
Abstract
Adoptive cellular therapy (ACT) after lymphodepletive conditioning can induce dramatic clinical responses, but this approach has been largely limited to melanoma due to a lack of reliable methods for expanding tumor-specific lymphocytes from the majority of other solid cancers. We have employed tumor RNA-pulsed dendritic cells (DCs) to reliably expand CD4+ and CD8+ tumor-reactive T lymphocytes for curative ACT in a highly-invasive, chemotherapy- and radiation-resistant malignant glioma model. Curative treatment of established intracranial tumors involved a synergistic interaction between myeloablative (MA) conditioning, adoptively transferred tumor-specific T cells, and tumor RNA-pulsed DC vaccines. Hematopoietic stem cells (HSCs), administered for salvage from MA conditioning, rapidly migrated to areas of intracranial tumor growth and facilitated the recruitment of tumor-specific lymphocytes through HSC-elaborated chemokines and enhanced immunologic rejection of intracranial tumors during ACT. Furthermore, HSC transplant under non-myeloablative (NMA) conditions also enhanced immunologic tumor rejection, indicating a novel role for the use of HSCs in the immunologic treatment of malignant gliomas and possibly other solid tumors.
Abbreviations:
- ACT, adoptive cellular therapy
- CAR, chimeric antigen receptor
- CBA, cytokine bead array
- CCL3, (MIP1α) macrophage inhibitory protein 1
- CXCL12, (SDF1) stromal derived factor 1
- DC, dendritic cell
- FACS, fluorescence activated cell sorting
- HSC, haematopoietic stem cell
- IL-7, interleukin 7
- IL-15, interleukin 15
- IFNγ, interferon gamma
- MA, myeloablative
- NMA, non-myeloablative
- OVA, ovalbumin
- SEM, standard error of mean
- TAA, tumor associated antigens
- TCR, T cell receptor
- TMZ, temozolomide
- TNFα, tumor necrosis factor α
- TTRNA-T cells, tumor-specific T cells activated ex vivo using RNA-pulsed DCs
- WBI, whole brain irradiation.
Introduction
ACT using tumor-infiltrating lymphocytes (TILs) following NMA and MA conditioning regimens has emerged as a remarkably effective treatment modality for refractory metastatic melanoma. The degree of lymphodepletion prior to ACT has correlated with clinical effectiveness, with greater than 70% of patients receiving MA(Citation1-4) demonstrating objective clinical responses to therapy, including a significant rate of durable complete responses of metastatic lesions within the central nervous system. Importantly, these studies have demonstrated that the brain is not refractory to effective and tolerable treatment by T cell-mediated immunotherapy. However, the extension of ACT to other solid tumors has been limited, at least in part, due to the lack of established and reliable protocols for expanding polyclonal tumor-specific lymphocytes ex vivo from the majority of solid tumors other than melanoma. While gene modification strategies employing chimeric antigen receptors (CARs) or tumor antigen specific T cell receptors (TCRs) have provided versatile platforms for engineered tumor-specific lymphocytes, these approaches are limited in dealing with the heterogeneity of antigen expression within malignant brain tumors and restricted in approach to a limited repertoire of well-characterized and tumor-specific target and receptor pairs. Tumor RNA-pulsed DCs vaccines have been shown to be capable of inducing CD4+ and CD8+ tumor-reactive lymphocytes(Citation5,6) and we have co-opted this platform to prime T cells ex vivo and expand a broad repertoire of tumor-specific T cells for use in ACT. In a relevant highly-invasive, temozolomide-resistant and radiation-resistant murine glioma model, we have demonstrated the curative capacity of ACT in the treatment of established intracranial tumors. Lymphodepletive conditioning alone including MA regimens had no impact on intracranial tumor growth; nor did vaccination with tumor RNA-loaded DC vaccines or adoptive transfer of tumor-specific lymphocytes after MA conditioning. In contrast, however, the combination of DC vaccination and adoptive T cell transfer after MA conditioning revealed synergistic cellular interactions that resulted in a doubling of median survival and up to 40% long term cures in tumor bearing animals. HSCs given for hematopoietic rescue facilitated a previously unrecognized role in facilitating intratumoral trafficking of tumor-specific lymphocytes and the immunologic rejection of invasive tumor cells.
MA host conditioning, typically with total body irradiation (TBI) or high-dose chemotherapy followed by autologous HSC rescue has clearly demonstrated a potentiating effect in adoptive immunotherapy against other solid tumors in both murine and human systems.Citation2-4,7 The lymphopenic host environment eliminates immunosuppressive cell populations and increases the bioavailability of a variety of cytokines, subsequently facilitating homeostatic proliferation of host and adoptively transferred lymphocytes.Citation8-10 The role of HSCs after MA conditioning beyond necessity for hematopoietic cell rescue has been largely unstudied, although a previous report highlighted the capacity for HSCs to impact on lymphocyte proliferation through the induction of homeostatic cytokines interleukin 7 (IL-7) and interleukin 15 (IL-15).Citation11 We found that intravenously administered HSCs rapidly trafficked to areas of intracranial tumor growth and were subsequently found co-localized with adoptively transferred tumor-specific lymphocytes in the tumor microenvironment. HSCs within the tumor mircroenvironment led to significant increases in antitumor reactive T cells at the tumor site. This was due to chemotactic interactions between HSCs and T cells mediated by HSC secretion of CCL3 (MIP-1α) in the tumor bed that facilitated the subsequent recruitment of activated tumor-specific T cells. These interactions between HSCs and T cells within malignant gliomas enhanced immunologic control of tumor growth even under NMA conditions, highlighting a previously unappreciated role for HSCs in mitigating intratumoral T cell trafficking and immunologic tumor rejection during ACT. To our knowledge, these are the first studies demonstrating a role for HSCs in facilitating intratumoral T cell trafficking and enhancing the immunologic rejection of malignant brain tumors.
These studies validate a highly efficacious ACT platform that is demonstrably divergent from other adoptive T cell immunotherapeutics and is capable of targeting intracranial tumors with a high degree of specificity. This ACT platform leverages highly novel observations that highlight the importance of previously uncharacterized role of HSCs in the immunologic rejection of tumors during adoptive immunotherapy. These findings have important implications for further assessing the role of HSC transplant in the physiologic antitumor immune response after MA therapy and for the novel use of HSCs in cellular immunotherapy targeting malignant brain tumors and other cancers.
