
Abstract
With the emergence of effective immunotherapeutics, which nevertheless harbor the potential for toxicity and are expensive to use, biomarkers are urgently needed for identification of cancer patients who respond to treatment. In this clinical-epidemiological study of 202 cancer patients treated with oncolytic adenoviruses, we address the biomarker value of serum high-mobility group box 1 (HMGB1) protein. Overall survival and imaging responses were studied as primary endpoints and adjusted for confounding factors in two multivariate analyses (Cox and logistic regression). Mechanistic studies included assessment of circulating tumor-specific T-cells by ELISPOT, virus replication by quantitative PCR, and inflammatory cytokines by cytometric bead array. Patients with low HMGB1 baseline levels (below median concentration) showed significantly improved survival (p = 0.008, Log-Rank test) and radiological disease control rate (49.2% vs. 30.0%, p = 0.038, χ2 test) as compared to high-baseline patients. In multivariate analyses, the low HMGB1 baseline status was a strong prognostic (HR 0.638, 95% CI 0.462–0.881) and the best predictive factor for disease control (OR 2.618, 95% CI 1.004–6.827). Indicative of an immune-mediated mechanism, antitumor T-cell activity in blood and response to immunogenic-transgene coding viruses associated with improved outcome only in HMGB1-low patients. Our results suggest that serum HMGB1 baseline is a useful prognostic and predictive biomarker for oncolytic immunotherapy with adenoviruses, setting the stage for prospective clinical studies.
Introduction
Recent years have provided the first breakthroughs of cancer immunotherapy.Citation1-5 With regards to oncolytic immunotherapy, tumor lytic viruses are used to direct patients’ own immune system against their cancer.Citation6 Treatment responses seen after immunotherapy, while often durable due to immunological memory, can take time to mount and remain hard to detect by imaging,Citation7 complicating planning and execution of clinical trials. With effective immunotherapeutics such as ipilimumab, adoptively transferred T-cells or oncolytic viruses, some patients experience complete remission, while others with similar clinico-pathological characteristics may rapidly succumb to their disease.Citation8 Therefore, predictive and prognostic factors are urgently needed for identification of patients possibly benefiting from immunotherapy. In this clinical-epidemiological study, we address the value of serum HMGB1 protein as a potential prognostic and predictive biomarker for oncolytic immunotherapy in patients with advanced metastatic solid tumors.
HMGB1 is a nuclear chromatin protein and a central cytokine in local inflammation. It is secreted by monocytes, macrophages, and dendritic cells, and also released from other cell types during necrotic and immunogenic cell death.Citation9 Similar to many cytokines, chronic production of HMGB1 can contribute to carcinogenesis, tumor progression, and immune tolerance through chronic inflammation,Citation9,10 whereas dynamic changes in HMGB1 levels can act as a damage-associated molecular pattern (DAMP) activating dendritic cells.Citation11,12 Thus, HMGB1 has a role in both acute immune response and chronic inflammation, with the former being relevant for therapeutic effects, while the latter might indicate the immune status of the tumor. Given the central role of HMGB1 in immunosuppression and immune activation, we hypothesized that in patients with advanced tumors baseline HMGB1 level is an indicator of immunosurveillance status: High baseline values would indicate prior immunological detection of the tumor, which has subsequently resulted in local immunosuppression given the progression of the neoplasia into a clinical entity. Such tumors might not be optimal candidates for “immunogenic” immunotherapy including oncolytic viruses, but could rather be good candidates for anti-immunosuppressive therapies such as checkpoint inhibiting monoclonal antibodies. In contrast, low HMGB1 baseline levels could be indicative of tumors not emphatically recognized by the immune system, correlating with lack of extensive immunoediting, evasion or suppression, a situation conducive to immunogenic immunotherapy.
The prognostic role of HMGB1 in cancer has previously been studied in the context of conventional therapies. Several studies suggest that high HMGB1 serum levelsCitation13-15 or expression at the tumor siteCitation16,17 correlate with poor prognosis and therapy outcome, while a few studies suggest the opposite.Citation18,19 In the context of cancer immunotherapy, however, which specifically acts on the immune system where HMGB1 is a key cytokine, the prognostic and predictive value of HMGB1 has not been previously studied. Here we show that serum HMGB1 baseline level is a useful prognostic and predictive biomarker for oncolytic immunotherapy. We also study mechanistic features behind the effect, and find clues that HMGB1 status predicts the ability of the adaptive immune system to mount effective antitumor immune responses. Importantly, we were able to identify patient subgroups that benefit from oncolytic immunotherapy.
Results
Low HMGB1 baseline levels associate with improved survival and disease control after oncolytic immunotherapy
The study population consisted of 202 patients with advanced metastatic solid tumors progressing after conventional therapies (). Serum HMGB1 baseline levels showed an inverse correlation to overall survival (). Since the “normal” serum level of HMGB1 for advanced cancer patients is unknown, we used the median concentration of 0.512 ng/mL as a cutoff for dividing patients into low and high HMGB1-baseline groups. Median overall survival in the low HMGB1-baseline group was 151 d (95% CI 120–181 d), significantly longer than the 102 d (95% CI 89–115 d) observed in the high HMGB1 group ().
Table 1. Patient characteristics in HMGB1 baseline groups
Figure 1. HMGB1 protein levels in serum correlate with survival and associate with changes in tumor effusion HMGB1. (A) Serum HMGB1 concentration at baseline was measured by ELISA and plotted against overall survival (total N = 202). Patients with lower HMGB1 serum levels at baseline tended to show prolonged survival. The dotted line indicates the median HMGB1 baseline level, which was used as a cutoff for study grouping. Data are shown with offset axes to clarify survival of patients with undetectable serum levels of HMGB1. (B) Patients with lower than median HMGB1 baseline level showed significantly prolonged survival as compared to high HMGB1-baseline patients (p = 0.008, n = 101 per group, Log-Rank test). While all patients had serum available, eight patients also had tumor-related ascites or pleural effusion allowing HMGB1 measurement: (C) Serum and ascites/pleural effusion samples collected on the same day before and after oncolytic adenovirus treatment were analyzed by HMGB1 ELISA. Colon (C164), pancreatic (H339), lung (K117), mesothelioma (M126 and M158), ovarian (O26), and cervical (X331) cancer patients all had corresponding changes in intracavitary fluids and serum HMGB1 levels, with only one exception (ovarian O314 patient), suggesting a close association between circulating and tumor level changes in HMGB1. Left y-axes present the serum HMGB1 levels (solid lines) while the right y-axes indicate ascites/pleural fluid concentrations (dotted lines) that were constantly higher, as expected for local HMGB1 production at the tumor.
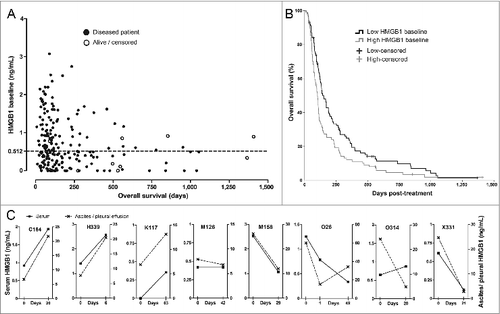
HMGB1 baseline groups showed similar distribution of patient characteristics and treatments ( and ). Notably, all five pediatric patients had low HMGB1 baselines, which is well in accord with our hypothesis, because rapidly-developing pediatric tumors may feature less immunosurveillance, evasion, and suppression.Citation20 Since none of the relevant characteristics or treatments correlated with HMGB1 status, which was unknown at the time of the treatment decision and follow-up, we conclude that clinical aspects did not influence the results. Importantly, serum HMGB1 changes in blood closely associated with tumor-related ascites or pleural effusion changes (), suggesting that measurement in blood is indicative of the HMGB1 state in tumors.
