
Abstract
Chronic myelogenous leukemia (CML) requires the BCR/ABL tyrosine kinase for disease onset and maintenance. As a result, CML can be successfully treated with tyrosine kinase inhibitors (TKIs) such as imatinib. Most patients are maintained in a disease-suppressed state on daily TKI therapy for several years and in many cases this treatment prevents progression to the blast phase. If the TKI is discontinued, CML redevelops in 95% of patients as a result of persisting leukemia initiating cells (LICs). There are several hypotheses that describe the potential mechanism(s) responsible for LIC persistence in CML, but supporting evidence is limited. Furthermore, of the few patients who discontinue TKI therapy and are “cured” (i.e., in treatment-free remission), most have residual BCR/ABL-expressing cells in their hematopoietic tissues. There are also healthy individuals without a CML diagnosis who express the BCR/ABL mutation in a fraction of their hematopoietic cells. Finally, mice that express BCR/ABL from the Bcr locus as a knockin mutation do not develop CML. These mice have lower BCR/ABL levels than retroviral or transgenic models of BCR/ABL that do develop CML. Understanding why mice with BCR/ABL expressed from the Bcr locus and some people that express BCR/ABL are not afflicted with CML will provide insights into therapies to prevent or cure this disease.
Keywords:
Abbreviations
| AML | = | acute myelogenous leukemia |
| BTT | = | bone marrow transduction and transplantation |
| CML | = | chronic myelogenous leukemia |
| CMP | = | common myeloid progenitor |
| GMP | = | granulocyte monocyte progenitor |
| HSC | = | hematopoietic stem cell |
| LIC | = | leukemia initiating cell |
| MPN | = | myeloproliferative neoplasm |
| qRT-PCR | = | real-time quantitative reverse-transcription polymerase chain reaction |
| SCT | = | stem cell transplant |
| TKI | = | tyrosine kinase inhibitor |
Introduction
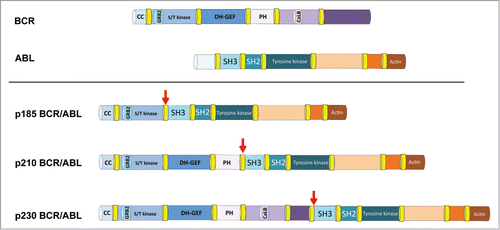
Janet Rowley's original observation that specific chromosomal abnormalities are associated with specific types of leukemia was pivotal for our current understanding of leukemia biology.Citation1 The first translocation that she identified was the t(9;22) Philadelphia chromosome, which was later discovered to express the BCR/ABL oncogene, in patients with chronic myeloid leukemia (CML).Citation2,3 Breakpoint cluster region (BCR) is a serine/threonine kinase with several interaction domains for proteins such as actin, lipids, and GTP. ABL is a tyrosine kinase that shuttles between the nucleus and cytoplasm and is expressed in many tissues. Under normal circumstances, ABL transmits signals from ligand-stimulated plasma membrane-bound growth factor receptors to influence cytoskeletal changes and other cellular phenotypes.Citation4 Specific breakpoints in the chromosomal translocation lead to distinct forms of the BCR/ABL protein (p185 BCR/ABL, p210 BCR/ABL, and p230 BCR/ABL), all of which have constitutive ABL kinase activity (). For unknown reasons, these are generally found in different types of leukemia; the p210 form is associated with CML, whereas p230 and p185 are associated with neutrophilic leukemia and acute lymphoblastic leukemia, respectively.Citation5
Figure 1. Schematic of key domains in the BCR and ABL proteins. BCR is a 143-kDa protein with actin and self-binding coiled-coil (CC), kinase, GRB2 binding, guanine nucleotide exchange (DH-GEF), lipid binding plekstrin homology (PH), and calcium binding (CalB) domains. ABL is a 123-kDa tyrosine kinase with 2 Src homology domains (SH3 and SH2) that regulate the kinase as well as an actin-binding domain. The p185 BCR/ABL fusion protein is found in acute lymphoblastic leukemia, the p210 BCR/ABL fusion protein is found in chronic myelogenous leukemia, and the p230 BCR/ABL fusion protein is found in chronic neutrophilic leukemia. The red arrows indicate the junction between the BCR and ABL sequences.