Results
Adoptive cellular therapy: synergistic interactions mediate antitumor efficacy
We enlisted a preclinical model of spontaneously arising astrocytoma from genetically engineered Nf1 +/−, p53 +/− mice previously described to give rise to high-grade malignant gliomas.Citation12 A cell line established from these mice glioma (KR158B) was injected into the frontal cortex using stereotactic injection in order to establish a transplantable and reliable model of high-grade and invasive glioma. The resulting tumor creates islands of invasive tumor infiltrate in bilateral cerebral hemispheres, closely recapitulating the invasive nature of human malignant gliomas ().
Figure 1. Highly invasive radiation and chemotherapy resistant glioma. (A) KR158B intracranial glioma is a highly invasive intracranial glioma that forms glioma islands and closely resembles primary human glioma. Shown is H&E stain of formalin-fixed, paraffin-embedded section (7 micron) taken from mice with established tumor. (B) Treatment with standard therapy with either whole brain radiation therapy (WBRT) or Temozolomide (TMZ) did not provide a survival benefit; however, the combination of WBRT+TMZ provided a modest increase in median survival, 38 d vs. 43 d, p = 0.0389 Gehan–Breslow–Wilcoxon test.
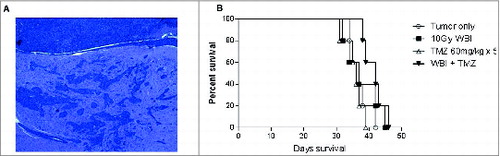
Standard treatment for high-grade gliomas involves the use of concomitant radiotherapy and temozolomide (TMZ) followed by adjuvant TMZ cycles, but treatment failure almost uniformly occurs due to inherent or induced tumor-resistance.Citation13 We explored the impact of whole brain irradiation (WBI) alone, TMZ alone, or concomitant WBI+TMZ on the KR158B glioma in order to assess the relative efficacy of immunotherapy compared to standard treatment modalities (). No survival benefit was provided by either chemotherapy alone or radiation alone, and the combination therapy led to a very modest improvement (gain of 5 d) in median survival (p = 0.0389, Gehan–Breslow–Wilcoxon test) with all animals succumbing to tumor growth. This model of a highly-invasive, radiation-resistant, and chemotherapy-resistant astrocytoma closely mimics the pathologic phenotype and treatment refractory nature of human malignant gliomas, thus making it an ideal pre-clinical model for exploration of the efficacy of ACT.
In order to generate antitumor specific T cells for ACT, total RNA isolated from KR158B glioma cells was pulsed onto DCs using electroporation. These RNA-pulsed DCs were co-cultured with syngeneic splenocytes from tumor antigen primed mice in the presence of exogenous IL-2, thus resulting in the activation of tumor-specific T lymphocytes. Ex vivo T cell expansion led to an overall enrichment of CD8+ T cells from 21.84% ± 3.19% (SEM) of total CD3+ splenocytes, to 86.58% ± 7.42% (SEM) of total CD3+ cells. To determine antitumor reactivity and specificity of these ex vivo expanded T cells, lymphocytes were co-cultured against target tumor cells or surrogate targets of DCs pulsed with tumor RNA vs. control RNA (OVA). Antigen-specific recognition was assessed by cytokine bead array (CBA) examining Th1 vs. Th2 cytokine production. Tumor-specific T lymphocytes demonstrated high level production of IFNγ and TNFα in response to syngeneic tumor cells and DCs pulsed with total tumor RNA but not OVA-expressing targets ().
Figure 2. In vitro expanded antitumor T cells target glioma cells with specificity. (A) In vitro expanded Ovalbumin (OVA) antigen specific T cells or total tumor antigen specific T cells (TTRNA-T cells) secrete pro-inflammatory cytokines TNFα and IFNγ after recognition of cognate antigen expressed by DC2.4 cells electroporated with either OVA RNA or KR158B RNA (TTRNA) or syngeneic tumor cells. (B) In vitro expanded TTRNA T cells were magnetically sorted into CD4+ and CD8+ TTRNA T cells after expansion, and both T cell populations secreted TNFα and IFNγ upon recognition of cells expressing TTRNA. N = 5 replicates.
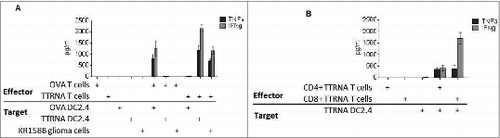
In order to determine whether both CD4+ and CD8+ tumor-specific lymphocytes were generated by the tumor RNA-pulsed DC platform, ex vivo expanded T cells were magnetically sorted for either CD4+ or CD8+ T cell populations and co-cultured with target cells expressing total tumor antigens. Both CD4+ and CD8+ cells secreted TNFα and IFNγ upon recognition of total tumor antigens demonstrating that this platform of ex vivo T cell expansion engenders both tumor-specific CD4+ helper T cells and CD8+ effector cells ().
Adoptive cellular therapy cures established intracranial glioma
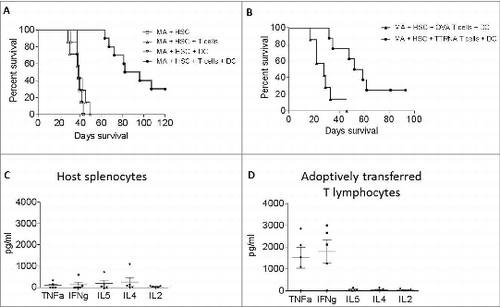
To determine if ex vivo expanded tumor-reactive T cells were efficacious in vivo, mice bearing established intracranial gliomas received MA conditioning (9G TBI) and HSC rescue followed by ACT which includes transfer of ex vivo expanded tumor-reactive T cells and 3 weekly DC vaccines (Fig. S1). This resulted in a doubling of median survival compared to tumor only controls, as well as cures in approximately 30% of treated mice (P < 0.0001) (). Most notably, synergistic interactions between the cellular components of ACT were required for antitumor efficacy (). MA conditioning with HSC rescue alone had no treatment effect, nor did MA+HSC followed by adoptive transfer of tumor-specific T cells alone or weekly DC vaccination alone. This is in stark contrast to the treatment effects of MA+HSC followed by adoptive T cell transfer and DC vaccination. These results strikingly demonstrate a potent interaction between the cellular components of this therapy that dramatically alters survival outcomes when combined in the conditioned host. To determine if antigen specificity of adoptively transferred T cells was required for efficacy, similarly activated and expanded T cells specific for non-tumor associated antigen, OVA, were adoptively transferred during ACT. No treatment benefit was observed, demonstrating that the antigen-specific T lymphocytes generated by ex vivo DC stimulation are required for the observed antitumor efficacy ().
