugs do not solely target tumor cells but also enhance antitumor immunity.Citation28 Additionally, these drugs can negatively regulate immunosuppressive MDSCs and Treg populations.Citation29,30 One such drug, used in the clinical setting for treatment of metastatic renal cell carcinoma,Citation31 is the receptor tyrosine kinase inhibitor sunitinib.
In this study, we demonstrate that immunization with a cancer vaccine combined with clinically relevant doses of sunitinibCitation32,33 strongly augments the levels of total and antigen-specific T cells in the circulation, tumor and spleen of tumor-bearing mice. Simultaneously, intratumoral, intrasplenic and circulating levels of MDSCs are strongly diminished. Overall, this combination therapy leads to a strong increase in the ratio of antitumor immune effector cells to immunosuppressive cells within the tumor. Moreover, combined sunitinib and sub-optimal vaccination strategies induce abrogation of tumor growth. This study shows that sunitinib can further enhance cancer vaccine-induced immune responses and antitumor effects.
A feature that makes sunitinib an attractive drug for cancer treatment is its reported lower toxicity as compared to similar tyrosine kinase inhibitors, such as the monoclonal humanized anti-VEGF antibody bevacizumab or the mTOR kinase inhibitor temsirolimus.Citation34 The most common grade 3/4 treatment related adverse events when using sunitinib in the pivotal Phase I clinical trial for the treatment of patients with metastatic renal cell carcinoma were fatigue and thrombocytopenia.Citation12 Sorafenib and pazopanib, 2 other novel tyrosine kinase inhibitors reported to reduce MDSC levels in murine models of colon and Lewis lung carcinoma,Citation35 may have reduced side-effects as compared to sunitinib.Citation34 Nevertheless, in addition to the direct tumor targeting and MDSC depleting effects in patients with renal cell carcinoma (RCC), sunitinib also enhanced restoration of T helper type 1 (Th1) cells and induced a stable decrease in Tregs.Citation36-38 Various studies have demonstrated the well-known antiangiogenic and direct tumoricidal activity of sunitinib,Citation39,40 developed through pleiotropic effects exerted on the receptors of vascular endothelial and platelet-derived growth factors, as well as Flt-3 and the c-kit oncogene.Citation33,41 However, the effect of sunitinib on activation status and levels of immune cells has not yet been fully elucidated.
Several groups have previously reported the effect of sunitinib on MDSCs. In an MCA26 mouse colon carcinoma model, treatment with sunitinib decreased the numbers and functionality of MDSCs and Tregs, while concurrently enhancing both intratumoral and intrasplenic CD8+ T cell levels and responses among tumor-infiltrating lymphocytes (TILs).Citation29 Similar results were observed in the murine Renca tumor model where the anti-angiogenic and depleting effects of sunitinib were mediated through inhibition of signal transducer and activator of transcription 3 (Stat3).Citation23 The selective effect of sunitinib in enhancing levels and activity of CD8+ TILs might be due to diminished expression of the negative co-stimulatory molecules programmed cell death 1 (PDCD1), programmed cell death ligand 1 (PDL1) and cytotoxic T lymphocyte-associated protein 4 (CTLA4).Citation29 Consistent with previous reports,Citation42,43 in our study sunitinib enhanced the intratumoral, intrasplenic and circulating numbers and activation status of CD8+ T lymphocytes. In 2 other murine tumor models (C26 colon and 4T1 mammary carcinomas), sunitinib only seems to deplete splenic, but not intratumoral MDSCs.Citation37,44
We expect that the molecular mechanisms of MDSC depletion by sunitinib involves Stat3 blockade as well as decreasing expression of vascular endothelial derived growth factor receptor (VEGFR) by MDSCs.Citation23,37,Citation45 Stat3 activation has been shown to stimulate expression of proliferative genes in immature myeloid cells, thus leading to their subsequent development into MDSCs. VEGFR interacts with its corresponding VEGF ligand expressed on tumor cells. Blockade of these 2 factors would thus result in a diminished number of circulating MDSCs, with a lower capacity to migrate to the tumor site. The effect of sunitinib observed in this study cannot be ascribed to an inhibition of Tregs as only a dosage of 60 mg/kg of sunitinib significantly suppressed the number of Tregs. In the combination studies of SFVeE6,7 and sunitinib this high dosage was not used. Furthermore, we have shown before that in this tumor model Tregs do not play a major role in intratumoral immune suppression.Citation46 Previously, it was reported that sunitinib has no effect on human dendritic cells (DCs), as incubation of sunitinib with DCs did not reduce their cytokine secretion or expression of CD1a and costimulatory molecules involved in immune response activation.Citation43 In our study, sunitinib treatment, alone or in combination with immunization, did not affect circulating levels of DCs.