There are a myriad of other chromosomal translocations that lead to oncogene overexpression or to the formation of leukemogenic fusion proteins like BCR/ABL. One example of overexpression is the t(14;18) translocation that results in BCL2 overexpression and is associated with follicular lymphoma. In this case, BCL2 is translocated to a site adjacent to the immunoglobulin promoter. Similar to BCR/ABL,Citation6-10 this oncogene has been found in the blood of healthy leukemia/lymphoma-free individuals.Citation11-13 Recently, the presence of the t(14;18) translocation in the blood was shown to be a putative marker for future development of follicular lymphoma.Citation14 Because follicular lymphoma probably develops over decades, the possibility that some of these cases were asymptomatic undiagnosed patients cannot be ruled out. However, because some of the patients that were ultimately diagnosed with follicular lymphoma did not receive the diagnosis until 20 years later, the authors concluded that it was unlikely that they were affected yet undiagnosed at the time of the original blood sample. There are numerous examples of the formation of oncogenic fusion genes from chromosomal translocations in acute leukemias, including the t(8;21) translocation that results in the fusion of AML1 with ETO to form the AML1/ETO oncogenic transcription.Citation15,16
Although not all humans with BCR/ABL transcripts identified in their hematopoietic system have disease,Citation6-10 all patients with classic CML have the BCR/ABL mutation in their hematopoietic system. The progression of BCR/ABL-associated CML is commonly divided into 3 disease stages designated chronic, accelerated, and blast crisis phases. A “pre-disease” or so-called “previvor” phase (defined as a person with a cancer predisposing mutation but no cancer diagnosis; in this case the mutation is not germline but somatic) has also been proposed to exist but has not been studied in depth.Citation6-10 Similar to the presence of the t(14;18) translocation in healthy individuals, a proportion of whom were diagnosed years later with follicular lymphoma,Citation14 it has been difficult to follow disease-free patients with the BCR/ABL fusion in their bone marrow because of technical challenges related to the accuracy of the blood test.
The prevalence of CML-associated p210 BCR/ABL transcripts in healthy individuals varies with age and reported values range from 10% to 30% of tested adults. Bose et al. found that 27% (n = 15) of healthy individuals had p210 BCR/ABL transcripts in their blood.Citation9 Biernaux et al. found a 30% (n = 73) positivity rate of BCR/ABL transcripts in healthy subjects aged between 20 and 80 years whereas only 2% (n = 44) of the younger subjects (0 to 13 years) expressed BCR/ABL transcripts. These age-related data were confirmed recently by Ismail et al., who noted that the p210 BCR/ABL transcript was detected in 10% of peripheral blood samples from 189 healthy volunteers in their cohort. In this study, adults were 6 times more likely than children to have BCR/ABL positivity.Citation10 The authors proposed that this age difference was due to accumulation of DNA damage with increasing age.
None of the studies reporting BCR/ABL positivity in healthy adults investigated which cell types were expressing the fusion gene. However, Bayraktar and Goodman (2010) recently reported the case of a healthy 39-year-old man who had normal blood counts and sustained positivity for BCR/ABL in his bone marrow over the course of 2 years as measured by fluorescent in situ hybridization (FISH) and real-time quantitative reverse-transcription polymerase chain reaction (qRT-PCR).Citation7 This suggests that the BCR/ABL mutation is present in a hematopoietic stem cell. Whether this patient is at risk for developing CML is unknown, and whether such patients should receive imatinib therapy is subject to debate.