Figure 3. In vitro expanded antitumor T cells are efficacious against intracranial glioma. (A) Prolonged survival in mice treated with ACT. Mice with established intracranial glioma received ACT, in the context of MA + HSC transplant. Mice received either MA + HSC alone, MA + HSC + adoptive transfer of TTRNA T cells, MA + HSC + DC vaccine, or MA + HSC + TTRNA T cells + DC vaccine. (B) Glioma bearing mice received ACT using either in vitro expanded OVA specific T cells followed by OVA-RNA pulsed DC vaccine, or tumor specific T cells (TTRNA) followed by total tumor RNA-pulsed DC vaccination. Antitumor efficacy was only observed using tumor-specific TTRNA T lymphocytes. ACT was conducted using TTRNA T cells from DsRed+ mice. Host vs. adoptively transferred lymphocytes in ACT treated mice were FACS sorted as DsRed− or DsRed+ lymphocytes respectively (n = 5 animals). (C) Cytokine secretion of host splenocytes after co-culture against glioma targets. (D) TNFα and IFNγ secretion of adoptively transferred antitumor T lymphocytes upon recognition of KR158B tumor cells.
Successful adoptive therapy in patients with malignant melanoma has been associated with capacity to maintain tumor-specific lymphocytes in a functionally active state for prolonged periods in vivo.Citation14,15 To determine if antitumor reactivity was maintained long-term in vivo, and whether the transferred lymphocytes were exclusively responsible for tumor recognition or whether host antitumor immune cells were also engendered by DC vaccination, we harvested spleens from intracranial tumor bearing mice 4 weeks after receiving ACT with T cells generated from DsRed transgenic mice. The adoptively transferred T cells were isolated using sterile FACS sorting as described in methods. Either the isolated T cells (DsRed+) or host splenocytes (DsRed-) were co-cultured in vitro against glioma cells, and antitumor reactivity of transferred and host lymphocytes measured by CBA. Measurement of cytokine secretion revealed that the isolated adoptively transferred T cells secreted TNFα and IFNγ in response to tumor cell recognition (); however, the host cells did not demonstrate any antitumor reactivity (). These results demonstrate that the ex vivo generated T cells convey the preponderance of antitumor immunity and that the role of DC vaccination is through the enhancement of antitumor reactivity of the transferred cells and not induction of immunity within recovering host lymphocytes.
DC vaccination maintains T cell engraftment in vivo
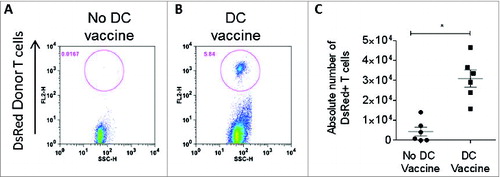
Based on our observation that a synergistic interaction is required between T cells, DCs, and HSCs, we further explored the role of DCs in the maintenance of tumor-specific T cells after adoptive transfer. Here, we demonstrate that DC vaccination facilitates the in vivo expansion of antigen specific T cells after adoptive transfer. Using tumor-specific T cells generated from syngeneic DsRed+ mice, we evaluated the presence of tumor-specific T cells in peripheral lymph nodes one week after adoptive transfer with our without total tumor RNA-pulsed DC vaccination. As shown in , DC vaccination was a requirement for the in vivo maintenance of tumor-specific T cells, indicating that post-transfer activation by DCs is critical for T cell persistence. Importantly, these results also demonstrated that efficacy was achieved without in vivo administration of exogenous cytokines such as IL-2, which has been associated with significant treatment associated toxicity in patients receiving immunotherapy. Treatment of normal mice with this regimen did not result in any signs of toxicity in animals followed for 100 d post-treatment (Fig. S2).
Figure 4. DC vaccine is required for T cell persistence. (A) In vivo frequency of adoptively transferred antitumor DsRed+ T lymphocytes was measured in lymph nodes one week after ACT without RNA-pulsed DC vaccine or (B) after RNA-pulsed DC vaccine. (C) Enumeration of DsRed+ adoptively transferred T cells in lymph nodes with or without DC vaccination (*p = 0.002, Mann–Whitney test, n = 6).
HSCs mediate T cell trafficking to glioma microenvironment
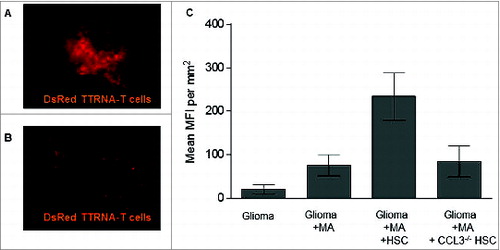
To investigate the role of HSC transfer in this treatment modality, we infused HSCs from DSRed+ donor mice prior to the transfer of tumor-specific lymphocytes derived from GFP transgenic mice and evaluated in vivo cell trafficking patterns and interactions. Tumor-bearing animals were sacrificed and intracranial tumor localization evaluated by fluorescent microscopy. Interestingly, cells derived from DsRed expressing HSCs were completely co-localized with GFP expressing tumor-specific T lymphocytes within the tumor microenvironment (), suggesting specific cellular interactions between these transferred cell types. Accumulation of lymphocytes or HSCs in adjacent normal cortex was not observed, indicating a tumor-specific co-localization of transferred T cells and HSCs and/or their differentiated progeny. To determine whether HSCs had an impact on T cell localization, mice received MA conditioning followed by adoptive T cell transfer with or without HSC rescue and the local accumulation of T cells within the tumor microenvironment enumerated 24 h and 1 week after cell transfer. A significantly higher number of tumor-specific T cells localized within the tumor microenvironment at both time points in mice that received T cells and HSCs vs. T cells alone (p = 0.0001, Wilcoxon test) (). These data demonstrated that HSC transplant prior to adoptive T cell transfer facilitated the trafficking of T cells to sites of intracranial tumor growth and were suggestive of an enhanced chemotactic function of HSCs on T cell migration.
