
Abstract
Recent studies have revealed the contribution of fibro-adipogenic progenitors (FAPs) to the pathogenesis and progression of Duchenne Muscular Dystrophy (DMD). While FAPs direct compensatory regeneration at early stages of disease, as the disease progresses they contribute to the progressive replacement of contractile myofibers with fibrotic scars and fatty infiltration. Using the mouse model of DMD – the mdx mice - we have recently reported that FAPs mediate the ability of HDAC inhibitors (HDACi) to promote muscle regeneration and prevent fibro-adipogenic degeneration at early stages of disease. This effect is mediated by the induction of myomiRs that, in turn, target the SWI/SNF components BAF60A and B, thereby favoring the formation of BAF60C-based SWI/SNF complex, which directs the switch from the fibro-adipogenic to the myogenic lineage. Here we show direct evidence of induction of miR-206 and BAF60C, and reduction of BAF60A, in FAPs isolated from mdx muscles exposed to the HDACi Trichostatin A (TSA). We also discuss how increased expression of myomiRs in dystrophic muscles can be integrated with circulating myomiRs to provide accurate biomarkers of disease progression and response to treatment.
Keywords:
Abbreviations
| FAPs | = | fibro-adipogenic progenitors |
| DMD | = | Duchenne Muscular Dystrophy |
| HDACi | = | HDAC inhibitors |
| TSA | = | Trichostatin A |
| MD | = | Muscular dystrophies |
| MuSCs | = | muscle stem cells |
| Mdx | = | murine model of DMD |
| NA-seq | = | nuclease accessibility site sequencing |
| BAFs | = | BRG1/BRM-associated factors |
| NAS | = | nuclease accessibility sites |
| LncRNA | = | long non-coding RNA |
| LINE | = | long interspersed non-coding elements |
| SINE | = | short interspersed non-coding elements |
| miRs | = | microRNA |
| HTS | = | high-throughput screening |
| CK | = | creatine kinase |
Muscular dystrophies (MD) comprise more than 30 inherited diseases characterized by progressive muscle weakness and degeneration. The most common and severe MD is the Duchenne Muscular Dystrophy (DMD).
DMD is caused by mutations in the dystrophin gene, located on the X-chromosome, that lead to the absence of dystrophin protein.Citation1 Dystrophin-deficient muscles are vulnerable to mechanical damage, leading to myofiber degeneration and necrosis following contractile activity.Citation2 At early stages of disease, the cycles of muscle contraction/degeneration are counterbalanced by “compensatory” repair, which is characterized by proliferation and differentiation of muscle (satellite) stem cells (MuSCs) to form new myofibers. These stages coincide with a “clinical latency” of DMD, with attenuated symptoms of disease – the so-called “Honeymoon” stage. The progression of the disease coincides with qualitative changes in muscle tissue composition, with ensuing deposition of fibrotic tissue and fat, and exhaustion of the regeneration potential that bias muscle repair toward a progressive replacement of contractile myofibers with fibrotic scars and fat infiltration.Citation3-5
While the progressive decline of compensatory regeneration has been historically attributed to the functional exhaustion of muscle satellite cells and is currently regarded as a key event in the pathogenesis of DMD, its relationship with fibrosis and fat deposition has only recently begun to be appreciated, owing to the seminal discovery of a population of muscle interstitial cells, originally termed fibro/adipogenic progenitors (FAPs).Citation6-9 These cells can contribute to either muscle regeneration and fibro-adipogenic degeneration. In healthy muscles, FAPs provide the support to satellite cell-mediated regeneration during acute injury, by integrating the signals released from inflammatory infiltrate with the activation of muscle satellite cells.Citation10 This response is typically active in muscles of the murine model of DMD (the mdx mice) at early stages of disease.Citation4 However, as the disease progresses, these cells become biased toward their intrinsic fibro-adipogenic activity and repress satellite cell-mediated regeneration, while promoting fibrosis and fat infiltration.Citation11,4 As such, FAPs are currently considered a heterogeneous population of functionally versatile cells that contribute to the pathogenesis of DMD and possibly other chronic degenerative muscle disorders,Citation12 and might provide valuable target for therapeutic interventions toward promoting compensatory regeneration, while inhibiting fibro-adipogenic degeneration of diseased muscles.