Current treatments for chronic phase CML are either maintenance on one of the tyrosine kinase inhibitors (TKI) such as imatinib, nilotinib, or desatinib (for a comprehensive review of such drugs see Khan and BixbyCitation17) or allogeneic stem cell transplantation (SCT) if the patient is unable to tolerate TKI therapy. Although CML can be cured with SCT, finding acceptable donors can be difficult and the high rate of SCT-related complications warrants the use of TKIs as the current standard of care for patients with chronic phase CML. The use of TKIs has revolutionized CML patient care over the past 10 years. Imatinib, the first TKI widely used to treat the disease, has led to a reduction in the 5-year CML mortality rate to that of healthy age-matched individuals.Citation18 However, only 5% of patients treated with TKIs are ultimately cured;Citation19-21 in the majority of patients the disease returns upon TKI discontinuation.Citation22 These data suggest that although TKIs eliminate the vast majority of CML cells and inhibit disease progression, they do not eliminate the leukemia initiating cells (LICs) that persist and maintain the disease state ().
Figure 2. Schematic of stem and progenitor ratios in normal hematopoiesis and hematopoiesis of chronic myelogenous leukemia (CML) cells in the presence or absence of tyrosine kinase inhibitors (TKIs). Cells with all shades of blue are normal; the lighter the shade, the more differentiated the cell. Burgundy (leukemia initiating cells [LICs]), orange (progenitors, e.g., common myeloid progenitora [CMPs], granulocyte monocyte progenitors [GMPs]), and beige cells (differentiated cells, e.g., neutrophils and monocytes) carry the BCR/ABL oncogene.
![Figure 2. Schematic of stem and progenitor ratios in normal hematopoiesis and hematopoiesis of chronic myelogenous leukemia (CML) cells in the presence or absence of tyrosine kinase inhibitors (TKIs). Cells with all shades of blue are normal; the lighter the shade, the more differentiated the cell. Burgundy (leukemia initiating cells [LICs]), orange (progenitors, e.g., common myeloid progenitora [CMPs], granulocyte monocyte progenitors [GMPs]), and beige cells (differentiated cells, e.g., neutrophils and monocytes) carry the BCR/ABL oncogene.](/cms/asset/4c504878-b0f5-4e1a-9d84-b4b3ad6a1b1d/kmco_a_963450_f0002_oc.gif)
Currently, very little is known about the molecular and cellular characteristics of the LICs that are refractory to TKI therapy. This gap in our understanding presents a barrier to developing a cure. One well-studied area of disease persistence is resistance to TKIs as a result of point mutations in the ABL kinase domain (reviewed elsewhereCitation23-25). However, the persistence of BCR–ABL-“wild type” LICs in patients taking TKIs indicates that in the majority of cases inhibition of the kinase activity is not sufficient to eradicate the LICs. There are several proposed reasons for LIC persistence and it is possible that no single mechanism is involved, but instead a combination of 2 or more. Possible mechanisms include high levels of TKI drug-efflux pumps in the LICs, non-addiction of LICs to the kinase as a result of a quiescent cellular state, either a lack of BCR/ABL expression or a level of expression that is too high for sufficient inhibition by TKIs, or the presence of additional cancer mutations that are not targeted by imatinib. Defective immune surveillance whereby the LIC avoids immune recognition could also contribute to one or more of these molecular or cellular mechanisms of persistence.
Here, we review current knowledge about the role of BCR/ABL in CML pathophysiology. In particular, we focus on the role of BCR/ABL in the earliest phases of the disease by discussing data supporting mechanisms that prevent or promote neoplastic transformation of BCR/ABL mutant cells and that allow them to persist in the presence of TKIs.
BCR/ABL is Necessary for Initiation, Maintenance, and Progression of CML
BCR/ABL is widely believed to be necessary for CML initiation and maintenance because it is a constitutively active tyrosine kinase that feeds into several pro-growth and survival signaling pathways. There are 3 main sets of data that support this idea. First, in structure-function studies using standard cell culture and retroviral models of CML, kinase-dead mutant forms of BCR/ABL are incapable of initiating CML.Citation26 Second, the ability of imatinib and other TKIs to suppress disease shows that BCR/ABL is required for disease maintenance.Citation18 Finally, most CML patients have BCR/ABL in their blood and bone marrow.