Table 2. Oncolytic adenovirus treatments and outcomes in HMGB1 baseline groups
To test if HMGB1 status would also predict treatment efficacy, we assessed imaging responses of all 65 and 50 evaluable (i.e. fulfilling RECIST1.1 or PET criteria) patients in low and high HMGB1 groups: 18.5% of low-baseline patients experienced tumor regression (minor response or better), while only 8.0% tumor-regression rate was observed in high-baseline patients (). Complete responses were seen in 6 and 0 cases in low and high baseline groups, respectively. Moreover, 49.2% of evaluable patients in low and 30.0% in high HMGB1-baseline group showed disease stabilization or better, which might also be clinically relevant given that patients were progressing prior to oncolytic virus treatments. Thus, the disease control rate was significantly higher in low HMGB1-baseline patients (p = 0.038, χ2 test).
HMGB1 baseline status is an independent prognostic and predictive factor for oncolytic immunotherapy
We next assessed the results in two multivariate analyses that included the relevant confounding factors possibly contributing to outcome. For overall survival, a Cox proportional hazards model was established, while imaging response data (disease control or not) were regressed using a logistic model. The prognostic value of HMGB1 remained significant in the Cox model (), and the adjusted survival and hazard functions appeared even more separated (Fig. S1A and B). The other prognostic factors that emerged were much as expected: WHO performance status, tumor type, and virus arming with granulocyte-macrophage colony stimulating factor (GMCSF). In the logistic regression model, HMGB1 baseline status emerged as the only independent predictive factor for treatment efficacy (). Interestingly, even the most important parameter for clinical decision making, performance status, failed to reach statistical significance in this analysis. Cross-validation of the model showed an overall good predictive power for disease control (Fig. S1C). In summary, serum HMGB1 baseline status proved to be an independent prognostic and predictive biomarker for oncolytic immunotherapy, and it was the most strongly predictive factor of treatment efficacy.
Table 3. Multivariate analysis for disease control and overall survival
Antitumor T-cell activity and treatment with immunostimulatory viruses correlate with improved survival only in low HMGB1-baseline patients
We analyzed antitumor T-cell activity in blood of 129 evaluable patients before and after therapy to test our hypothesis of an immune-mediated mechanism behind the prognostic and predictive value of serum HMGB1. Altogether 64 patients featured T-cell inductions against the tested tumor-epitope survivin in ELISPOT analysis, and 48 of these were coupled with simultaneous unspecific interferon-γ inductions, which may represent T-cells reactive to other tumor epitopes.Citation21,22 While the frequency of T-cell induction was similar in both groups (46.7% in low and 52.2% in high HMGB1 group), only the low-HMGB1 patients seemed to benefit from it (): the median survival was doubled from 111 d in high-HMGB1 group (95% CI 101–121 d) into 221 d in low-HMGB1 group (95% CI 113–329 d), but lacked statistical significance (). Next, we also assessed decreases in circulating antitumor T-cell counts, which has been indicated compatible with trafficking of T-cells from blood into tumors,Citation21,23,24 thus constituting another indication of T-cell reactivity. It should be noted, however, that documented fratricide killing among survivin-specific T-cells may limit the tumor homing of this cell type.Citation25 Low-HMGB1 patients with both T-cell “induction” and “trafficking” survived significantly longer than the corresponding high-HMGB1 patients (), and importantly, they also had significantly longer survival over the remaining low-HMGB1 patients (p = 0.038, Log-Rank test, not shown), thus representing the most immunologically active subgroup.
Figure 2. Antitumor T-cell activity in blood correlates with improved survival in HMGB1-low, but not in HMGB1-high patients. Antitumor T-cell activity in peripheral blood was measured by interferon-γ ELISPOT and correlated with overall survival of (A) 60 low-baseline and (B) 69 high-baseline HMGB1 patients. The longest surviving patients among (A) the low HMGB1-baseline group were those who had both induction and decrease in antitumor T-cells in their blood, which have been suggested compatible with cell amplification and trafficking to target tissues respectively (p = 0.075 as compared to “Anergy," Log-Rank test), whereas in (B) the high HMGB1-baseline group the antitumor T-cell activity did not seem to correlate with survival. (C) Patients with induction of antitumor T-cells in blood were compared based on their HMGB1 baseline status, but without significant difference (p = 0.111, Log-Rank test). When a decrease in antitumor T-cell counts in blood, a phenomenon compatible with trafficking of T-cells into tumors, was studied together with induction, (D) the low HMGB1-baseline patients had significantly improved survival as compared to high-baseline patients (p = 0.043, Log-Rank test). In panels A and B, respectively, n = 9 and 11 in “induction and decrease,” n = 19 and 25 in “induction,” n = 19 and 20 in “decrease,” and n = 13 both in “anergy;” In panel C, n = 28 in low and n = 36 in high HMGB1-baseline group; In panel D, n = 9 in low and n = 11 in high HMGB1-baseline group.
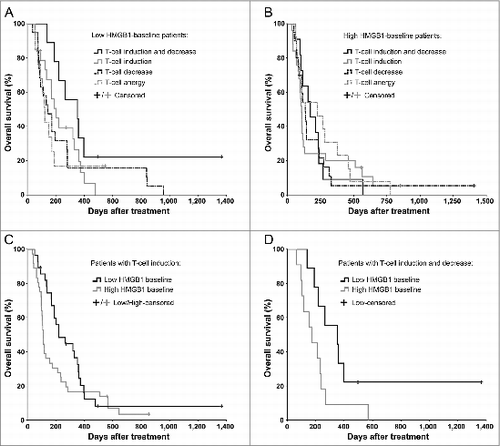
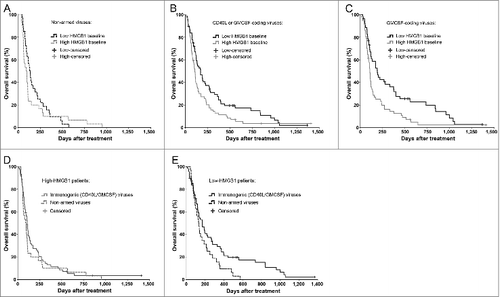
Further supporting an immune-mediated mechanism, patients who received oncolytic viruses coding for immunostimulatory transgenes (CD40-ligand or GMCSF) only benefited if they had low HMGB1 baseline (). Notably, while the median survival was almost doubled in low as compared to the high-HMGB1 patients treated with GMCSF-coding viruses (), the proportion of long-term survivors was particularly increased, which is compatible with the long-term advantage associated also with other immunotherapeutic agents.Citation1,7
Figure 3. Immunogenic transgene coding viruses improve the survival of low-HMGB1 patients only. To test if serum HMGB1 baseline status plays even greater role in highly immunogenic treatments using oncolytic adenoviruses armed with granulocyte-macrophage colony stimulating factor (GMCSF) or CD40-ligand (CD40L), only patients with a particular first-treatment virus class were selected. Kaplan–Meier analysis based on HMGB1 baseline status revealed (A) no survival difference between the groups when treated with non-armed, less-immunogenic viruses (p = 0.315, Log-Rank test), while (B) a clear improvement in survival was observed in favor for HMGB1-low patients when treated with immunogenic transgene (CD40L or GMCSF) coding viruses (p = 0.016, Log-Rank test) and even more so with (C) GMCSF-coding viruses (p = 0.004, Log-Rank test). When comparing treatment-virus classes within the HMGB1 baseline groups, (D) high-HMGB1 patients did not show any survival differences between the virus types, while (E) low-HMGB1 patients treated with immunogenic transgene coding viruses showed a significant survival advantage (p = 0.042, Log-Rank test). Panel A, n = 33 in low, and n = 30 in high HMGB1 group; panel B, n = 68 in low and n = 71 in high HMGB1 group; panel C, n = 53 in low and n = 57 in high HMGB1 group. In panels D and E, respectively, n = 30 and n = 33 in “non-armed viruses” group, and n = 71 and n = 68 in “immunogenic (CD40L/GMCSF) viruses” group. CD40L, CD40-ligand; GMCSF, granulocyte macrophage-colony stimulating factor.