A recent study by Alizadeh et al. showed that doxorubicin, in addition to diminishing levels of MDSCs, also blocks the immunosuppressive functions of the residual, non-depleted MDSCs.Citation42 Because of this, we investigated the potential effects of sunitinib on the functional activity of the residual, non-depleted fraction of intratumoral MDSCs. The most commonly encountered factors responsible for the suppressive activity of MDSCs are arginase1 (ARG1), inducible nitric oxide synthase (iNOS) and transforming growth factor β (TGFβ). Various studies have shown that ARG1 and iNOS deplete the nonessential amino acid L-arginine, a nutrient crucial for T-cell differentiation and function.Citation8,21 Additionally, the immunosuppressive factor TGFβ has been reported to negatively affect functions of cytotoxic T cells.Citation47 In our study, no significant changes were observed in the mRNA levels of these and other enzymes and factors between MDSCs isolated from tumors of sunitinib- or PBS-treated mice. In contrast to doxorubicin treatment, sunitinib did not change the intrinsic functional activity of residual MDSCs. Furthermore, sunitinib treatment did not change the levels of MDSC-induced immunosuppressive enzymes and factors.Citation8
There are only very few studies to date that combined sunitinib and vaccination therapy. Our study is most consistent with a study performed in a murine model of MC38 colon carcinoma showing that sequential administration of sunitinib and a poxvirus-based vaccine enhanced antitumor activity as compared to either treatment alone.Citation48 In their study sunitinib was given orally and preceded and overlapped with the priming and booster immunizations. We additionally demonstrate that sunitinib treatment starting one day after vaccine administration increased the numbers of intratumoral E7-specific CTLs and decreased MDSCs. In a previous study, we determined the effector cell types responsible for the antitumor effect generated by SFVeE6,7 immunization. The CD8+, CD4+ or both T cells subsets were depleted in vivo with monoclonal antibodies. In the group of mice immunized with SFVeE6,7 with or without CD4+ T-cell depletion, all mice were protected from tumor outgrowth. In contrast, in the group of immunized mice depleted of CD8+ T cells all mice developed tumors within two weeks.Citation49 Thus, the immunomodulatory effects of the combined sunitinib and immunization treatment seem to change the intratumoral immune balance towards antitumor immunity.
We further show that immunization following a 9-day sunitinib treatment strongly enhances the antitumor responses. This latter observation contrasts with findings of a recent study of Jaini R et al. using a 4T1 mammary tumor model in which they found sunitinib treatment inhibited immune priming due to a decrease in antigen-presenting cells and only immunization prior to sunitinib resulted in antitumor immune responses.Citation50 This difference is remarkable but might be due to the vaccine employed (alpha-lactalbumin) or the tumor model involved. Considering that sunitinib has such a marked effect on the immunization efficacy of our SFVeE6,7 vaccine, further studies seem warranted to unravel this difference in response. In our study, the intratumoral depletion of MDSCs and the concomitant increase in E7-specific T cells translated into a substantial increase in the ratio of effector antigen-specific CD8+ T cells to MDSCs. In addition, we show that these effector antigen-specific CD8+ T cells upregulate CD69, indicating activation of these cells. These findings may, at least partially, explain the in vivo antitumor results showing that combination of sunitinib with sub-optimal immunization abrogates tumor growth.
Taken together, our results report a synergistic effect of combined sunitinib treatment with cancer vaccine therapy in generating an immunological environment depleted of MDSCs and enriched in activated antitumoral E7-antigen specific cytotoxic T lymphocytes, favorable to tumor regression. Thus, this bimodal therapy changed the immunological balance in favor of antitumor immunity by simultaneously enhancing the tumor antigen-specific fraction as well as depleting protumoral immune cells, an immunological response beneficial to treatment outcome. These results provide compelling rationale for the clinical implementation of sunitinib and therapeutic antitumoral immunizations, to enhance the effectiveness of immunotherapy.