In recent studies, we have investigated FAPs as potential cellular mediators of the beneficial effects of HDAC inhibitors (HDACi) that have been observed in animal models of DMD, such as mdx miceCitation13-16 and Zebrafish.Citation17 Interestingly, HDACi exert beneficial effects in mdx mice at early, but not late, stages of disease and this coincides with striking changes of FAP function and phenotype observed along with disease progression.Citation4 Within the “permissive” regenerative environment of young mdx mice, FAPs display a latent pro-myogenic phenotype that is fully expressed upon the exposure to HDACi, in concomitance with the inhibition of the fibro-adipogenic potential. By contrast, at late stages of DMD progression, FAPs adopt a constitutive fibro-adipogenic phenotype that appears to be both “dominant” over the pro-myogenic potential and resistant to HDACi.Citation4
The mutually exclusive phenotypes adopted by FAPs at different stages of disease progression and the related differential response to HDACi can be explained by the activation of alternative transcription networks. In our recent work,Citation18 we have performed an integrated genome-wide approach to address this issue. Combinatorial analysis of gene expression microarray, genome-wide chromatin remodeling by nuclease accessibility site sequencing (NA-seq), and small RNA sequencing (RNA-seq) revealed that HDACi derepress the “latent” myogenic program in FAPs from muscles of mdx mice at early stages, while FAPs from muscles of mdx mice at late stages of disease are refractory to HDACi. In particular, we observed reduced chromatin accessibility in response to HDACi at muscle-specific loci in FAPs from old mdx mice, as compared to FAPs isolated from muscles of young mdx mice. This finding suggests that in FAPs from old dystrophic muscles the chromatin adopts a configuration that precludes the reprogramming by epigenetic drugs, such as HDACi. It also indicates that changes in chromatin remodeling activity, observed at early stages of disease, are regulated by HDAC-mediated control of the chromatin remodeling machinery.
Chromatin remodeling is typically catalyzed by specialized complexes that are endowed with specific enzymatic activities. The SWI/SNF chromatin remodeling complex has been previously shown to regulate lineage determination in many cell typesCitation19,20 and to be required for the activation of skeletal myogenesis in muscle progenitors.Citation21,22 SWI/SNF complexes show heterogeneous composition, with mutually exclusive incorporation of 2 enzymatic subunits (the ATPases BRG1 and BRM) and a number of structural subunits, collectively referred to as BRG1/BRM-associated factors (BAFs)Citation23. Among them, there are 3 alternative variants of the 60 kDa subunit - BAF60a, b, and c - which confer the affinity for tissue-specific transcription factors.Citation24 Despite the large sequence homology shared by BAF60 variants, they appear to exert specialized functions in lineage determination. While BAF60c plays a key role in both cardiac and skeletal muscle,Citation22,24-26 BAF60a and BAF60b appear to be involved in the activation of alternative pathways, including lipid metabolism.Citation27 Thus, the finding that pharmacological blockade of HDAC in FAPs of young mdx mice induces BAF60c and the muscle-specific transcriptional activator MyoD, while down-regulating the expression of BAF60a and b,Citation18 supports the conclusion that HDAC-mediated control of SWI/SNF composition regulates chromatin remodeling and lineage determination in FAPs.
Interestingly, the vast majority of HDACi-induced nuclease accessibility sites (NAS) annotated in FAPs from young mdx mice associates with non-coding regions of the genome, including long non-coding RNA (LncRNA), long and short interspersed non-coding elements (LINE and SINE), and microRNA (miRs). Previous work from the Bozzoni lab has shown an HDAC-dependent control of miR expression in mdx mice.Citation28 Based on this report, and with the support of microRNA high-throughput screening (HTS) against SWI/SNF BAF60 variants, we identified HDACi-induced miRs that establish a reciprocal network with the SWI/SNF complex to promote “localized” chromatin remodelling at muscle loci and impart the pro-myogenic phenotype in FAPs of young mdx mice exposed to HDACi. Specifically, we have shown that HDACi upregulate muscle-specific miRs - the myomiRs miR-1, miR-133, and miR-206 - that promote muscle progenitor proliferation and differentiation,Citation29-31 and could be detected in the serum in patients affected by DMD and other muscular dystrophies.Citation32-37 Indeed, studies from our group and othersCitation18,38 have established a direct relationship between myomiRs and selection of BAF60 subunits, by showing that myomiRs selectively target BAF60a and b.