In patients with low HMGB1 baseline, antitumor T-cell activity correlates with disease control in imaging
Overall survival of patients receiving treatment is an endpoint influenced both by predictive and prognostic factors. To focus on the predictive effects of HMGB1 and T-cells, we correlated imaging responses to antitumor T-cell changes in blood. The disease control rates of patients with T-cell data available were similar to that of the study population overall (48.9% in low and 28.6% in high HMGB1 group). For patients with T-cell induction, the rates were 55.0% in low and 27.3% in high HMGB1 group (p = 0.068, χ2 test). In concordance with survival data, the best disease control rate of 66.7% was seen in low-HMGB1 patients who experienced both induction and the previously discussed trafficking of antitumor T-cells. Meanwhile, high-HMGB1 patients featuring both phenomena evidently failed to benefit from it, possibly due to the local immunosuppression, as they had a dismal disease control rate of 20% (p = 0.040, χ2 test). Taken together, our results suggest that the frequency of antitumor T-cell activation is not affected by baseline HMGB1 status, but favorable consequences with regard to survival or tumor imaging require low HMGB1 baseline.
Additional correlative analyses are compatible with the proposed immunological role of HMGB1 baseline status
To gain broader mechanistic insight, we addressed other clinical and biological variables potentially associated with the observed HMGB1 baseline status. First, therapy tolerance was equally well-tolerated in both HMGB1 groups as seen in reported adverse reactions (Table S1), and appeared to be in line with previous reports.Citation21,22,26–31 Second, virus replication – as estimated by virus genomes in blood – was assessed without differences between the groups (Fig. S2). Third, inflammatory cytokine titers in serum showed no statistical differences, although pro-inflammatory interleukin-6 (IL-6) at baseline and early after therapy trended toward higher levels in the HMGB1-high group (Fig. S3). Conversely, the immunologically naïve or less immunosuppressed state in HMGB1-low patients might explain lower IL-6 levels.
Since tumor cells and the associated immune cells are the dominant source of HMGB1, we studied if baseline tumor burden (whole-body scans) correlated with serum HMGB1, but found no overall differences (Fig. S4). Interestingly, however, a subgroup of very large bulky tumors showed a linear correlation with HMGB1 baseline levels.
Finally, since secretion of HMGB1 from immune cells or release from dying tumor cells might have different circulation kinetics, we examined if post-treatment changes in serum HMGB1 levels would correlate with outcome variables. As seen in Fig. S5, HMGB1 elevation at later time points (15–28 d post-treatment) trended for shorter median survival. This was especially evident in patients with low HMGB1 baseline, suggesting that sustained low circulating levels of HMGB1 would be beneficial. A similar trend was seen with regards to treatment efficacy: the sustained low-HMGB1 patients showed an impressive disease control rate of 66.7% (two complete responses, one partial response, one minor response, six stable diseases, and five progressive diseases) as compared to 43.9% in low-baseline patients with post-treatment HMGB1 elevation (p = 0.131, χ2 test).
In summary, our correlative analyses revealed no mechanistic links to “classical” virotherapy parameters such as virus replication, but instead further supported the proposed immune-mediated mechanism behind the prognostic and predictive value of baseline serum HMGB1.
Discussion
This is the first large-scale biomarker study for oncolytic immunotherapy. Our results indicate that a low baseline level of serum HMGB1 is an independent positive prognostic and predictive factor for oncolytic immunotherapy in advanced cancer patients.
We performed several correlative analyses (T-cell activity in blood, virus replication, inflammatory cytokines, and tumor load at baseline) to examine biological mechanisms behind the finding. Clear indications of immune-mediated mechanisms were seen: The prognosis and imaging outcome of low HMGB1-baseline patients was further improved by dynamic changes in antitumor T-cells in peripheral blood. In contrast, high HMGB1-baseline patients failed to benefit from T-cell activity, probably due to immunoevasion of tumors featuring significant immunosuppression. Thus, our HMGB1 data help interpret antitumor T-cell data, which have previously been confusing as no correlation to treatment outcomes have been seen.Citation21,22,31 Adding to immune mechanistic observation, only the low-HMGB1 patients showed further benefit from highly immunogenic treatments with GMCSF or CD40-ligand coding viruses. Moreover, high-HMGB1 patients showed a trend for higher levels of pro-inflammatory IL-6 at baseline, which has been linked to advanced disease, immunosuppression, and poor effectiveness of immunotherapy.Citation32,33
Extracellular HMGB1 mediates tumor immune suppression e.g. by recruiting myeloid-derived suppressor cells,Citation34 activating regulatory T-cells,Citation35 and inhibiting tumor-infiltrating dendritic cells,Citation10 all of which are expected to result in antitumor T-cell suppression and poor outcome after immunogenic immunotherapy. Recently, a smaller-scale study using oncolytic vaccinia virus identified immunoglobulin-like transcript 2 (ILT2) expression on peripheral blood T-cells as a candidate predictive biomarker in melanoma patients.Citation36 In accord with our T-cell data, the authors found no differences between responders and non-responders in overall cytotoxic T-cell frequency, but the immunosuppressive subset of T-cells (CD8+FoxP3+ILT2+) was increased in the non-responding patients. Thus, the only other clinical biomarker study that exists for oncolytic viruses also proposes an immune-mediated mechanism via T-cell suppression.
Another notable mechanistic insight arises from our analysis of baseline tumor loads. While HMGB1 values at baseline lacked direct correlation with tumor burden in the overall population, the highest tumor loads (score >10) linearly correlated with increasing HMGB1 levels. Similarly, trends for higher virus replication, GMCSF production and acute phase IL-6 production were observed in the high HMGB1-baseline group. These parameters may be connected, since larger tumors are fertile ground for a high magnitude of virus replication leading to more transgene (GMCSF) expression and subsequently increased antiviral cytokine response. Nevertheless, large bulky tumors can also be quite immunosuppressive,Citation37 which may dampen virus-induced antitumor immunity.