Figure 5. HSC transplant increases T cell trafficking to the glioma site. (A) GFP+ adoptively transferred antitumor T cells and DsRed+ HSCs were observed co-localizing at the glioma bed in mice that received ACT (image is 10× magnification). (B) T cell migration to intracranial glioma in the presence or absence of HSCs was enumerated one week post adoptive transfer (*p = 0.0015, Wilcoxon test, n = 12). (C) Chemokine array determined that KR158B glioma cells do not secrete crucial lymphocyte recruiting chemokine CCL3. (D) The capacity of glioma specific T cells to migrate toward glioma cells with or without HSCs was assessed using in vitro migration assays. HSC derived CCL3 was blocked using neutralizing antibodies. (**p = 0.0055, Wilcoxon test, n = 5).

To determine if HSCs could facilitate T cell trafficking, we conducted in vitro migration assays using Matrigel coated transwell systems to test the effects of HSCs and glioma cells on T cell migration. We observed that while ex vivo expanded tumor-specific T cells migrated toward both glioma tumor cells and HSCs, they exhibited much greater chemotactic attraction to HSCs either alone or mixed with the tumor cells (). This led to the hypothesis that HSCs may be secreting chemoattractants that lead to increased T cell accumulation at the tumor site. HSCs have been characterized to secrete numerous growth factors and chemokines that can modulate lymphoid cell growth and migration, most notably CXCL12 and CCL3 (SDF-1 and MIP-1α respectively).Citation16,17 Using a multi-chemokine protein array, we examined chemokine production by KR158B tumor cells in order to identify factors differentially produced by tumor cells and HSCs that might account for enhanced migratory capacity. This screen demonstrated that while the glioma cells produced CXCL12 they did not produce appreciable levels of CCL3 (). To test the hypothesis that CCL3 could mediate enhanced T cell migration toward HSCs, we evaluated the impact of blocking antibodies to CCL3 on in vitro T cell migration. Neutralizing antibodies to CCL3 had no effect on migration toward tumor cells, consistent with our observation that tumor cells did not produce appreciable levels of this chemokine. However, blockade of CCL3 almost completely abrogated migration in response to HSCs in vitro (), implicating this chemokine as the major mediator of lymphocyte migration.
In order to determine whether CCL3 played a role in T cell trafficking toward HSCs in vivo at the tumor site, we injected HSCs isolated from either wild-type mice () or CCL3 knockout mice (CCL3−/− HSC)() directly into established intracranial tumors, followed by peripheral intravenous adoptive transfer of tumor specific lymphocytes. Tumors were excised 24 h later and trafficking of tumor-specific lymphocytes from the periphery to sites of intratumoral growth was enumerated using fluorescent microscopy (). T cell migration to intracranial tumors was greatly enhanced in the presence of HSCs from wild-type mice compared to mice that received no HSC transfer at all (p < 0.02). Transfer of CCL3−/− HSCs completely abrogated the enhancing effects of HSC transfer, demonstrating that CCL3 produced by HSCs was the key mediator in enhancing lymphocyte trafficking to CNS tumors in vivo.
Figure 6. HSC-derived CCL3 mediates T cell trafficking to intracranial glioma. (A) KR158B glioma bearing mice received either wild type HSCs or (B) HSCs derived from CCL3 knockout mice directly into the glioma bed and intravenous injection of DsRed tumor-specific T cells. Intratumoral T cell infiltration was measured using fluorescent microscopy 24 h after adoptive transfer. (C) Quantification of adoptively transferred T cells within the tumor site (n = 5; Two-tailed t test, *p = 0.0219). HSCs from wildtype mice show enhanced T cell trafficking to tumor.
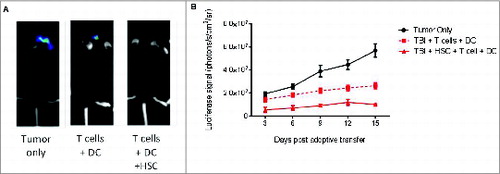
Collectively, these results demonstrated that HSCs migrate to areas of intracranial tumor growth and mediate the enhanced recruitment of tumor-specific lymphocytes through secretion of the chemokine CCL3. To determine whether or not the recruitment of tumor-specific lymphocytes resulted in enhanced antitumor activity, we conducted bioluminescent imaging of the impact of HSC transplant on suppression of tumor growth in mice receiving ACT with or without HSC transplant (). Since mice receiving MA conditioning without HSC transplant succumb to early bone marrow failure (not shown), assessment of the impact of HSC transplant was evaluated using NMA conditioning (5G TBI). Results demonstrated a significant impact of HSC transplant on the suppression of tumor growth, indicating a novel role for HSCs in facilitating immunologic rejection of malignant gliomas during ACT, even under NMA conditions (Mixed effects linear model analysis, p = 0.007).
Figure 7. HSC transfer enhances immunologic glioma rejection. (A) Intracranial astrocytoma bearing mice received adoptive immunotherapy consisting of antitumor T lymphocytes followed by DC vaccine, with or without HSC transfer. Bioluminescent imaging of tumor growth in untreated animals (tumor only), animals receiving tumor-specific T cells plus DC vaccine only (T cells + DC), or lymphoyctes, DC vaccination, and HSC transfer (T cells + DC + HSC) was evaluated at day 14. (B) Impact of HSC transplant on tumor growth was evaluated in mice receiving NMA conditioning and ACT with or without HSCs by bioluminescent imaging (n = 3, Mixed effects linear model analysis, *p = 0.007).
Our results demonstrate the safety and potency of ACT targeting invasive malignant gliomas using total tumor RNA-pulsed DCs as a platform for expansion of tumor-specific lymphocytes. Importantly, they reveal a synergistic interaction between adoptive T cell transfer, DC vaccination, and HSC transplant, wherein HSCs play a previously unappreciated role in facilitating the successful migration of tumor-specific lymphocytes to sites of invasive malignant glioma growth and enhancing immunologic rejection of invasive tumor cells. These results have significant potential to improve immunotherapeutic treatments for patients with high-grade brain tumors and also open new avenues of research into the role of HSCs in antitumor immunity and their novel use in cellular immunotherapy protocols.