We sought to correlate the relative expression of BAF60 variants with the expression of myomiRs in FAPs from mdx mice exposed to HDACi. shows that after exposure to the HDACi Trichostatin A (TSA), cultured FAPs isolated from young dystrophic muscles show an increased nuclear expression of BAF60c (), reduced levels of BAF60a (), and up-regulation of miR-206 (). These events underline the activation of the myogenic program, at the expense of the fibro-adipogenic one, as FAPs from young mdx mice exposed to TSA and cultured in adipogenic medium form miR-206 and MyHC-positive myotubes (, bottom panel), rather than differentiating into Oil red O-positive adipocytes (, upper panel). The activation of the MyoD/BAF60c/myomiR network is also required for the increased ability of FAPs to support MuSC differentiation after TSA treatment.Citation4,18 The relationship between HDAC-mediated control of muscle lineage and the ability of FAPs to promote satellite cell-mediated muscle regeneration is currently unclear and deserves further investigation. Likewise, how HDAC-regulated chromatin remodelling and gene expression control soluble cues that mediate functional interactions between FAPs and satellite cells is still unclear. The increasing evidence on miR-mediated cell-to-cell communication process in skeletal muscle and, in particular, their clustering into exosomes,Citation39,40 suggest that miR can also mediate functional interactions between FAPs, MuSCs and possibly other cell types that contribute to the regeneration process (). Although highly speculative, we postulate that myomiRs released by degenerating fibers and/or FAPs, could act locally as pro-myogenic signals in the regenerative environment at initial stages of muscle repair.
Figure 1. Expression of BAF60a, BAF60c and miR-206 in FAPs isolated from young mdx mice treated with TSA (Trichostatin A). (A) Expression of BAF60c (Green), in growth medium (GM), was assessed by immunofluorescence. Nuclei were counterstained with DAPI (blue) (Left panel). The percentage of BAF60c positive cells per field was measured and reported as graphed (Right panel). The error bars indicate SEM. (B) Representative images of BAF60a (Red) expression assessed by immunofluorescence in GM (Left panel). Nuclei were counterstained with DAPI (blue). The right panel shows the percentage of BAF60a positive cells per field (Right panel). The error bars indicate SEM. (C) miR-206 expression in GM was evaluated by immunohistochemical staining of miR-206 (Blue/Purple). Nuclei were counterstained with DAPI (Blue for immunofluorescence or Yellow for phase contrast) (Left panel). (D) Myogenic differentiation potential was assessed by myosin heavy chain (MHC) immunofluorescence (Green) and adipogenic differentiation potential by Oil-Red O staining (Red). miR-206 expression in DM was evaluated by immunohistochemical staining. Nuclei were counterstained with DAPI (Blue for immunofluorescence or Yellow for phase contrast).
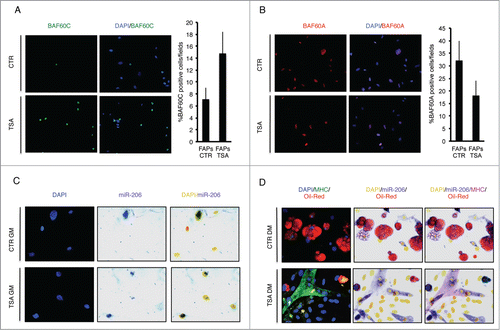
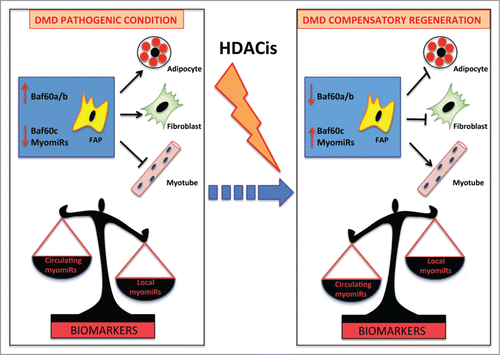
Figure 2. myomiRs as biomarkers of HDACi activity in DMD. FAPs are key cellular determinants of DMD progression. In pathologic conditions these cells differentiate into adipocytes and fibroblasts leading to muscle degeneration. This process is driven by BAF60a and BAF60b-based SWI/SNF complex that promotes the expression of fibro-adipogenic genes. As a consequence of muscle wasting, myomiRs are released in the circulation and have been proposed as biomarkers of DMD disease (Left panel). HDACi treatment at early stages promotes compensatory regeneration by inducing in FAPs BAF60c, which directs myogenesis, and up-regulating myomiRs, which suppress the fibro- adipogenesis In this context, a local increase of myomiRs inversely correlates with the decrease of circulating myomiRs, suggesting that the ratio between circulating/local myomiRs could provide an accurate biomarker of disease progression and response to HDACi (Right panel).