The observation that post-treatment elevation of HMGB1 did not associate with T-cell activity or improved outcome deserves special attention, since the role of HMGB1 as an immunogenic DAMP molecule has been well-established in animals.Citation12 We and others have shown that immunogenic cell death occurs rapidly after anticancer therapy including oncolytic adenoviruses,Citation11,22,38,39 rendering it likely that transient local release of HMGB1 is capable of activating dendritic cells and boosting immune response against the tumor. Explaining some of the conflicting findings reported in cancer patients,Citation15,17-19 this phenomenon may be too spatially and temporally dynamic to be resolved solely by “snapshot” ELISA or immunohistochemistry based assays. In contrast, constitutive secretion of HMGB1 from immune cells at steady state could be an indicator of harmful immunosuppression,Citation10,34 as trended in our ΔHMGB1 analyses at late time points (Fig. S5). Of note, immunogenic cell death would result in also other danger signals such as adenosine triphosphate and calreticulin,Citation10,22 which could help tip the scales toward immune response instead of suppression.
In order to rigorously assess possible confounding factors, we selected median HMGB1 baseline level as a cutoff point. Thereby we ended up with two groups of patients that were readily comparable in multivariate analyses. Because a median cutoff cannot be used prospectively for patient selection e.g., in the context of a trial, we therefore also analyzed the survival data with two numerical cutoffs: 0.25 and 1.0 ng/mL. As seen in Fig. S6, a higher cutoff at around 1.0 ng/mL would allow more patients to be treated while still excluding the patients least likely to respond (p < 0.0001, Log-Rank test). However, these tentative results should be investigated in multivariate and prospective studies. Potential limitations of the present study are a heterogeneous patient population and some variation in treatment schemes. While the HMGB1-baseline groups were adjusted for patient characteristics and treatments in multivariate models (), we recognize that the tumor-specific characteristics could not be included in the models due to limited number of factors. Our setting does, however, represent actual clinical situations, and together with the fact that HMGB1 status was unknown at the time of treatment decision and follow-up, these aspects can in fact increase the applicability of our findings to real life patients.
Our data sets the stage for prospective clinical studies using predefined HMGB1 levels as putative predictive and prognostic biomarkers. Further, it will be interesting to see if these results can be reproduced in the context of other oncolytic viruses and cancer immunotherapy platforms such as cancer vaccines and adoptive T-cell therapy. Theoretically, with all of these “immunogenic immunotherapies,” patients with low serum HMGB1 at baseline are likely to benefit. In contrast, checkpoint inhibiting antibodies such as ipilimumab and nivolumab might be expected to work in the high-HMGB1 patients, as they combat immunosuppression but need antitumor T-cells to already be active. Identifying biomarkers for immunotherapy will help select the right patients for each therapy, thus sparing costs and human suffering.
Patients and Methods
Study design and conduction
The objective of our clinical-epidemiological study was to examine the prognostic and predictive role of serum HMGB1 in advanced cancer patients treated with oncolytic immunotherapy. We hypothesized that serum HMGB1 level could serve as a simple biomarker for prognosis and predicting therapy outcome. Accumulating evidence and the biological role of extracellular HMGB1 led us to the pre-specified hypothesis of an immune-mediated mechanism behind the effect. We performed the study and present the data here according to the REMARK guidelines.Citation40 Patient population consisted of 202 cancer patients with available non-hemolytic baseline serum samples, who were treated with oncolytic adenoviruses in the context of an Advanced Therapy Access Program (ATAP; see below). See CONSORT diagram for patient selection (Fig. S7). All patients gave written informed consent, and the clinical-epidemiological study was approved by the local Ethics Committee (HUS 62/13/03/02/2013). Patients received their first oncolytic adenovirus treatment at the start of the follow-up, which was given, in the absence of contraindications, together with concomitant low-dose cyclophosphamide,Citation28 and in a subset of patients with low-dose oral temozolomideCitation22 (see ). Since a normal serum HMGB1 level for advanced cancer patients is unknown, the significance testing was preceded by dividing patients into HMGB1 baseline groups based on the overall median HMGB1 concentration in serum at baseline (cutoff at 0.512 ng/mL, n = 101 per group). Thereby we ended up with two corresponding, comparable groups. Relevant medical history, baseline characteristics and follow-up data for each patient were collected from medical records and population registry until death or study conduction. Overall survival and radiological evaluations of treatment responses were considered as preplanned primary endpoints.
Patient samples (including HMGB1, T-cell, virus titer, and cytokine data) were analyzed retrospectively, and therefore, the HMGB1 status was unknown at the time of treatment decisions and follow-up, rendering clinical decision making and follow-up, including radiological outcome assessment, between the HMGB1 groups unbiased. Moreover, HMGB1 ELISA analyses were concurrently performed by one lab technician who had no information about the pre-specified hypotheses. Technical replicates for serum ELISA and cytokine analyses proved unnecessary due to negligible variance in readings (same plate with same conditions for every sample of a respective patient), whereas the interferon-γ ELISPOT analysis was performed in triplicates. With regards to data analysis, all patient characteristics, biological variables, study grouping, and results were rigorously assessed in statistical and multivariate analyses by investigators consulting an independent statistician, and the data are overtly presented in this manuscript.
Patient population and general information
Patients with advanced solid tumors refractory to conventional treatment modalities were treated with oncolytic adenoviruses in the context of an ATAP (see below) between November 2007 and January 2012. Before virotherapy, all patients had progressing solid tumors after standard therapies, with WHO performance score ≤ 3 and no major organ dysfunction (). Exclusion criteria were: organ transplant or severe malfunction of vital organ systems, known brain metastasis, HIV or other major immunosuppression, elevated bilirubin, ALT or AST increased over x3 upper limit of normal, severe thrombocytopenia, and other severe disease. The principles of treatments including possible side-effects were explained verbally and in writing, and all patients gave written informed consent.
Two hundred and two out of total 290 patients treated in the ATAP had baseline serum samples available and were thus included in this clinical-epidemiological study (Fig. S7). HMGB1 baseline groups were determined retrospectively based on the measured median HMGB1 concentration in serum at baseline (n = 101 in both groups). The most common tumor types included colorectal, breast, pancreatic, and ovarian cancer (). Median age at baseline was 58 y in low, and 57 in high HMGB1-baseline group. Study population included five pediatric patients, all of which had low HMGB1 baseline levels (mean 0.382 ng/mL +/– 0.049 SEM). WHO performance status at baseline showed mild discrepancy between the HMGB1 groups, with more poor-performing patients in high-baseline group (not significant). Patients were heavily pretreated with a median of one previous surgery and three chemotherapy regimens in both groups. Median virus treatment date was 9th of March 2009 in low, and 4th of May 2009 in high HMGB1-baseline group. Patient characteristics and previous therapies were well-balanced between the HMGB1 baseline groups (), and were taken into account in multivariate analyses ().
ATAP was a personalized therapy program, not a clinical trial, and treatment decisions were based on individual characteristics of the patients, their tumors and what had been learned from earlier patients. ATAP was regulated by Finnish Medicines Agency (FIMEA) as determined by the European Committee (EC) Regulation No 1394/2007 on advanced therapy medicinal products, amending Directive 2001/83/EC and Regulation (EC) No 726/2004. ATAP treatments were based on Article 35 of Helsinki Declaration of World Medical Association, and were performed according to Good Clinical Practice. Patient sample analyses and the epidemiological techniques utilized in this study were approved by the Helsinki University Central Hospital Operative Ethics Committee (HUS 62/13/03/02/2013).