Discussion
Immunotherapy holds the promise of eradicating invasive malignancies with exquisite precision through leveraging cytotoxic capabilities of tumor antigen reactive lymphocytes. Approaches with adoptive transfer of tumor-specific T cells have demonstrated a high degree of efficacy against solid tumors in both murine and human systems. Although the brain is a relatively immune-privileged site with respect to the presence of endogenous immune effector cells, previous studies have demonstrated that the CNS is privy to peripheral immune surveillance by activated lymphocytes, lending credence that adoptive T cell immunotherapy can elicit antitumor responses against intracranial tumors. Previous studies have demonstrated that adoptive T cell therapy can target the gp100 tumor-associated antigen in the GL261 preclinical glioma model,Citation18 while clinical investigations using the adoptive transfer of tumor infiltrating lymphocytes (TIL) in melanoma have demonstrated up to a 41% complete response in the treatment of brain metastases.Citation19 These studies further substantiate that brain cancers should not excluded from the evaluation of immune-based therapies for perceived safety or efficacy constraints.
We have developed an adoptive cellular transfer platform that targets a highly invasive, radiation- and chemotherapy-resistant intracranial astrocytoma with precision. Our studies reveal an important synergistic interplay between the adoptively transferred T cells, DC vaccine, and HSCs in the ACT platform that is required for antitumor efficacy, while each component alone is not sufficient to provide any appreciable survival benefit.
In this current study, we demonstrate that previously uncharacterized intercellular interactions between tumor-reactive T cells and HSCs critically drive antitumor immunity. Myeloablation requires HSC rescue to support host bone marrow reconstitution, lymphocyte expansion, and host T cell regeneration. In the context of adoptive immunotherapy, this subsequently allows homeostatic proliferative advantage of adoptively transferred tumor specific lymphocytes prior to host reconstitution, thus antitumor T cells gain a competitive advantage to become disproportionately over-represented in the recovering lymphocyte population as shown in murine models and in humans.Citation11 Eloquent adoptive T cell transfer studies in metastatic melanoma have clearly demonstrated that HSC co-transfer significantly enhances serum levels of T cell proliferative cytokines IL-7 and IL-15, while supporting the expansion of both host and adoptively transferred T cells.Citation11 This data, coupled with our novel observation that antitumor efficacy is dependent on the synergistic interactions between adoptively transferred T cells, DC vaccines, and HSCs, demonstrates that the role of HSCs in the context of adoptive immunotherapy is greater than the previously appreciated. Our exploration of T cell and HSC interactions was predicated on the observed co-localization of HSCs and tumor-specific lymphocytes specifically within the tumor microenvironment. Subsequent analysis revealed that HSCs enhanced the efficacy of ACT by recruiting antitumor T cells through HSC-derived chemokines. Chemokines in the context of tumor biology play a paramount role in tumor cell metastasis and lymphocyte migration, driving T cells to infiltrate solid tumors.Citation20 CCL3 has been well-characterized as a chemokine involved in the migration of hematopoietic cells toward sites of inflammation.Citation16,17 HSC transplant directly facilitated increased recruitment of tumor-specific lymphocytes and led to the enhanced immunologic rejection of invasive malignant glioma growth (). Our data demonstrate that HSC-derived CCL3 is largely responsible for enhanced migration and persistence of adoptively transferred tumor-reactive T cells to intracranial gliomas. These studies, for the first time, implicate HSC transfer directly in the suppression of tumor growth through augmentation of antitumor immunity and lymphocyte-mediated tumor rejection. While these mechanisms were shown to be operative in the context of adoptive transfer of tumor-specific lymphocytes and DC vaccination, it would be of interest to examine whether HSC transplant may facilitate the recruitment of endogenous lymphocytes and perhaps directly modulate recovering host antitumor immunity as well.
It has previously been characterized that HSCs are recruited to intracranial glioma via tumor-secreted CXCL12Citation21,22 which is the ligand for CXCR4, a receptor characteristic of HSCs.Citation23,24 Studies have demonstrated that CXCR4 blockade inhibits bone marrow-derived progenitor cell migration to glioma, indicating that CXCR4 is required for their chemotaxis to tumor.Citation21 It has been suggested that HSC-derived pericytes may be recruited by tumors to contribute to tumor vasculature.Citation25 This intracranial recruitment of HSCs is further increased by radiation and the hypoxic environment in the brain.Citation26 The efficacy of our ACT platform is dependent on the degree of host conditioning. Here, we leverage the previously undesirable effects HSC recruitment to tumor by using the HSCs to increase intratumoral trafficking of tumor-reactive T cells to the glioma. Although, we have demonstrated the increase in antitumor T cell frequency at the tumor site in response to HSCs, we are currently conducting studies to further investigate additional effects of HSCs on T cell effector function, proliferation, and survival.
The platform employing total tumor RNA-loaded DCs to generate tumor-specific lymphocytes holds significant versatility when compared to other approaches that have gained considerable traction such as TIL therapy for melanoma and genetic engineering of T cells with high affinity TCRs or CARs.Citation27 Both high avidity/affinity TCRs and CAR T cells require the identification of tumor associated antigens (TAA) more highly or exclusively expressed in tumor cells than in normal tissue. In particular, through the engineering of high affinity receptors coupled with augmented intracellular signaling domains, cross reactivity with normal tissues expressing low levels of TAAs targeted by genetically modified T cells have been the cause of significant toxicities in some treated patients.Citation28-30 With the exception of EGFRvIII, truly tumor-specific TAAs in brain tumors have not been readily identified, thus the targeting of a broad repertoire of antigens with the use of total tumor RNA overcomes the limitations of antigen selection in brain tumors. Furthermore, the heterogeneity of expression of tumor antigens and patient-specific mutations that likely contribute to the unique antigenic milieu within glioma tumors is not currently addressed by genetically engineered T cell immunotherapy strategies. Thus, the use of tumor RNA-pulsed APCs as a patient-specific platform for ACT holds considerable potential to overcome the limitations of other approaches.