Collectively these data indicate that HDAC-regulated myomiR-BAF60 variant network “shapes” the composition of the SWI/SNF complex and directs its activity to determine whether FAPs supports compensatory regeneration or fibro-adipogenic degeneration of dystrophic muscles. This model is in striking analogy with previously reported miR-mediated control of BAF53 and 45 variants that regulate sequential stages of neurogenic differentiation.Citation41,42
While this data provide new insight into the molecular pathogenesis of DMD, they also shed new light on the detection and use miRs as clinical biomarkers of disease progression. Beyond their active role in regulating myogenesis and skeletal muscle homeostasis, myomiRs are passively released in the blood as a consequence of degeneration of dystrophic myofibers.Citation34,43 Circulating myomiRs can be detected in the peripheral blood of dystrophic patients and their increase correlates with the severity of the disease. Recently, myomiR quantification in blood has been proposed as a diagnostic and prognostic marker more accurate than the conventional detection of creatine kinase (CK) in DMD patientsCitation34, because miRs are more stable and less susceptible to variables, such as exercise.Citation43 The stability of circulating miRs is likely to be preserved by liposomes,Citation44 protein complexes,Citation45 and exosome vesicles.Citation46 In particular, exosomes are emerging as soluble mediators of cell-to-cell communications that might broadcast regulatory signals in a protected environment insulated from the oxidative stress generated during tissue regeneration.Citation47,48
While the accurate detection of circulating myomiRs and their diagnostic and prognostic interpretation in DMD is under evaluation,Citation49,50 we propose that FAP-specific expression of miRs could also be considered as an independent biomarker that correlates with compensatory regeneration of DMD muscles at early stages of disease or following therapeutic interventions, such as treatment with HDACi. We speculate that changes in the expression profile of miRs in FAPs could reflect histological changes during disease progression and be used to monitor the transition from the compensatory early stages to the fibrotic late stages of DMD. As FAPs are key targets of therapeutic interventions in DMD,Citation4,18 and possibly other degenerative muscular disorders, measuring myomiR levels in FAPs could also predict the response to treatments aimed at promoting regeneration and reducing fibrosis. Thus, we propose that integrating the levels of circulating myomiRs with the expression levels of myomiRs in FAPs might offer a reliable marker of disease progression, by correlating myofiber degeneration (circulating miRs) with muscle potential to regenerate or undergo fibrosis and fat infiltration (local, intramuscular miRs). Importantly, the levels of circulating miRs have been proposed as a non-invasive biomarker of myofiber degeneration in DMD patients and their reduction appears to correlate with dystrophin re-expression by exon skipping.Citation33,34,45,51 By contrast, the measurement of myomiR expression in FAPs is currently relying on the availability of muscle biopsies, and future studies should explore the possibility to detect FAP myomiRs using semi-invasive methods – e.g., taking advantage of their interstitial position to isolate FAPs from muscle interstitial bio-fluids by fine needle aspiration. The availability of FAPs from muscles of DMD patients and the relative measurement of myomiRs levels would offer an unprecedented molecular readout of muscle tendency to regenerate or undergo fibro-adipogenic degeneration that can be used not only to monitor the disease progression, but also as an inclusion criteria to select DMD patients for clinical trials and as biomarkers of treatment efficacy. Overall, the integration of passively released (circulating) and locally produced (intramuscular) myomiRs should be regarded as a novel outcome measure of DMD progression and response to therapeutic intervention of potential interest for the upcoming clinical trials.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
PLP is an Investigator of Sanford Children's Health Research Center. This work has been supported by the following grants to PLP: R01AR056712, R01AR052779 and P30 AR061303 from the National Institute of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), EPIGEN, FILAS and AFM-Telethon. This work has benefited from research funding from the European Community's Seventh Framework Program in the project FP7-Health – 2009 ENDOSTEM 241440 (Activation of vasculature associated stem cells and muscle stem cells for the repair and maintenance of muscle tissue). LG was supported by a fellowship from Fondazione Sovena.