Treatments and follow-up
Patients received oncolytic adenovirus intratumorally in ultrasound or CT guidance, when applicable (). In case of peritoneal or pleural disease, the “intratumoral” injection was performed intracavitary, usually preceded by aspiration of effusion when present. Some patients received virus intravenously, as previously published.Citation26 To boost immunogenicity of the oncolytic virotherapy, immunostimulatory transgenes, i.e., GMCSF or CD40-ligand (CD40L), encoding oncolytic adenoviruses were featured in most treatments. In the absence of contraindications, patients received concomitant low-dose cyclophosphamide for selective reduction of immunosuppressive regulatory T-cells.Citation28 In addition, low-dose oral temozolomide, which induces autophagy in combination treatments with oncolytic adenoviruses,Citation22 was used in a subset of treatments (). Oncolytic adenovirus and concomitant chemotherapeutic doses were adjusted for pediatric patients (n = 5). 48% of patients in low and 56% in high HMGB1-baseline group received oncolytic immunotherapy as a serial treatment of three consecutive treatment cycles, since in 2010 this became the standard way of giving treatments in ATAP.Citation21 Treatment schemes were well-balanced between the HMGB1 baseline groups (), and were taken into account in multivariate analyses ().
Patient follow-up started on the day of the virus treatment. Patients were monitored for 24 h in the hospital and 4 weeks as outpatients for possible adverse reactions. Clinical status and laboratory data were recorded intermittently. Adverse reactions were reported according to Common Terminology Criteria for Adverse Events v3.0 (Table S1). Pre-existing symptoms were recorded only if worsened, and in these cases scored according to final severity. Adverse reactions were further classified as either being serious adverse events leading to patient hospitalization, malformation, life-threatening condition, or death (any of these constituting a “serious adverse event”), or not. Since previous reports indicate that transient decrease of lymphocytes in blood seen after immunotherapy, compatible with lymphocyte redistribution, does not appear as an actual adverse event but rather a phenomenon contributing to treatment efficacy,Citation22,23 we excluded it from adverse reaction analyses. Median follow-up time (overall survival from the first virus treatment) was 151 d in low and 102 d in high HMGB1-baseline group (). At the time of study conduction (August 2013), nine patients in low and four patients in high-HMGB1 group were still alive, and thus are considered as censored cases.
Radiological imaging was performed by contrast-enhanced computed tomography (CT), magnetic resonance imaging (MRI), or positron emission tomography (PET-CT) scanning before and 3–12 weeks after the first treatment, with a median post-treatment imaging day of 49.5 and 50, in low and high HMGB1-baseline groups, respectively. In case of a serial treatment, comprising of three consecutive treatment cycles at 3–4 week intervals,Citation21 post-treatment imaging was performed after the complete treatment series (median post-treatment imaging day of 76.5 in low, and 69.5 in high HMGB1-baseline group, range in both: 9–14 weeks). Response evaluations were performed by professional radiologists by applying RECIST v1.1 criteria to overall disease status including injected and non-injected tumors (for CT and MRI scans): CR, complete response; PR, partial response (≥ 30% reduction in the sum of tumor diameters); MR, minor response (MR, 10–29% reduction); PD, progressive disease (≥ 20% increase or appearance of new metastatic lesions); SD, stable disease (tumor measurements not fulfilling the criteria for response or progression). In case of PET-CT, the same percentages were used, but evaluation was based on change in SUVmax sum values, a modification of the PET Response Criteria in Solid Tumors, as previously described.Citation41
Oncolytic adenoviruses
Oncolytic adenoviruses given to 202 patients as their first virus treatment have been previously published.Citation24,26,27,29,30,38,42-44 To briefly characterize, ICOVIR-7 and Ad5-Δ24-RGD-GMCSF are serotype 5 adenoviruses with a capsid modification of the RGD motif in fiber region.Citation27,29 Ad5/3-Cox2L-Δ24, Ad5/3-Δ24-GMCSF, and Ad5/3-hTERT-E1A-CD40L are serotype 5 adenoviruses featuring a capsid-modification of serotype 3 knob.Citation26,42,43 Ad3-hTERT-E1A is a fully serotype 3 oncolytic adenovirus.Citation31,44 As a transgene, Ad5-Δ24-GMCSF, Ad5/3-Δ24-GMCSF, Ad5-RGD-Δ24-GMCSF, and Ad5/3-E2F-Δ24-GMCSF viruses express granulocyte-macrophage colony-stimulating factor (GMCSF),Citation24,26,43 Ad5/3-hTERT-E1A-CD40L expresses human CD40 ligand,Citation30,38 and Ad5/3-Δ24-hNIS encodes a sodium iodide symporter protein (hNIS).Citation45 The latter is used for virus tracking and radiotherapy adjuvant, not for immune-modulating purposes, and thus is not considered as a transgene in this study. Ad5/3-hTERT-E1A-CD40L and Ad3-hTERT-E1A have E1A under the human telomerase promoter.Citation38,44 The tumor selectivity of these viruses is based on a deletion (Δ24) in the retinoblastoma binding site of E1A and/or tumor-specific promoter (E2F, hTERT), thus targeting replication to cancer cells. Virus production was done according to the current good manufacturing practice.
Clinical-epidemiological data analysis
Patient characteristics, treatments (both previous and ATAP), reported adverse reactions, survival information, and imaging data were collected from medical records. Overall survival was calculated from the first oncolytic adenovirus treatment date until death or study conduction (dead/alive information was obtained from the population registry).
Tumor load score (total tumor burden at baseline) was assessed from pretreatment whole-body imaging scans available for 55 low and 40 high HMGB1-baseline patients. Tumor masses in liver, lungs, peritoneal cavity, bones, lymph nodes, and other sites were graded from 0 to 3 (none to high tumor burden), bulky tumor at any location gave an additional three points, and the total tumor load score was calculated (possible range: 0–21 points). In addition, presence of pleural/ascites effusion was recorded (Fig. S4), but this did not affect the solid tumor load score.
HMGB1 ELISA
Patient blood samples were collected at normal hospital visits (baseline samples before the first virus treatment). After venipuncture, whole blood was allowed to clot in non-heparinized tubes at room temperature for 0.5–1 h, followed by separation by centrifugation at 3,000 rpm, and the resulting serum was removed and immediately stored at −20°C. Samples collected during November 2007–January 2012 were handled identically and showed no signs of HMGB1 degradation over time during freezer storage (see Fig. S8). Accordingly, others have found serum HMGB1 apparently stable in long-term storage under various freezing conditions.Citation46,47 Frozen serum samples were thawed at 25°C and analyzed with HMGB1 ELISA Kit (ST51011; IBL International, Hamburg, Germany) according to manufacturer's recommendations, using a high sensitive range protocol: 50 μL of serum, controls and standards were pipetted into 96-well microtiter ELISA plate containing 50 μL of diluent buffer. Plate was shaken for 30 sec, and incubated for 23 h at 37°C under adhesive foil. After washing x5, 100 μL of enzyme conjugate was added and incubated for 2 h at 25°C under adhesive foil, followed again by x5 washing. Analysis was performed by multipipetting color solution (100 μL), incubating 30 min at 25°C, adding 100 μL of stop solution, and measuring optical density with spectrophotometer at 450 nm. Internal controls were used to monitor assay quality and to check the consistency between the plates. Hemolytic serum samples were considered unsuitable for testing. Identical ELISA protocol was used for ascites/pleural effusion samples, which were also collected at hospital visits, aliquoted and stored at −20°C. Assay of all serum and effusion samples of a respective patient was performed on the same plate.