Materials and Methods
Cell lines
Baby hamster kidney cells (BHK-21) were obtained in 1996 from the American Type Culture Collection (# CCL-10). The TC-1 cell line, generated from C57Bl/6 primary lung epithelial cells with a retroviral vector and expresses human papillomavirus 16 (HPV16) E6E7, was obtained in 1998 from Prof. dr. Cornelis Melief (Leiden University Medical Center, Leiden, The Netherlands). The C3 cell line was obtained in 1998 from Dr. Mariet Feltkamp and Prof. dr. Jan ter Schegget (Leiden University Medical Center, Leiden, The Netherlands). Cell lines were authenticated by morphology and growth characteristics, tested for mycoplasma and frozen, and cells were cultured for less than 3 months. The TC1 cell line was also tested for E6, E7 by western blot and for MHC Class I RAHYNIVTF expression by FACS analysis prior to freezing. After thawing, cells were cultured as previously describedCitation22 and growth kinetics were recorded and validated twice weekly.
Cell viability assay
To determine cell viability of TC-1 tumor cells in the absence or presence of different concentrations of sunitinib, TC-1 tumor cells were plated into 96-well plates at a density of 104 cells per well and left to adhere overnight in IMDM medium (Iscove's modified Dulbecco's medium, Gibco, Invitrogen, catalog nr. 31980030) supplemented with 10% fetal calf serum (FCS). Subsequently, cells were incubated with 1.25 μM, 2.5 μM, 5 μM, 10 μM, 20 μM, 30 μM or 40 μM sunitinib solution, for a period of 24 h. Next, viability was quantified using a standard MTT assay kit (Sigma Aldrich, catalog nr. TOX1-1KT). The results were expressed as mean viable cells relatively to viability of TC-1 alone (considered as 100% viability) ± SD. The assay was performed in replicates of 6 and repeated three times.
Mice
Specified pathogen-free female C57BL/6 mice were used at 8 to 10 weeks of age. The mice were purchased from Harlan CPB and kept according to institutional guidelines. The local Animal Experimentation Ethical Committee approved all animal experiments.
Production, purification and titer determination of SFVeE6,7 particles
Production, purification and titer determination of SFVeE6,7 was performed as described previously. Citation51 In brief, SFVeE6,7 particles were produced by co-electroporation of BHK-21 cells with an RNA encoding the SFV replicase and the transgene (the E6E7 fusion protein) and a helper RNA encoding the structural proteins of SFV. Recombinant SFV replicon particles produced by transfected BHK-21 cells were purified on a discontinuous sucrose density gradient. rSFV particles were titrated on BHK-21 cells using a polyclonal rabbit anti-replicase (nsP3) antibody [a gift from Dr. T. Ahola (Biocentre Viikki, Helsinki)].
Tumor inoculation, SFVeE6,7 immunizations, and sunitinib treatment
Mice were inoculated subcutaneously in the neck with 2×104 TC-1 cellsCitation52 suspended in 0.2 mL Hank's Balanced Salt Solution (Invitrogen, catalog nr. 14025092). As tumor growth slightly varies, mice were, at the start of treatments, divided into groups such that each group contained mice with tumors of equal variations in size. Sunitinib 20, 40 or 60 mg/kg body weight was administered intraperitoneally for 9 consecutive days, starting day 7 or 15 after tumor inoculation.Citation37 Some groups received intramuscular SFVeE6,7 immunizations in a dosage of 106 or 5×106 particles. All SFVeE6,7 immunizations were administered in the hind legs of the mice in a total volume of 50 μL (25 μL/ hind leg). Tumor dimensions were assessed using calipers, and the tumor volume was determined using the formula: volume = [length × (width)2] × 0.5. According to the guidelines of the local ethical committee, mice were sacrificed when losing more than 15% of weight, when tumors exceeded 1000 mm3, protruded through the skin or at the end of experiments.
Tumor digestion and splenocyte isolation procedures
At the end of experiments tumor-bearing mice were sacrificed and tumors and spleens were isolated. Tumors were weighed, cut into small pieces and re-suspended in digestion medium [1 mg/mL Collagenase A (Roche, catalog nr. 10103578001) in WiIliam's E medium (Gibco, Invitrogen, catalog nr. 32551-020) pre-warmed to 37oC. Tumors were homogenized using the gentleMACS™ Dissociator (MiltenyiBiotec) according to the manufacturer's instructions and incubated for 30 min at 37oC on a shaker (120 rpm). Next, tumors were homogenized one more time and incubated for another 30 min at 37oC on a shaker (120 rpm). After digestion, cells were filtered through a 70 μm Falcon cell strainer (BD Biosciences).