References
- Mendell JR, Boué DR, Martin PT. The con- genital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006; 9:427-43; PMID:17163796; http://dx.doi.org/10.2350/06-07-0127.1
- Dalkilic I, Kunkel LM Muscular dystrophies: genes to pathogenesis. Curr Opin Genet Dev 2003; 13:231-38; PMID:12787784; http://dx.doi.org/10.1016/S0959-437X(03)00048-0
- Serrano AL, Mann CJ, Vidal B, Ardite E, Perdiguero E, Muñoz-Cánoves P. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr Top Dev Biol 2011; 96:167-201; PMID:21621071; http://dx.doi.org/10.1016/B978-0-12-385940-2.00007-3
- Mozzetta C, Consalvi S, Saccone V, Tierney M, Diamantini A, Mitchell KJ, Marazzi G, Borsellino G, Battistini L, Sassoon D, et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol Med 2013; 5(4):626-39; PMID:23505062; http://dx.doi.org/10.1002/emmm.201202096
- Desguerre I, Mayer M, Leturcq F, Barbet JP, Gherardi RK, Christov C. Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol 2009; 68(7):762-73; PMID:19535995; http://dx.doi.org/10.1097/NEN.0b013e3181aa31c2
- Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM. Muscle injury activates resident fibroadipogenic progenitors that facilitate myogenesis. Nat Cell Biol 2010; 2:153-63; PMID:20081841; http://dx.doi.org/10.1038/ncb2015
- Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol 2010; 2:143-52; PMID:20081842; http://dx.doi.org/10.1038/ncb2014
- Judson RN, Zhang RH, Rossi FM. Tissue-resident mesenchymal stemprogenitor cells in skeletal muscle: collaborators or saboteurs? FEBS J 2013; 280(17):4100-8; PMID:23763717; http://dx.doi.org/10.1111/febs.12370
- Malecova B, Puri PL. “Mix of Mics”- phenotypic and biological heterogeneity of “Multipotent” muscle interstitial cells (MICs). J Stem Cell Res Ther 2012; (Suppl 11):004; PMID:24634800; http://dx.doi.org/10.4172/2157-7633.S11-004
- Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, Chawla A. Type 2 innate signals stimulate fibroadipogenic progenitors to facilitate muscle regeneration. Cell 2013;153(2):376-88; PMID:23582327; http://dx.doi.org/10.1016/j.cell.2013.02.053
- Uezumi A, Ito T, Morikawa D, Shimizu N, Yoneda T, Segawa M, Yamaguchi M, Ogawa R, Matev MM, Miyagoe-Suzuki Y, et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J Cell Sci 2011; 124:3654-64; PMID:22045730; http://dx.doi.org/10.1242/jcs.086629
- Giordani L, Puri PL. Epigenetic control of skeletal muscle regeneration: integrating genetic determinants and environmental changes. FEBS J 2013; 280(17):4014-25; PMID:23745685; http://dx.doi.org/10.1111/febs.12383
- Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med 2006; 12(10):1147-50; PMID:16980968; http://dx.doi.org/10.1038/nm1479
- Colussi C, Mozzetta C, Gurtner A, Illi B, Straino S, Ragone G, Pescatori M, Zaccagnini G, Rosati G, Minetti G, et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci 2008; 105(49):19183-7; PMID:19047631; http://dx.doi.org/10.1073/pnas.0805514105
- Consalvi S, Mozzetta C, Bettica P, Germani M, Fiorentini F, Del Bene F, Rocchetti M, Leoni F, Monzani V, Mascagni P, et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol Med 2013; 19:79-87; PMID:23552722; http://dx.doi.org/10.2119/molmed.2013.00011
- Vianello S, Consolaro F, Bich C, Cancela JM, Roulot M, Lanchec E, Touboul D, Brunelle A, Israël M, Benoit E, et al. Low doses of arginine butyrate derivatives improve dystrophic phenotype and restore membrane integrity in DMD models. FASEB J 2014; 28(6):2603-19; PMID:24604079; http://dx.doi.org/10.1096/fj.13-244798
- Johnson NM, Farr GH, Maves L. The HDAC inhibitor TSA ameliorates a zebrafish model of duchenne muscular dystrophy. PLoS Curr 2013; PMID:24459606; http://dx.doi.org/10.1371/currents.md.8273cf41db10e2d15dd3ab827cb4b027