For data analysis, raw values were plotted on the high sensitive range standard curve. High methodological accuracy of the respective HMGB1 ELISA has been previously reported,Citation48 and accordingly, we found technical replicates for serum analyses unnecessary due to low variance in intra-assay readings (average of +/– 0.040 SEM of ng/mL; n = 51). Occasional negative values were considered as undetectable levels of HMGB1 and regarded as zero, and reliable limit of detection of the assay is reported at 0.2 ng/mL. Serum HMGB1 baseline levels showed a relatively large variance, ranging from 0 to 3.07 ng/mL. The mean HMGB1 concentration at baseline was 0.72 ng/mL (+/– 0.65 SD), which is on the upper limit of reported normal variation in healthy individuals (0.39 ng/mL +/– 0.42 SD; HMGB1 data sheet, IBL International). Change in serum HMGB1 after treatment (ΔHMGB1) was assessed by subtracting individual baseline level from post-treatment values. For ΔHMGB1 assessment, only patients with ≥ 2 (non-hemolytic) post-treatment samples were included in the analysis (n = 172, see Fig. S5).
Detection of viral DNA
Patient blood samples were collected, treated, and stored as mentioned above. Detection of specific viral DNA of serotype 5 and serotype 3 oncolytic adenoviruses in serum and clots by quantitative PCR was performed as previously described.Citation30,31,43,49
Cytokine analysis
Serum cytokine analysis was performed using BD Cytometric Bead Array (CBA) Human Soluble Protein Master Buffer Kit for serum samples and BD CBA Human IL-6, IL-8, IL-10, tumor necrosis factor-α, and GM-CSF Flex sets (BD Biosciences, San Diego, CA) according to manufacturer's instructions. BD FACSArray Bioanalyzer, BD FACS Array System software and FCAP Array v1.0.2 software (BD Biosciences) were used for data analysis.
ELISPOT
Peripheral whole blood samples were collected similar to serum samples, peripheral blood mononuclear cells (PBMC) were extracted by Percoll gradient centrifugation, and PBMCs were immediately stored in CTL-CryoABC serum-free medium (Cellular Technology Ltd., Cleveland, OH) at −140°C freezer. Pre- and post-treatment PBMC samples were available for 129 patients who also had serum HMGB1 data, and T-cell reactivity against a ubiquitous tumor-epitope survivin was measured by interferon-γ ELISPOT as previously described,Citation26,43 and presented here according to the MIATA guidelines. All available samples were analyzed, and since T-cell responses can take time to establish after immunotherapy, we analyzed PBMC samples following consecutive treatment cycles as well. Of note, ELISPOT assays were performed without pre-stimulation or clonal expansion of PBMCs in order to avoid artificial or incorrect signals, and thus results indicate the actual frequency of these cells in blood. ELISPOT was conducted according to manufacturer's instructions using the h-INFγ ELISPOT PRO 10 plate kit (MABtech, Stockholm, Sweden). Immediately after thawing, alive cells (non-stained with Trypan Blue) were manually counted under a light microscope. Blocking medium contained 10% fetal calf serum. To detect tumor-specific antigen responses, PBMCs were stimulated in triplicates with a tumor-associated BIRC5 PONAB peptide survivin (ProImmune, Oxford, UK), together with appropriate controls, for 20 h. Dried plates were analyzed using an automated AID-ELISpot reader (Autoimmun Diagnostika, Strassberg, Germany), and results were expressed as means of triplicates. Unspecific interferon-γ T-cell responses were also observed, which might include other T-cells reactive to unknown tumor epitopes, and these were therefore not subtracted as previously reported.Citation21,22 We had predefined a change of ≥ 20% in anti-survivin ELISPOT units from baseline together with an absolute count of ≥ 10 spots (per 1 million cells) as a true positive T-cell activity (induction/decrease), and otherwise deemed it as anergy.Citation22 To test the impact of sole T-cell induction on prognosis, data were first compared to overall survival but without correlation (p = 0.521, Log-Rank test, not shown).
Statistical analysis
Adverse reaction and imaging response comparisons were performed by χ2 (chi-squared) tests and patient characteristic comparisons by χ2 and two-tailed Student's t tests (GraphPad Software Inc., La Jolla, CA). Blood qPCR and serum cytokine data were analyzed by Kruskal–Wallis test, one-way analysis of variance (ANOVA) with Dunn's multiple comparison test, and/or by Mann–Whitney U test, as stated in the figure legends (GraphPad). Tumor load data was analyzed by linear correlation, group scores were compared by Mann–Whitney test, and the frequency of pleural/ascites effusion was compared by χ2 test (GraphPad). Patient survival data was plotted into a Kaplan–Meier curve and groups were compared by Log-Rank test (SPSS 21.0; SPSS, Chicago, IL). Multivariate analyses (Cox and logistic regression models) were performed by SPSS 21.0. For Cox proportional hazards model, assumption of proportional hazards for each factor was tested by evaluating parallelity of lines in log minus log survival plot, and by counting the Schoenfield's partial residuals for uncertain factors and linearly regressing them against natural logarithm of survival time. Candidate confounding variables included in the models are listed in and described above. There were no missing data in multivariate models. Receiver operating characteristic (ROC) curve was calculated from the predicted probabilities of each individual case in the logistic regression model (SPSS 21.0). To avoid overfitting, Cox and logistic regression models were also tested with fewer number of relevant parameters, which rendered similar results for HMGB1 status and equal predictive power. All tests were two-sided and values of p < 0.05 were considered statistically significant.
Disclosure of Potential Conflicts of Interest
AH and OH are shareholders in Oncos Therapeutics, Ltd. OH is shareholder in and AH is an employee and shareholder in TILT Biotherapeutics Ltd.
Author Contributions
I.L. collected and interpreted patient data, designed and performed laboratory and statistical analyses, and wrote the paper. A.Ko. contributed to qPCR and tumor load assessment analyses and helped in data interpretation. M.M-S. contributed to HMGB1 ELISA analyses. O.H. contributed to ELISPOT analyses and interpreted data. M.O. collected and interpreted patient data, and provided statistical support. K.K. performed patient imaging and response evaluations. T.J. contributed to patient care and data analysis. A.Ka. helped in interpretation of immunological data and suggested experiments. A.H. designed and supervised the project, interpreted data and wrote the manuscript. All authors read, commented, and approved the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
989771_Supplementary_Materials.zip
Download Zip (1.2 MB)Acknowledgments
We thank Raita Heiskanen, Saija Kaikkonen, Saila K. Pesonen, and Susanna Grönberg-Vähä-Koskela for expert technical assistance.
Funding
This study was supported by Helsinki Biomedical Graduate Program, Finnish Cultural Foundation, HUCH Research Funds (EVO), Sigrid Juselius Foundation, Academy of Finland, Biocentrum Helsinki, Biocenter Finland, Finnish Cancer Organizations, and University of Helsinki.
References
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711-23; PMID:20525992; http://dx.doi.org/10.1056/NEJMoa1003466
- Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, Spitler LE, Puzanov I, Doleman S, Ye Y et al. OptiM: a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol 2013; 31 (suppl; abstr LBA9008).