Spleens were weighed, cut into small pieces and re-suspended in IMDM medium with 5% FCS, 100 U/mL penicillin and 100 μg/mL streptomycin. The suspension was filtered through a 70 μm Falcon cell strainer, centrifuged and resuspended by ticking. Erythrocytes were lysed for 5 min at room temperature with 5 mL of sterile erythrocyte lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2-EDTA pH = 7.2–7.4). The reaction was stopped with 15 mL IMDM medium; after centrifugation, the supernatant was discarded, cells were resuspended in IMDM medium with 5% FCS.
Cell sorting and co-culture
Freshly isolated tumor cells from TC-1 tumors were stained with the following fluorophore-conjugated antibodies: PeCy7-conjugated anti-CD11b, FITC-conjugated anti-Gr1 and APC-conjugated anti-CD3 (eBioscience, catalog nr. 25-0112-82, 11-5931-82, 17-0031-82). Cells were washed and sorted on a FACS MoFloAstrios (Beckman Coulter) based on forward and sideward scatter. Cells were positively sorted into CD11b+Gr1+ MDSCs and negatively sorted into CD3+ T cells. As determined by flow cytometric re-analysis, the purity of the sorted MDSCs was >92%.
Spleens cells isolated from donor mice immunized twice with 5 × 106 SFVeE6,7 particles were cultured in 96-well round bottom plates for 7 d. On day 0 of culture, flow-sorted MDSCs, isolated from TC-1 tumors, were added at different ratios to these cultures. On day 4 of culture, cells were labeled with carboxyfluorescein succinimidyl ester (CFSE; Invitrogen, catalog nr. C34554) for 10 min at 37oC. On day 5 of co-culture, recombinant IL-2 was added to the co-culture at a concentration of 5U/mL. As negative controls, splenocytes were cultured without stimulant. As positive controls, splenocytes were stimulated and cultured in the absence of MDSCs. At the end of the co-culture period, cells were collected and used for analysis.
MDSCs, MHC Class I tetramer staining and cytofluorimetric analysis
To determine the numbers of MDSCs, cells were stained with PE-Cy7-conjugated anti-CD11b, and FITC-conjugated anti-Gr1, (clone RB6-8C5; eBioscience, catalog nr. 25-0112-82, 11-5931-82) that binds with high affinity to the Ly-6G epitope and a lower affinity to the Ly-6C epitope.Citation53 For Treg analysis, cells were stained with PE-conjugated anti-CD4 and intracellular with Alexa700-conjugated anti-FoxP3 (eBioscience, catalog nr. 12-0043-82, 56-5773-82). To characterize CD8+ T cells specific for the antigenic epitope HPV16 E749-57 peptide RAHYNIVTF, 1×106 splenocytes or 1–2 × 106 cells isolated from tumors were washed with FACS buffer (PBS containing 0.5% bovine serum albumin [BSA]) and stained with PE-conjugated H2-Db RAHYNIVTF tetramers. Subsequently, cells were stained with anti-CD8 conjugated to PE-Cy7 (eBioscience) and FITC or Alexa700-conjugated anti-CD69 (eBioscience, catalog nr. 25-0081-82, 17-0691-82) antibodies. Next, cells were washed twice and analyzed by cytofluorimetry using a LSR-II (BD Biosciences) flow cytometer. Dead cells were excluded either by LIVE/DEAD® Fixable Dead Cell Stain Kit (Invitrogen, catalog nr. L34955) or by 4, 6-diamino-2-phenylindole (DAPI, Invitrogen, catalog nr. D3571). Pe-Cy7-conjugated anti-CD11b and PE-conjugated anti-CD11c (eBioscience, catalog nr. 25-0112-82, 12-0114-82) were used to determine neutrophil and dendritic cell levels, respectively.