The use of high-dose chemotherapy, including MA regimens coupled with HSC rescue, in the management of several malignancies including, non-Hodgkin's lymphoma, high risk neuroblastoma, and medulloblastomas offers a unique window to leverage the myelosuppressive toxicities of these clinically relevant regimens to develop and augment novel immunotherapeutic approaches vital for improving therapeutic outcomes in these aggressive tumors.Citation31 Moreover, MA chemo has been reported to be effective in children with medulloblastomasCitation32-34 and to offer some benefit in the treatment of high grade gliomas,Citation35,36 epitomizing the capacity to piggyback immunotherapeutic regimens as promising considerations for adjuvant therapies. For example, we have demonstrated that MA doses of TMZ can effectively be used as a conditioning regimen to enhance the efficacy of immunotherapy in a preclinical model system.Citation37 Since clinical care for many high-grade brain tumors utilizes MA therapy plus HSC rescue, particularly in the treatment of pediatric brain cancers, we can potentially leverage the existing clinical treatment approaches as host conditioning regimens to further enhance the antitumor efficacy of ACT. Such combinatorial approaches that leverage the myelosuppressive toxicity of standard chemotherapy regimens as host conditioning regimens for subsequent ACT are currently the focus of ongoing clinical studies within our program. Importantly, our observation that HSC transplant enhances tumor rejection even under NMA conditions () opens the door to consideration of the use of HSCs as a cellular component of ACT platforms without the constraints and associated toxicities conveyed with MA treatment. This may have particular relevance in the treatment of many solid tumors in adults where, in contrast to more frequent use in the pediatric setting, MA treatment plus stem cell rescue is less often routinely employed.
In summary, we demonstrate synergistic cellular interactions between T cells, DCs, and HSCs in a curative ACT platform targeting a highly-invasive and treatment-resistant malignant astrocytoma. These studies reveal a novel role for HSCs in mediating intratumoral T cell trafficking and immunologic rejection of intracranial tumors and suggest that HSCs may be co-opted to enhance ACT protocols targeting malignant brain tumors and potentially other solid cancers.
Materials and Methods
Adoptive cell therapy (ACT)
C57BL/6 mice (Jackson Laboratories) were stereotactically implanted with 104 KR158B astrocytoma cells into the right caudate nucleus on Day 0. Mice then received a single dose of NMA 5Gy or MA 9Gy TBI on Day 4. Mice receiving HSC rescue were given intravenous injection of 5 × 104 lin− bone marrow derived stem cells within 6 h of TBI. HSCs were derived from bone marrow of C57BL/6 mice and magnetically depleted for lin− bone marrow stem cells (Miltenyi Biotec). Intravenous injection of 107 tumor specific T lymphocytes was administered between 16 and 24 h after TBI. This was immediately followed by an intradermal vaccination of 2.5 × 105 total tumor RNA-pulsed DCs. DC vaccines 2 and 3 were administered at weekly intervals and mice followed for survival and sacrificed when moribund (Fig. S1). Intracranial tumor growth was evaluated using bioluminescent imaging where indicated.
Tumor cells
KR158B-luc cells were a kind gift from Dr. Tyler Jacks (Massachusetts Institute of Technology, Boston, MA). This tumor cell line was originally isolated from a spontaneously arising astrocytoma in an Nf1;Trp53 mutant mouse on a C57BL/6 background.Citation12 Culture media consist of DMEM without sodium pyruvate (LifeTechnologies), supplemented with 10% heat inactivated fetal bovine serum (FBS)(LifeTechnologies), 5.5 mL Penicillin/Streptomycin (LifeTechnologies). Cells were kept in a 37°C incubator with 5% CO2 levels.
Isolation of RNA
Total RNA from tumor cells was isolated using commercially available RNeasy mini kits (Quiagen, cat#74104) as per the manufacturer instructions.
Dendritic cell vaccine
DCs were isolated from the bone marrow of C57BL/6 mice using an altered previously published protocol.Citation38 Briefly, femurs and tibias of C57BL/6 mice were harvested and bone marrow flushed with RPMI (LifeTechnologies) +10% FBS (LifeTechnologies). Red cells were lysed with 10 mL Pharmlyse (BD Bioscience) and mononuclear cells were re-suspended in CDCM (RPMI-1640, 5% FBS, 1 M HEPES (LifeTechnologies), 50 μM, 55 mM βmercaptoethanol (LifeTechnologies), 100 mM Sodium pyruvate (LifeTechnologies), 10 mM Nonessential amino acids (LifeTechnologies), 200 mM L-glutamine (LifeTechnologies), 10 μg GM-CSF (R&D Systems), 10 μg IL-4 (R&D Systems), 5.5 mL Penicillin/Streptomycin (LifeTechnologies)) and plated into tissue culture treated six well plates at a density of 106 cells/mL in a total volume of 3 mL/well. Non-adherent cells were discarded at day 3. At day 7, non-adherent cells were collected and re-plated onto 100 mm tissue treated culture dishes at a density of 106 cells/mL in a total volume of 5 mL/dish. Twenty four hours later, resulting cells were electroporated with 25 μg of total RNA isolated from KR158B-luc cells (RNeasy, Qiagen). RNA-pulsed DCs were collected the following day and suspended in PBS at a final concentration of 1.25 × 106 cells, and 200 μL cell suspension was administered via intraperitoneal injection.
Generation of tumor-specific T cells
Total RNA was isolated from KR158B-luc tumor and electroporated into bone marrow derived DCs using BTX Single Waveform Electroporation System (Harvard Apparatus). Naïve mice received intradermal vaccination with total tumor RNA-pulsed DCs, their spleens harvested 7 d later, and the splenocytes expanded ex vivo using RNA-pulsed DCs and 100IU IL-2 (R&D Systems) for 7 d. T cells were expanded from primed spleens of either wildtype C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME, stock #000664), DsRed transgenic mice on a C57BL/6 background (Jackson Laboratories, stock #006051) or GFP transgenic mice on a C57BL/6 background (Jackson Laboratories, stock #004353). Tumor-reactive T cells were adoptively transferred intravenously after 5–7 d of in vitro activation.
Mice
Four to 8 week old C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME, stock#000664). Transgenic mice used were all on the C57BL/6 background and purchased from Jackson Laboratories: DsRed transgenic mice (Jackson laboratories, stock #006051); GFP transgenic mice (Jackson Laboratories, stock#004353); MIP1α−/− mice (Jackson Laboratories, stock #002687). The investigators adhered to the “Guide for the Care and Use of Laboratory Animals” as proposed by the committee on care of Laboratory Animal Resources Commission on Life Sciences, National Research Council. The facilities at the Duke Cancer Center Isolation Facility are fully accredited by the American Association for Accreditation of Laboratory Animal Care, and all studies were approved by the Duke University Institutional Animal Care and Use Committee.