- Saccone V, Consalvi S, Giordani L, Mozzetta C, Barozzi I, Sandoná M, Ryan T, Rojas-Muñoz A, Madaro L, Fasanaro P, et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev 2014; 28(8):841-57; PMID:24682306; http://dx.doi.org/10.1101/gad.234468.113
- Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell 2009; 136(2):200-6; PMID:19167321; http://dx.doi.org/10.1016/j.cell.2009.01.009
- Puri PL, Mercola M. BAF60 A, B, and Cs of muscle determination and renewal. Genes Dev 2012; 26(24):2673-83; PMID:23222103; http://dx.doi.org/10.1101/gad.207415.112
- de la Serna IL, Ohkawa Y, Imbalzano AN. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet 2006; 7:461-73; PMID:16708073; http://dx.doi.org/10.1038/nrg1882
- Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat Genet 2004; 36:738-43; PMID:15208625; http://dx.doi.org/10.1038/ng1378
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWISNF complexes. Genes Dev 1996; 10(17):2117-30; PMID:8804307; http://dx.doi.org/10.1101/gad.10.17.2117
- Albini S, Coutinho P, Malecova B, Giordani L, Savchenko A, Forcales SV, Puri PL. Epigenetic reprogramming of human embryonic stem cells into skeletal muscle cells and generation of contractile myospheres. Cell Rep 2013; 3:661-70; PMID:23478022; http://dx.doi.org/10.1016/j.celrep.2013.02.012
- Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 2004; 432:107-112; PMID:15525990; http://dx.doi.org/10.1038/nature03071
- Forcales SV, Albini S, Giordani L, Malecova B, Cignolo L, Chernov A, Coutinho P, Saccone V, Consalvi S, Williams R, et al. Signal-dependent incorporation of MyoDBAF60c into Brg1-based SWISNF chromatin-remodelling complex. EMBO J 2011; 31:301-16; PMID:22068056; http://dx.doi.org/10.1038/emboj.2011.391
- Li S, Liu C, Li N, Hao T, Han T, Hill DE, Vidal M, Lin JD. Genome-wide coactivation analysis of PGC-1a identifies BAF60a as a regulator of hepatic lipid metabolism. Cell Metab 2008; 8:105-117; PMID:18680712; http://dx.doi.org/10.1016/j.cmet.2008.06.013
- Cacchiarelli D, Martone J, Girardi E, Cesana M, Incitti T, Morlando M, Nicoletti C, Santini T, Sthandier O, Barberi L, et al. MicroRNAs involved in molecular circuitries relevant for the duchenne muscular dystrophy pathogenesis are controlled by the dystrophinnNOS pathway. Cell Metab 2010; 12:341-51; PMID:20727829; http://dx.doi.org/10.1016/j.cmet.2010.07.008
- Sokol NS, Ambros V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev 2005; 19(19):2343-54; PMID:16166373; http://dx.doi.org/10.1101/gad.1356105
- Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 2006; 38(2):228-33; PMID:16380711; http://dx.doi.org/10.1038/ng1725
- Kim HK, Lee YS, Sivaprasad U, Malhotra A, Dutta A. Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol 2006; 174(5):677-87; PMID:16923828; http://dx.doi.org/10.1083/jcb.200603008
- Vignier N, Amor F, Fogel P, Duvallet A, Poupiot J, et al. Distinctive serum miRNA profile in mouse models of striated muscular pathologies. PLoS One 2013; 8(2):e55281; PMID:23418438; http://dx.doi.org/10.1371/journal.pone.0055281
- Roberts TC, Blomberg KEM, McClorey G, Andaloussi SE, Godfrey C, et al. Expression analysis in multiple muscle groups and serum reveals complexity in the MicroRNA transcriptome of the mdx mouse with implications for therapy. Mol Ther - Nucleic Acids 2012 1:e39; PMID:23344181
- Cacchiarelli D, Legnini I, Martone J, Cazzella V, D’Amico A, Bertini E, Bozzoni I. miRNAs as serum biomarkers for Duchenne muscular dystrophy. EMBO Mol Med 2011; 3(5):258-65; PMID:21425469; http://dx.doi.org/10.1002/emmm.201100133