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013; 19:329-36; PMID:23396206; http://dx.doi.org/10.1038/nm.3089
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363:411-22; PMID:20818862; http://dx.doi.org/10.1056/NEJMoa1001294
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368:1509-18; PMID:23527958; http://dx.doi.org/10.1056/NEJMoa1215134
- Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Molecular therapy : the journal of the American Society of Gene Therapy 2011; 19:1008-16; PMID:21505424; http://dx.doi.org/10.1038/mt.2011.65
- Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbe C, Maio M, Binder M, Bohnsack O, Nichol G et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15:7412-20; PMID:19934295; http://dx.doi.org/10.1158/1078-0432.CCR-09-1624
- Kyi C, Postow MA. Checkpoint blocking antibodies in cancer immunotherapy. FEBS Lett 2014; 588:368-76; PMID:24161671; http://dx.doi.org/10.1016/j.febslet.2013.10.015
- Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Ann Rev Immunol 2010; 28:367-88; PMID:20192808; http://dx.doi.org/10.1146/annurev.immunol.021908.132603
- Kang R, Zhang Q, Zeh HJ, 3rd, Lotze MT, Tang D. HMGB1 in cancer: good, bad, or both? Clin Cancer Res 2013; 19:4046-57; PMID:23723299; http://dx.doi.org/10.1158/1078-0432.CCR-13-0495
- Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, Chaput N, Mira JP, Delaloge S et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 2007; 220:47-59; PMID:17979839; http://dx.doi.org/10.1111/j.1600-065X.2007.00573.x
- Guo ZS, Liu Z, Bartlett DL, Tang D, Lotze MT. Life after death: targeting high mobility group box 1 in emergent cancer therapies. Am J Cancer Res 2013; 3:1-20; PMID:23359863
- Fahmueller YN, Nagel D, Hoffmann RT, Tatsch K, Jakobs T, Stieber P, Holdenrieder S. Immunogenic cell death biomarkers HMGB1, RAGE, and DNAse indicate response to radioembolization therapy and prognosis in colorectal cancer patients. Int J Cancer 2013; 132:2349-58; PMID:23047645; http://dx.doi.org/10.1002/ijc.27894
- Wittwer C, Boeck S, Heinemann V, Haas M, Stieber P, Nagel D, Holdenrieder S. Circulating nucleosomes and immunogenic cell death markers HMGB1, sRAGE and DNAse in patients with advanced pancreatic cancer undergoing chemotherapy. Int J Cancer 2013; 133:2619-30; PMID:23729200
- Stoetzer OJ, Fersching DM, Salat C, Steinkohl O, Gabka CJ, Hamann U, Braun M, Feller AM, Heinemann V, Siegele B et al. Circulating immunogenic cell death biomarkers HMGB1 and RAGE in breast cancer patients during neoadjuvant chemotherapy. Tumour Biol 2013; 34:81-90; PMID:22983919; http://dx.doi.org/10.1007/s13277-012-0513-1
- Li T, Gui Y, Yuan T, Liao G, Bian C, Jiang Q, Huang S, Liu B, Wu D. Overexpression of high mobility group box 1 with poor prognosis in patients after radical prostatectomy. BJU Int 2012; 110:E1125-30; PMID:22672360; http://dx.doi.org/10.1111/j.1464-410X.2012.11277.x
- He W, Tang B, Yang D, Li Y, Song W, Cheang T, Chen X, Li Y, Chen L, Zhan W et al. Double-positive expression of high-mobility group box 1 and vascular endothelial growth factor C indicates a poorer prognosis in gastric cancer patients. World J Surg Oncol 2013; 11:161; PMID:23866030; http://dx.doi.org/10.1186/1477-7819-11-161
- Bao G, Qiao Q, Zhao H, He X. Prognostic value of HMGB1 overexpression in resectable gastric adenocarcinomas. World J Surg Oncol 2010; 8:52; PMID:20579387; http://dx.doi.org/10.1186/1477-7819-8-52
- Yamazaki T, Hannani D, Poirier-Colame V, Ladoire S, Locher C, Sistigu A, Prada N, Adjemian S, Catani JP, Freudenberg M et al. Defective immunogenic cell death of HMGB1-deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ 2014; 21:69-78; PMID:23811849; http://dx.doi.org/10.1038/cdd.2013.72
- Vakkila J, Jaffe R, Michelow M, Lotze MT. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: a major nosologic difference with adult tumors. Clin Cancer Res 2006; 12:2049-54; PMID:16609014; http://dx.doi.org/10.1158/1078-0432.CCR-05-1824
- Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, Tahtinen S, Oksanen M, Heiskanen R, Pesonen S et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res 2013; 19:2734-44; PMID:23493351; http://dx.doi.org/10.1158/1078-0432.CCR-12-2546
- Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, Hemminki O, Diaconu I, Pesonen S, Koski A et al. Oncolytic adenovirus with temozolomide induces autophagy and antitumor immune responses in cancer patients. Mol Ther 2013; 21:1212-23; PMID:23546299; http://dx.doi.org/10.1038/mt.2013.51
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010; 28:3167-75; PMID:20516446; http://dx.doi.org/10.1200/JCO.2009.26.7609
- Hemminki O, Hemminki A. Patients treated with oncolytic adenovirus Ad5/3-hTERT-E2F-GMCSF induce immunological responses against the tumor. 12th CIMT Annual Meeting (abstr 20). Mainz, Germany: The Association for Cancer Immunotherapy 2014.