Intracranial tumors
KR158B cells were harvested with 0.25% trypsin (LifeTechnologies), washed once in serum-containing medium, and washed twice in Dulbecco's phosphate-buffered saline (DPBS) (LifeTechnologies). Cell pellets were suspended in DPBS at the appropriate concentration of viable cells as determined by trypan blue dye exclusion, mixed with an equal volume of 10% methylcellulose (R&D Systems) in DPBS and loaded into a 250-μL Hamilton syringe (Hamilton, Reno, NV) with an attached 25-gauge needle. In C57BL/6 mice, the tip of the needle was positioned at bregma and 2 mm to the right of the cranial midline suture and 4 mm below the surface of the cranium using a Kopf stereotactic frame (David Kopf Instruments, Tujunga, CA).
Host conditioning
Host conditioning was achieved with TBI using a cesium source. Mice received single doses of 5Gy or 9Gy radiation as approved by the institutional IACUC and Radiation Safety protocols.
Flow cytometric analysis
Live cells were analyzed using a BD Bioscience FACS Calibur gated by FSC/SSC. Fluorescent transgenic GFP or DsRed T cells were detected at the FL1 or FL2 channel respectively. Sterile fluorescent activated cell sorting for DsRed+ T cells was conducted on the FACS Aria II (BD Biosciences).
Fluorescent microscopy
Fluorescent microscopy was conducted on freshly isolated perfused brain tissue that was sliced into 300 μm with a Leica VT1000s vibratome (Leica Microsystems) and plated onto uncoated glass slides. Brain slices were immediately imaged with a Nikon Eclipse TE2000-E inverted fluorescent microscope and analyzed using MetaMorph Microscopy Automation and Image Analysis Software.
Bioluminescent in vivo imaging
Intracranial KR158B-luc tumors were imaged in vivo using an IVIS Kinetic (Perkin Elmer). Mice were injected with 100 μL luciferin substrate (Sigma) and imaged between 10–15 min after injection for optimal signal output. Bioluminescence for all animals was read at 1sec exposure.
Functional stimulation assay
To determine antitumor T cell function, effector T cells were co-cultured with target cells overnight and supernatant was collected. A BD Cytometric Bead Array kit was used to determine mouse Th1/Th2 cytokine release in the supernatant (BD Bioscience) as per manufacturer instructions.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
994374_Supplementary_Materials.zip
Download Zip (480.4 KB)Acknowledgment
The authors would like to thank Dan Neal of the University of Florida Department of Neurosurgery for help with some statistical analysis.
Funding
This work was supported by grants from the Pediatric Brain Tumor Foundation (PBTF) of the United States (PBTF Institute at Duke, D.D.B., J.H.S., and D.A.M.), SRC on Primary and Metastatic Tumors of the CNS (P50-NS20023, D.D.B. and J.H.S), and National Brain Tumor Society (D.A.M.). Additional support in part was provided by Duke University's Clinical & Translational Science Awards grant 1UL2 RR024128-01 from the National Institutes of Health National Center for Research Resources.
References
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17(13):4550-7; PMID:21498393; http://dx.doi.org/10.1158/1078-0432.CCR-11-0116
- Rosenberg SA, Kochenderfer JN. Personalized cell transfer immunotherapy for B-cell malignancies and solid cancers. Mol Ther 2011; 19(11):1928-30; PMID:22051601; http://dx.doi.org/10.1038/mt.2011.223
- Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med 2012; 4(127):127ps8; PMID:22461638; http://dx.doi.org/10.1126/scitranslmed.3003634
- Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer–what clinicians need to know. Nat Rev Clin Oncol 2011; 8(10):577-85; PMID:21808266; http://dx.doi.org/10.1038/nrclinonc.2011.116
- Heiser A, Maurice MA, Yancey DR, Wu NZ, Dahm P, Pruitt SK, Boczkowski D, Nair SK, Ballo MS, Gilboa E et al. Induction of polyclonal prostate cancer-specific CTL using dendritic cells transfected with amplified tumor RNA. J Immunol 2001; 166(5):2953-60; PMID:11207244; http://dx.doi.org/10.4049/jimmunol.166.5.2953
- Melhem NM, Liu XD, Boczkowski D, Gilboa E, Barratt-Boyes SM. Robust CD4+ and CD8+ T cell responses to SIV using mRNA-transfected DC expressing autologous viral Ag. Eur J Immunol 2007; 37(8):2164-73; PMID:17615585; http://dx.doi.org/10.1002/eji.200636782
- Wrzesinski C, Paulos CM, Kaiser A, Muranski P, Palmer DC, Gattinoni L, Yu Z, Rosenberg SA, Restifo NP. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. J Immunother 2010; 33(1):1-7; PMID:19952961; http://dx.doi.org/10.1097/CJI.0b013e3181b88ffc
- Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, Veloso E, Zheng Z, Westphal S, Mair R et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med 2005; 11(11):1230-7; PMID:16227990; http://dx.doi.org/10.1038/nm1310
- Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol 2012; 189(11):5147-54; PMID:23100512; http://dx.doi.org/10.4049/jimmunol.1200274
- Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 2005; 202(7):907-12; PMID:16203864; http://dx.doi.org/10.1084/jem.20050732
- Wrzesinski C, Paulos CM, Gattinoni L, Palmer DC, Kaiser A, Yu Z, Rosenberg SA, Restifo NP. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J Clin Invest 2007; 117(2):492-501; PMID:17273561; http://dx.doi.org/10.1172/JCI30414
- Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 2000; 26(1):109-13; PMID:10973261; http://dx.doi.org/10.1038/79075
- Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol 2012; 2012:831090; PMID:23133490
- Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, Wunderlich JR, Mixon A, Farid S, Dudley ME et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014; 124(5):2246-59; PMID:24667641; http://dx.doi.org/10.1172/JCI73639
- Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013; 19(6):747-52; PMID:23644516; http://dx.doi.org/10.1038/nm.3161