- Eisenberg I, Alexander MS, Kunkel LM. miRNAS in normal and diseased skeletal muscle. J Cell Mol Med 2009; 13(1):2-11; PMID:19175696; http://dx.doi.org/10.1111/j.1582-4934.2008.00524.x
- Williams AH, Liu N, van Rooij E, Olson EN. MicroRNA control of muscle development and disease. Curr Opin Cell Biol 2009; 21(3):461-9; PMID:19278845; http://dx.doi.org/10.1016/j.ceb.2009.01.029
- Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, Cardani R, Perbellini R, Isaia E, Sale P, et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemia. FASEB J 2009; 23(10):3335-46; PMID:19528256; http://dx.doi.org/10.1096/fj.08-128579
- Goljanek-Whysall K, Mok GF, Fahad Alrefaei A, Kennerley N, Wheeler GN, Münsterberg A. MyomiR-dependent switching of BAF60 variant incorporation into Brg1 chromatin remodeling complexes during embryo myogenesis. Development 2014; 141(17):3378-87; PMID:25078649; http://dx.doi.org/10.1242/dev.108787
- Forterre A, Jalabert A, Chikh K, Pesenti S, Euthine V, Granjon A, Errazuriz E, Lefai E, Vidal H, Rome S. Myotube-derived exosomal miRNAs downregulate Sirtuin1 in myoblasts during muscle cell differentiation. Cell Cycle 2014; 13(1):78-89; PMID:24196440; http://dx.doi.org/10.4161/cc.26808
- He WA, Calore F, Londhe P, Canella A, Guttridge DC, Croce CM. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc Natl Acad Sci U S A 2014; 111(12):4525-9; PMID:24616506; http://dx.doi.org/10.1073/pnas.1402714111
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 2007; 55:201-15; PMID:17640523; http://dx.doi.org/10.1016/j.neuron.2007.06.019
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, Crabtree GR. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 2011; 476:228-31; PMID:21753754; http://dx.doi.org/10.1038/nature10323
- Mizuno H, Nakamura A, Aoki Y, Ito N, Kishi S, Yamamoto K, Sekiguchi M, Takeda S, Hashido K. Identification of muscle-specific microRNAs in serum of muscular dystrophy animal models: promising novel blood-based markers for muscular dystrophy. PLoS One 2011; 6(3):e18388; PMID:21479190; http://dx.doi.org/10.1371/journal.pone.0018388
- Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 2011 13:423-33; PMID:21423178; http://dx.doi.org/10.1038/ncb2210
- Roberts TC, Godfrey C, McClorey G, Vader P, Briggs D, et al. Extracellular microRNAs are dynamic non-vesicular biomarkers of muscle turnover. Nucl Acids Res 2013 41:9500-13; PMID:23945935; http://dx.doi.org/10.1093/nar/gkt724
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 2008; 105:10513-8; PMID:18663219; http://dx.doi.org/10.1073/pnas.0804549105
- Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta 2012; 1820:940-8; PMID:22503788; http://dx.doi.org/10.1016/j.bbagen.2012.03.017
- Borges FT, Melo SA, Ozdemir BC, Kato N, Revuelta I, Miller CA, Gattone VH, Lebleu VS, Kalluri R. TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol. 2013; 24:385-92; PMID:23274427; http://dx.doi.org/10.1681/ASN.2012101031
- Roberts TC, Coenen-Stass AM, Betts CA, Wood MJ. Detection and quantification of extracellular microRNAs in murine biofluids. Biol Proced Online 2014; 16(1):5; PMID:24629058; http://dx.doi.org/10.1186/1480-9222-16-5
- Roberts TC, Coenen-Stass AM, Wood MJ. Assessment of RT-qPCR normalization strategies for accurate quantification of extracellular microRNAs in murine serum. PLoS One 2014; 9(2):e89237; PMID:24586621; http://dx.doi.org/10.1371/journal.pone.0089237
- Goyenvalle A, Babbs A, Wright J, Wilkins V, Powell D, et al. Rescue of severely affected dystrophinutrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum Mol Genet 2012; 21:2559-71; PMID:22388933; http://dx.doi.org/10.1093/hmg/dds082