Immunohistochemistry
Immunohistochemistry was performed as described before.Citation2 Briefly, 4-micrometer thick frozen tumor sections were fixed in cold acetone for 10 min. To identify MDSCs, the primary antibody used was a rat anti-mouse Ly6G-Ly6C (lymphocyte antigen 6 complex, locus C/G) monoclonal antibody (BD Biosciences, catalog nr. 562737). To identify CD8+ T cells, the primary antibody used was a rat anti-mouse CD8 antibody (Abcam, catalog nr. ab3081). Sections were incubated with the primary antibody for 60 min. The secondary antibody used was a goat anti-rat, HRP labeled antibody (AbD Serotec, catalog nr. STAR112P) diluted to a ratio of 1:100 in PBS with 1% BSA. Sections were incubated with the secondary antibody for 30 min. Development of the color reaction was performed with the commercially available AEC kit and background was stained with Mayers Hematoxylin (Sigma Aldrich, catalog nr. MHS1-100ML). ImmunoHistoMount (Sigma Aldrich, catalog nr. I1161-30ML) mounting medium was applied. Slides were covered with cover slips and scanned using a Hamamatsu NanoZoomer 2.0HT.
Real-time polymerase chain reaction
RNA from MDSCs previously isolated from TC-1 tumors and sorted using a MoFloAstrios cell sorter (Beckman Coulter) was extracted with an RNeasy Mini kit (Qiagen, catalog nr. 74104), following manufacturers' protocol. The amount of RNA per sample was equalized to 0.05 μg/μL in RNase-free water. The Verso Reverse Transcriptase kit (Thermo Scientific, catalog nr. # AB-1453/A) was used to synthesize cDNA, according to the manufacturer's protocol. Real-time PCR was performed using a StepOne real-time PCR system from Applied Biosystems. The reaction was conducted with 1 μL cDNA, 12.5 μL Absolute QPCR SYBR Green ROX Mix (Thermo Scientific, catalog nr. # AB-4105/A) and targeted gene-specific primers (for primer sequences [Eurogentec] see Table S1). Quantification of β-actin was used for sample normalization. As negative controls, RNase-free water was used instead of sample cDNA. The 2(−ddCt) method was used to calculate the relative induction of chemokines.Citation54
Statistical analysis
Data presented as mean ± standard deviation (SD) are representative of at least two independent experiments. Statistical differences between two groups were determined by a 2-tailed Mann-Whitney U test or unpaired t test. Statistical differences between several groups were determined by a non-parametric rank sum test for one-way ANOVA, followed by the multiple comparison procedure Kruskal-Wallis. All p-values of 0.05 or lower were considered significant. All statistical analyses were performed using GraphPad Prism software, version 5.0.0.288 (GraphPad software, San Diego).
Disclosure of Potential Conflicts of Interest
T. Daemen and H.W.Nijman are co-founders of ViciniVax, a spin-off company from the UMCG developing cancer vaccines.
Supplemental Materials
Supplemental data for this manuscript are available online at the publisher's website.
989764_Supplementary_Materials.zip
Download Zip (11.2 MB)Funding
We thank the Dutch Cancer Society who funded this work, Grant RuG-2009-4549.
References
- Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005; 174:7516-23; PMID:15944250; http://dx.doi.org/10.4049/jimmunol.174.12.7516
- Draghiciu O, Walczak M, Hoogeboom BN, Franken KL, Melief KJ, Nijman HW, Daemen T. Therapeutic immunization and local low-dose tumor irradiation, a reinforcing combination. Int J Cancer 2014; 134(4):859-72; PMID:23922012; http://dx.doi.org/10.1002/ijc.28418
- Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med 2004; 200:771-82; PMID:15381730; http://dx.doi.org/10.1084/jem.20041130
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:22437938; http://dx.doi.org/10.1038/nri3175
- Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res 2012; 72:876-86; PMID:22174368; http://dx.doi.org/10.1158/0008-5472.CAN-11-1792