- Liesveld JL, Harbol AW, Belanger T, Rosell KE, Abboud CN. MIP-1alpha and TGF-beta production in CD34+ progenitor-stromal cell coculture systems: effects of progenitor isolation method and cell-cell contact. Blood Cells Mol Dis 2000; 26(4):261-75; PMID:11042027; http://dx.doi.org/10.1006/bcmd.2000.0305
- Menten P, Saccani A, Dillen C, Wuyts A, Struyf S, Proost P, Mantovani A, Wang JM, Van Damme J. Role of the autocrine chemokines MIP-1alpha and MIP-1beta in the metastatic behavior of murine T cell lymphoma. J Leukoc Biol 2002; 72(4):780-9; PMID:12377948
- Prins RM, Odesa SK, Liau LM. Immunotherapeutic targeting of shared melanoma-associated antigens in a murine glioma model. Cancer Res 2003; 63(23):8487-91; PMID:14679014
- Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, Sherry RM. Successful treatment of melanoma brain metastases with adoptive cell therapy. Clin Cancer Res 2010; 16(19):4892-8; PMID:20719934; http://dx.doi.org/10.1158/1078-0432.CCR-10-1507
- Nakasone Y, Fujimoto M, Matsushita T, Hamaguchi Y, Huu DL, Yanaba M, Sato S, Takehara K, Hasegawa M. Host-derived MCP-1 and MIP-1alpha regulate protective anti-tumor immunity to localized and metastatic B16 melanoma. Am J Pathol 2012; 180(1):365-74; PMID:22037251; http://dx.doi.org/10.1016/j.ajpath.2011.09.005
- Xu Q, Yuan X, Xu M, McLafferty F, Hu J, Lee BS, Liu G, Zeng Z, Black KL, Yu JS. Chemokine CXC receptor 4–mediated glioma tumor tracking by bone marrow–derived neural progenitor1487;stem cells. Mol Cancer Ther 2009; 8(9):2746-53; PMID:19723878; http://dx.doi.org/10.1158/1535-7163.MCT-09-0273
- Tabatabai G, Bähr O, Möhle R, Eyüpoglu IY, Boehmler AM, Wischhusen J, Rieger J, Blümcke I, Weller M, Wick W. Lessons from the bone marrow: how malignant glioma cells attract adult haematopoietic progenitor cells. Brain 2005; 128(Pt 9):2200-11; PMID:15947066; http://dx.doi.org/10.1093/brain/awh563
- Ishii T, Nishihara M, Ma F, Ebihara Y, Tsuji K, Asano S, Nakahata T, Maekawa T. Expression of stromal cell-derived factor-11487;pre-B cell growth-stimulating factor receptor, CXC chemokine receptor 4, on CD34+ human bone marrow cells is a phenotypic alteration for committed lymphoid progenitors. J Immunol 1999; 163(7):3612-20; PMID:10490954
- Voermans C, Kooi ML, Rodenhuis S, van der Lelie H, van der Schoot CE, Gerritsen WR. In vitro migratory capacity of CD34+ cells is related to hematopoietic recovery after autologous stem cell transplantation. Blood 2001; 97(3):799-804; PMID:11157500; http://dx.doi.org/10.1182/blood.V97.3.799
- Bababeygy SR, Cheshier SH, Hou LC, Higgins DM, Weissman IL, Tse VC. Hematopoietic stem cell-derived pericytic cells in brain tumor angio-architecture. Stem Cells Dev 2008; 17(1):11-8; PMID:18240955; http://dx.doi.org/10.1089/scd.2007.0117
- Tabatabai G, Frank B, Möhle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-beta-dependent HIF-1alpha-mediated induction of CXCL12. Brain 2006; 129(Pt 9):2426-35; PMID:16835250; http://dx.doi.org/10.1093/brain/awl173
- Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989; 86(24):10024-8; PMID:2513569; http://dx.doi.org/10.1073/pnas.86.24.10024
- Xu XJ, Tang YM Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett 2014; 343(2):172-8; PMID:24141191; http://dx.doi.org/10.1016/j.canlet.2013.10.004
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010; 18(4):843-51; PMID:20179677; http://dx.doi.org/10.1038/mt.2010.24
- Buning H, Uckert W, Cichutek K, Hawkins RE, Abken H. Do CARs need a driver's license? Adoptive cell therapy with chimeric antigen receptor-redirected T cells has caused serious adverse events. Hum Gene Ther 2010; 21(9):1039-42; PMID:20626322; http://dx.doi.org/10.1089/hum.2010.131
- Marachelian A, Butturini A, Finlay J. Myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for childhood central nervous system tumors. Bone Marrow Transplant 2008; 41(2):167-72; PMID:18176620; http://dx.doi.org/10.1038/sj.bmt.1705953
- Bergthold G, El Kababri M, Varlet P, Dhermain F, Sainte-Rose C, Raquin MA, Kieffer V, Goma G, Grill J, Valteau-Couanet D et al. High-dose busulfan-thiotepa with autologous stem cell transplantation followed by posterior fossa irradiation in young children with classical or incompletely resected medulloblastoma. Pediatr Blood Cancer 2014; 61(5):907-12; PMID:24470384; http://dx.doi.org/10.1002/pbc.24954
- Perez-Martinez A, Lassaletta A, González-Vicent M, Sevilla J, Díaz MA, Madero L. High-dose chemotherapy with autologous stem cell rescue for children with high risk and recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors. J Neurooncol 2005; 71(1):33-8; PMID:15719272; http://dx.doi.org/10.1007/s11060-004-4527-4
- Dunkel IJ, Gardner SL, Garvin JH Jr, Goldman S, Shi W, Finlay JL. High-dose carboplatin, thiotepa, and etoposide with autologous stem cell rescue for patients with previously irradiated recurrent medulloblastoma. Neuro Oncol 2010; 12(3):297-303; PMID:20167818; http://dx.doi.org/10.1093/neuonc/nop031
- Chen B, Ahmed T, Mannancheril A, Gruber M, Benzil DL. Safety and efficacy of high-dose chemotherapy with autologous stem cell transplantation for patients with malignant astrocytomas. Cancer 2004; 100(10):2201-7; PMID:15139065; http://dx.doi.org/10.1002/cncr.20223
- Finlay JL, Goldman S, Wong MC, Cairo M, Garvin J, August C, Cohen BH, Stanley P, Zimmerman RA, Bostrom B et al. Pilot study of high-dose thiotepa and etoposide with autologous bone marrow rescue in children and young adults with recurrent CNS tumors. The Children's Cancer Group. J Clin Oncol 1996; 14(9):2495-503; PMID:8823328
- Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, Schmittling RJ, Snyder D, Herndon JE 2nd, Bigner DD et al. Myeloablative temozolomide enhances CD8(+) T-cell responses to vaccine and is required for efficacy against brain tumors in mice. PLoS One 2013; 8(3):e59082; PMID:23527092; http://dx.doi.org/10.1371/journal.pone.0059082
- Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, Brinster C. Isolation of dendritic cells. Curr Protoc Immunol 2009; Chapter 3:Unit 3 7; PMID:19653207; http://dx.doi.org/10.1002/0471142735.im0307s86