- Leisegang M, Wilde S, Spranger S, Milosevic S, Frankenberger B, Uckert W, Schendel DJ. MHC-restricted fratricide of human lymphocytes expressing survivin-specific transgenic T cell receptors. J Clin Invest 2010; 120:3869-77; PMID:20978348; http://dx.doi.org/10.1172/JCI43437
- Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M, Guse K et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther 2010; 18:1874-84; PMID:20664527; http://dx.doi.org/10.1038/mt.2010.161
- Nokisalmi P, Pesonen S, Escutenaire S, Sarkioja M, Raki M, Cerullo V, Laasonen L, Alemany R, Rojas J, Cascallo M et al. Oncolytic adenovirus ICOVIR-7 in patients with advanced and refractory solid tumors. Clin Cancer Res 2010; 16:3035-43; PMID:20501623; http://dx.doi.org/10.1158/1078-0432.CCR-09-3167
- Cerullo V, Diaconu I, Kangasniemi L, Rajecki M, Escutenaire S, Koski A, Romano V, Rouvinen N, Tuuminen T, Laasonen L et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther 2011; 19:1737-46; PMID:21673660; http://dx.doi.org/10.1038/mt.2011.113
- Pesonen S, Diaconu I, Cerullo V, Escutenaire S, Raki M, Kangasniemi L, Nokisalmi P, Dotti G, Guse K, Laasonen L et al. Integrin targeted oncolytic adenoviruses Ad5-D24-RGD and Ad5-RGD-D24-GMCSF for treatment of patients with advanced chemotherapy refractory solid tumors. Int J Cancer 2012; 130:1937-47; PMID:21630267; http://dx.doi.org/10.1002/ijc.26216
- Pesonen S, Diaconu I, Kangasniemi L, Ranki T, Kanerva A, Pesonen SK, Gerdemann U, Leen AM, Kairemo K, Oksanen M et al. Oncolytic immunotherapy of advanced solid tumors with a CD40L-expressing replicating adenovirus: assessment of safety and immunologic responses in patients. Cancer Res 2012; 72:1621-31; PMID:22323527; http://dx.doi.org/10.1158/0008-5472.CAN-11-3001
- Hemminki O, Diaconu I, Cerullo V, Pesonen SK, Kanerva A, Joensuu T, Kairemo K, Laasonen L, Partanen K, Kangasniemi L et al. Ad3-hTERT-E1A, a fully serotype 3 oncolytic adenovirus, in patients with chemotherapy refractory cancer. Mol Ther 2012; 20:1821-30; PMID:22871667; http://dx.doi.org/10.1038/mt.2012.115
- Zarogoulidis P, Yarmus L, Darwiche K, Walter R, Huang H, Li Z, Zaric B, Tsakiridis K, Zarogoulidis K. Interleukin-6 cytokine: a multifunctional glycoprotein for cancer. Immunome Res 2013; 9:16535; PMID:24078831
- Burdelya L, Kujawski M, Niu G, Zhong B, Wang T, Zhang S, Kortylewski M, Shain K, Kay H, Djeu J et al. Stat3 activity in melanoma cells affects migration of immune effector cells and nitric oxide-mediated antitumor effects. J Immunol 2005; 174:3925-31; PMID:15778348; http://dx.doi.org/10.4049/jimmunol.174.7.3925
- Li W, Wu K, Zhao E, Shi L, Li R, Zhang P, Yin Y, Shuai X, Wang G, Tao K. HMGB1 recruits myeloid derived suppressor cells to promote peritoneal dissemination of colon cancer after resection. Biochem Biophys Res Commun 2013; 436:156-61; PMID:23707808; http://dx.doi.org/10.1016/j.bbrc.2013.04.109
- Liu Z, Falo LD, Jr., You Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J Immunol 2011; 187:118-25; PMID:21642542; http://dx.doi.org/10.4049/jimmunol.1003378
- Zloza A, Kim DW, Kim-Schulze S, Jagoda MC, Monsurro V, Marincola FM, Kaufman HL. Immunoglobulin-like transcript 2 (ILT2) is a biomarker of therapeutic response to oncolytic immunotherapy with vaccinia viruses. J Immun Cancer 2014; 2:1; PMID:24829758; http://dx.doi.org/10.1186/2051-1426-2-1
- Fialova A, Partlova S, Sojka L, Hromadkova H, Brtnicky T, Fucikova J, Kocian P, Rob L, Bartunkova J, Spisek R. Dynamics of T-cell infiltration during the course of ovarian cancer: the gradual shift from a Th17 effector cell response to a predominant infiltration by regulatory T-cells. Int J Cancer 2013; 132:1070-9; PMID:22865582; http://dx.doi.org/10.1002/ijc.27759
- Diaconu I, Cerullo V, Hirvinen ML, Escutenaire S, Ugolini M, Pesonen SK, Bramante S, Parviainen S, Kanerva A, Loskog AS et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res 2012; 72:2327-38; PMID:22396493; http://dx.doi.org/10.1158/0008-5472.CAN-11-2975
- Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, Edwards MR, Michelsen KS, Kroeger KM, Liu C et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med 2009; 6:e10; PMID:19143470
- McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Statistics Subcommittee of the NCIEWGoCD. REporting recommendations for tumor MARKer prognostic studies (REMARK). Nat Clin Pract Oncol 2005; 2:416-22; PMID:16130938; http://dx.doi.org/10.1038/ncponc0252
- Koski A, Ahtinen H, Liljenback H, Roivainen A, Koskela A, Oksanen M, Partanen K, Laasonen L, Kairemo K, Joensuu T et al. [(18)F]-fluorodeoxyglucose positron emission tomography and computed tomography in response evaluation of oncolytic adenovirus treatments of patients with advanced cancer. Hum Gene Ther 2013; 24:1029-41; PMID:24099555; http://dx.doi.org/10.1089/hum.2013.123
- Pesonen S, Nokisalmi P, Escutenaire S, Sarkioja M, Raki M, Cerullo V, Kangasniemi L, Laasonen L, Ribacka C, Guse K et al. Prolonged systemic circulation of chimeric oncolytic adenovirus Ad5/3-Cox2L-D24 in patients with metastatic and refractory solid tumors. Gene Ther 2010; 17:892-904; PMID:20237509; http://dx.doi.org/10.1038/gt.2010.17
- Cerullo V, Pesonen S, Diaconu I, Escutenaire S, Arstila PT, Ugolini M, Nokisalmi P, Raki M, Laasonen L, Sarkioja M et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res 2010; 70:4297-309; PMID:20484030; http://dx.doi.org/10.1158/0008-5472.CAN-09-3567
- Hemminki O, Bauerschmitz G, Hemmi S, Lavilla-Alonso S, Diaconu I, Guse K, Koski A, Desmond RA, Lappalainen M, Kanerva A et al. Oncolytic adenovirus based on serotype 3. Cancer Gene Ther 2010; 18:288-96; PMID:21183947; http://dx.doi.org/10.1038/cgt.2010.79
- Rajecki M, Sarparanta M, Hakkarainen T, Tenhunen M, Diaconu I, Kuhmonen V, Kairemo K, Kanerva A, Airaksinen AJ, Hemminki A. SPECT/CT imaging of hNIS-expression after intravenous delivery of an oncolytic adenovirus and 131I. PloS One 2012; 7:e32871; PMID:22412937; http://dx.doi.org/10.1371/journal.pone.0032871
- Uzawa A, Mori M, Masuda S, Muto M, Kuwabara S. CSF high-mobility group box 1 is associated with intrathecal inflammation and astrocytic damage in neuromyelitis optica. J Neurol Neurosurg Psychiatry 2013; 84:517-22; PMID:23255728; http://dx.doi.org/10.1136/jnnp-2012-304039
- Hoshina T, Kusuhara K, Ikeda K, Mizuno Y, Saito M, Hara T. High mobility group box 1 (HMGB1) and macrophage migration inhibitory factor (MIF) in Kawasaki disease. Scand J Rheumatol 2008; 37:445-9; PMID:18720262; http://dx.doi.org/10.1080/03009740802144143
- Lehner J, Wittwer C, Fersching D, Siegele B, Holdenrieder S, Stoetzer OJ. Methodological and preanalytical evaluation of an HMGB1 immunoassay. Anticancer Res 2012; 32:2059-62; PMID:22593488
- Escutenaire S, Cerullo V, Diaconu I, Ahtiainen L, Hannuksela P, Oksanen M, Haavisto E, Karioja-Kallio A, Holm SL, Kangasniemi L et al. In vivo and in vitro distribution of type 5 and fiber-modified oncolytic adenoviruses in human blood compartments. Ann Med 2011; 43:151-63; PMID:21261555; http://dx.doi.org/10.3109/07853890.2010.538079
- Rangel-Moreno J, Moyron-Quiroz JE, Carragher DM, Kusser K, Hartson L, Moquin A, Randall TD. Omental milky spots develop in the absence of lymphoid tissue-inducer cells and support B and T cell responses to peritoneal antigens. Immunity 2009; 30:731-43; PMID:19427241; http://dx.doi.org/10.1016/j.immuni.2009.03.014