- Yu J, Du W, Yan F, Wang Y, Li H, Cao S, Yu W, Shen C, Liu J, Ren X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol 2013; 190:3783-97; PMID:23440412; http://dx.doi.org/10.4049/jimmunol.1201449
- Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 2007; 117:1155-66; PMID:17476345; http://dx.doi.org/10.1172/JCI31422
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162-74; PMID:19197294; http://dx.doi.org/10.1038/nri2506
- Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother 2013; 62:909-18; PMID:23589106; http://dx.doi.org/10.1007/s00262-013-1396-8
- Srivastava MK, Zhu L, Harris-White M, Kar UK, Huang M, Johnson MF, Lee JM, Elashoff D, Strieter R, Dubinett S, et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PLOS ONE 2012; 7:e40677; PMID:22815789; http://dx.doi.org/10.1371/journal.pone.0040677
- Molina AM, Jia X, Feldman DR, Hsieh JJ, Ginsberg MS, Velasco S, Patil S, Motzer RJ. Long-term response to sunitinib therapy for metastatic renal cell carcinoma. Clin Genitourin Cancer 2013; 11:297-302; PMID:23707221; http://dx.doi.org/10.1016/j.clgc.2013.04.001
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007; 356:115-24; PMID:17215529; http://dx.doi.org/10.1056/NEJMoa065044
- Bayraktar UD, Bayraktar S, Rocha-Lima CM. Molecular basis and management of gastrointestinal stromal tumors. World J Gastroenterol 2010; 16:2726-34; PMID:20533592; http://dx.doi.org/10.3748/wjg.v16.i22.2726
- Potapova O, Laird AD, Nannini MA, Barone A, Li G, Moss KG, Cherrington JM, Mendel DB. Contribution of individual targets to the antitumor efficacy of the multitargeted receptor tyrosine kinase inhibitor SU11248. Mol Cancer Ther 2006; 5:1280-9; PMID:16731761; http://dx.doi.org/10.1158/1535-7163.MCT-03-0156
- Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol 2010; 185:2273-84; PMID:20644162; http://dx.doi.org/10.4049/jimmunol.1000901 [doi]
- Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM, Chen SH. Reversion of immune tolerance in advanced malignancy: Modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood 2008; 111:219-28; PMID:17885078; DOI: http://dx.doi.org/10.1182/blood-2007-04-086835 [pii].
- Kato M, Takeda K, Kawamoto Y, Tsuzuki T, Hossain K, Tamakoshi A, Kunisada T, Kambayashi Y, Ogino K, Suzuki H, et al. C-kit-targeting immunotherapy for hereditary melanoma in a mouse model. Cancer Res 2004; 64:801-6; PMID:14871802; http://dx.doi.org/10.1158/0008-5472.CAN-03-2532
- Voron T, Marcheteau E, Pernot S, Colussi O, Tartour E, Taieb J, Terme M. Control of the immune response by pro-angiogenic factors. Front Oncol 2014; 4:70; PMID:24765614; http://dx.doi.org/10.3389/fonc.2014.00070
- Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 2008; 68:5439-49; PMID:18593947; http://dx.doi.org/10.1158/0008-5472.CAN-07-6621
- Lindsey WB, Lowdell MW, Marti GE, Abbasi F, Zenger V, King KM, Lamb LS,Jr. CD69 expression as an index of T-cell function: Assay standardization, validation and use in monitoring immune recovery. Cytotherapy 2007; 9:123-32; PMID:17453964; http://dx.doi.org/10.1080/14653240601182838
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253-68; PMID:22437938; http://dx.doi.org/10.1038/nri3175
- Daemen T, Riezebos-Brilman A, Regts J, Dontje B, van der Zee A, Wilschut J. Superior therapeutic efficacy of alphavirus-mediated immunization against human papilloma virus type 16 antigens in a murine tumour model: Effects of the route of immunization. Antivir Ther 2004; 9:733-42; PMID:15535411
- Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 2009; 69:2506-13; PMID:19244102; http://dx.doi.org/10.1158/0008-5472.CAN-08-4323
- Sorolla A, Yeramian A, Valls J, Dolcet X, Bergada L, Llombart-Cussac A, Marti RM, Matias-Guiu X. Blockade of NFkappaB activity by sunitinib increases cell death in bortezomib-treated endometrial carcinoma cells. Mol Oncol 2012; 6:530-41; PMID:22819259; http://dx.doi.org/10.1016/j.molonc.2012.06.006 [doi].
- Martinho O, Silva-Oliveira R, Miranda-Goncalves V, Clara C, Almeida JR, Carvalho AL, Barata JT, Reis RM. In vitro and in vivo analysis of RTK inhibitor efficacy and identification of its novel targets in glioblastomas. Transl Oncol 2013; 6:187-96; PMID:23544171; http://dx.doi.org/10.1593/tlo.12400
- Bronte V, Serafini P, Apolloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother 2001; 24:431-46; PMID:11759067; http://dx.doi.org/10.11207/jimmunother.1524-9557
- Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J Immunol 2001; 166:678-89; PMID:11123353; http://dx.doi.org/10.4049/jimmunol.166.1.678
- Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: Immunostimulation by anticancer drugs. Nat Rev Drug Discov 2012; 11:215-33; PMID:22301798; http://dx.doi.org/10.1038/nrd3626
- Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res 2009; 69:2514-22; PMID:19276342; http://dx.doi.org/10.1158/0008-5472.CAN-08-4709
- Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 2005; 11:6713-21; PMID:16166452; http://dx.doi.org/10.1158/1078-0432.CCR-05-0883
- Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G, et al. Sunitinib in patients with metastatic renal cell carcinoma. JAMA 2006; 295:2516-24; PMID:16757724; http://dx.doi.org/10.1001/jama.295.21.2516
- Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 2006; 24:25-35; PMID:16314617; http://dx.doi.org/10.1200/JCO.2005.02.2194
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 2003; 9:327-37; PMID:12538485; http://dx.doi.org/10.1158/1078-0432.CCR-02-2832
- Molina AM, Motzer RJ, Heng DY. Systemic treatment options for untreated patients with metastatic clear cell renal cancer. Semin Oncol 2013; 40:436-43; PMID:23972707; http://dx.doi.org/10.1053/j.seminoncol.2013.05.013
- Cao M, Xu Y, Youn JI, Cabrera R, Zhang X, Gabrilovich D, Nelson DR, Liu C. Kinase inhibitor sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab Invest 2011; 91:598-608; PMID:21321535; http://dx.doi.org/10.1038/labinvest.2010.205
- Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 2009; 15:2148-57; PMID:19276286; http://dx.doi.org/10.1158/1078-0432.CCR-08-1332
- Ko JS, Rayman P, Ireland J, Swaidani S, Li G, Bunting KD, Rini B, Finke JH, Cohen PA. Direct and differential suppression of myeloid-derived suppressor cell subsets by sunitinib is compartmentally constrained. Cancer Res 2010; 70:3526-36; PMID:20406969; http://dx.doi.org/10.1158/0008-5472.CAN-09-3278
- Porta C, Paglino C, Imarisio I, Ganini C, Pedrazzoli P. Immunological effects of multikinase inhibitors for kidney cancer: A clue for integration with cellular therapies? J Cancer 2011; 2:333-8; PMID:21716852; http://dx.doi.org/10.1074/jc.v2.3119398
- de Bouard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, Guillamo JS. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol 2007; 9:412-23; PMID:17622648; http://dx.doi.org/10.1215/15228517-2007-024
- Bampi VF, Gomes CF, De Oliveira LB, Da Silva JL. The effect of the anti-angiogenic drug sunitinib malate on the vascular architecture of oral squamous cell carcinoma. Microsc Res Tech 2014; 77(4):250-6; PMID:24458724; http://dx.doi.org/10.1002/jemt.22336
- Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer 2010; 10:130-7; PMID:20094047; http://dx.doi.org/10.1038/nrc2787
- Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E, Larmonier N. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res 2014; 74:104-18; PMID:24197130; http://dx.doi.org/10.1158/0008-5472.CAN-13-1545
- Hipp MM, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, Weinschenk T, Singh-Jasuja H, Brossart P. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune responses. Blood 2008; 111:5610-20; PMID:18310500; http://dx.doi.org/10.1182/blood-2007-02-075945
- Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti-angiogenic therapy. Int Immunopharmacol 2011; 11:856-61; PMID:21315783; http://dx.doi.org/10.1016/j.intimp.2011.01.030
- Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J Clin Invest 2008; 118:3367-77; PMID:18776941; http://dx.doi.org/10.1172/JCI35213
- Walczak M, Regts J, van Oosterhout AJ, Boon L, Wilschut J, Nijman HW, Daemen T. Role of regulatory T-cells in immunization strategies involving a recombinant alphavirus vector system. Antivir Ther 2011; 16:207-18; PMID:21447870; http://dx.doi.org/10.3851/IMP1751
- Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, Chen W, Wahl SM, Ledbetter S, Pratt B, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J Exp Med 2003; 198:1741-52; PMID:14657224; http://dx.doi.org/10.1084/jem.20022227
- Farsaci B, Higgins JP, Hodge JW. Consequence of dose scheduling of sunitinib on host immune response elements and vaccine combination therapy. Int J Cancer 2012; 130:1948-59; PMID:21633954; http://dx.doi.org/10.1002/ijc.26219
- Riezebos-Brilman A, Walczak M, Regts J, Rots MG, Kamps G, Dontje B, Haisma HY, Wilschut J, Daemen T. A comparative study on the immunotherapeutic efficacy of recombinant semliki forest virus and adenovirus vector systems in a murine model for cervical cancer. Gene Ther 2007; 14:1695-704; PMID:17928874; http://dx.doi.org/10.1038/sj.gt.3303036
- Jaini R, Rayman P, Cohen PA, Finke JH, Tuohy VK. Combination of sunitinib with anti-tumor vaccination inhibits T cell priming and requires careful scheduling to achieve productive immunotherapy. Int J Cancer 2014; 134:1695-705; PMID:24105638; http://dx.doi.org/10.1002/ijc.28488
- Lambeck AJ, Nijman HW, Hoogeboom BN, Regts J, de Mare A, Wilschut J, Daemen T. Role of T cell competition in the induction of cytotoxic T lymphocyte activity during viral vector-based immunization regimens. Vaccine 2010; 28:4275-82; PMID:20434555; http://dx.doi.org/10.1016/j.vaccine.2010.04.033
- Lin KY, Guarnieri FG, Staveley-O'Carroll KF, Levitsky HI, August JT, Pardoll DM, Wu TC. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res 1996; 56:21-6; PMID:8548765
- Ribechini E, Leenen PJ, Lutz MB. Gr-1 antibody induces STAT signaling, macrophage marker expression and abrogation of myeloid-derived suppressor cell activity in BM cells. Eur J Immunol 2009; 39:3538-51; PMID:19830733; http://dx.doi.org/10.1002/eji.200939530
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262
- Nasser MW, Qamri Z, Deol YS, Ravi J, Powell CA, Trikha P, Schwendener RA, Bai XF, Shilo K, Zou X, et al. S100A7 enhances mammary tumorigenesis through upregulation of inflammatory pathways. Cancer Res 2012; 72:604-15; PMID:22158945; http://dx.doi.org/10.1158/0008-5472.CAN-11-0669
- Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. IL-10 gene-deficient mice lack TGF-beta/smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic enterococcus faecalis. J Immunol 2005; 174:2990-9; PMID:15728512; http://dx.doi.org/10.4049/jimmunol.174.5.2990
- He YF, Zhang GM, Wang XH, Zhang H, Yuan Y, Li D, Feng ZH. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J Immunol 2004; 173:4919-28; PMID:15470033; http://dx.doi.org/10.4049/jimmunol.173.8.4919
- Meng Q, Yang P, Li B, Zhou H, Huang X, Zhu L, Ren Y, Kijlstra A. CD4+PD-1+ T cells acting as regulatory cells during the induction of anterior chamber-associated immune deviation. Invest Ophthalmol Vis Sci 2006; 47:4444-52; PMID:17003438; http://dx.doi.org/10.1167/iovs.06-0201
- Janakiram NB, Rao CV. iNOS-selective inhibitors for cancer prevention: Promise and progress. Future Med Chem 2012; 4:2193-204; PMID:23190107; http://dx.doi.org/10.4155/fmc.12.168
- Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: Do their functions overlap? Mol Genet Metab 2004; 81 Suppl 1:S38-44; PMID:15050972; http://dx.doi.org/10.1016/j.ymgme.2003.10.012
- Sousa MS, Latini FR, Monteiro HP, Cerutti JM. Arginase 2 and nitric oxide synthase: Pathways associated with the pathogenesis of thyroid tumors. Free Radic Biol Med 2010; 49:997-1007; PMID:20542107; http://dx.doi.org/10.1016/j.freeradbiomed.2010.06.006
- Sevko A, Umansky V. Myeloid-derived suppressor cells interact with tumors in terms of myelopoiesis, tumorigenesis and immunosuppression: Thick as thieves. J Cancer 2013; 4:3-11; PMID:23386900; http://dx.doi.org/10.7150/jca.5047
- Juhasz A, Ge Y, Markel S, Chiu A, Matsumoto L, van Balgooy J, Roy K, Doroshow JH. Expression of NADPH oxidase homologues and accessory genes in human cancer cell lines, tumours and adjacent normal tissues. Free Radic Res 2009; 43:523-32; PMID:19431059; http://dx.doi.org/10.1080/10715760902918683