Furthermore, because patients who are maintained on TKIs do not usually progress to acute leukemia, the tyrosine kinase activity of BCR/ABL is also thought to be required for CML progression. However, this could reflect disease progression that results from continuous proliferation of BCR/ABL-expressing cells. Unfortunately, some patients maintained on TKIs do progress to the blastic phase, thus treatment with TKIs is not a panacea. The fact that some patients progress while on TKIs suggests that, at least in some cases, progression involves a BCR/ABL-independent mechanism. Moreover, BCR/ABL levels have been reported to increase upon progression from the chronic phase of the disease to the blast crisis phase.Citation27,28 A requirement for the BCR/ABL kinase for progression is illustrated by the fact that TKI therapy not only induces molecular remissions in patients, but frequently halts the progression of chronic CML into accelerated phases.Citation29,30 It has been proposed that TKIs prevent disease progression by specifically eliminating cells with the highest levels of BCR/ABL. However, the fact that the acute phases of CML are not effectively treated with TKIs suggests that either high BCR/ABL levels are not necessary for the maintenance of these advanced stages, inhibition by TKIs is insufficient because of the high levels of BCR/ABL that accompany these phases, or simply that progression is independent of BCR/ABL.
Many model systems, including cell culture, support the hypothesis that BCR/ABL is an oncogene that is necessary (and sufficient) for initiation and maintenance of CML. These models are important tools for understanding how BCR/ABL promotes hematopoietic neoplasia and may help identify new drugs for therapy. However, these models select for the proleukemic properties of BCR/ABL and would not be expected to assay for any opposing tumor suppressor activities. Cultured cells are a commonly used model to assay for transformation by BCR/ABL. Expression of BCR/ABL transforms IL3-dependent hematopoietic cell lines such as Ba/F3 or 32D cells and early primary human hematopoietic progenitors to IL-3 independence, whereas kinase-dead mutants of BCR/ABL do not.Citation31 It is hoped that the properties of human hematopoietic progenitor cells expressing kinase active BCR/ABL mimic the properties of primary CML cells in patients. However, the formation of leukemia is complex and likely involves not only the effect of BCR/ABL expression within the hematopoietic cells, but also the interplay between BCR/ABL expressing cells and their environmental niche, which contains several stromal cell types.
Animal models of CML are more representative of human disease than cultured cells. These in vivo models are necessary for a comprehensive understanding of CML biology and to identify new therapeutic strategies. The most commonly used animal models are retroviralCitation32-36 and non-targetedCitation36-42 transgenics. Expression of BCR/ABL in mouse bone marrow by retroviral transduction and bone-marrow transplantation (BTT) consistently induces a myeloproliferative neoplasm (MPN) that resembles CML.Citation32,34 The biggest strength of the BTT model is that structure-function studies of mutant forms of BCR/ABL can be efficiently performed to define which domains of BCR/ABL are necessary for leukemogenesis. For example, mice that express kinase-dead BCR/ABL (point mutation at the ATP-binding site of ABL) do not develop leukemia.
In addition to the ABL kinase, certain domains in the BCR portion of BCR/ABL have been found to be important for disease initiation and maintenance. One example is the amino-terminal coiled-coil (CC) oligomerization domain of BCR. This domain is required for activation of the ABL kinase and for the association of BCR–ABL with actin fibers ().Citation43,44 A mutant form of BCR/ABL that lacks the BCR-CC domain (ΔCC-BCR/ABL) is kinase dead and does not induce MPN.Citation45,46 However, reactivation of the kinase activity of ABL by mutating its SH3 kinase inhibitory domain (through deletion or a point mutation) rescues the defects of the ΔCC-BCR/ABL and MPN can then be induced.Citation47 These results demonstrate that the BCR coiled-coil domain is essential for the induction of MPN by BCR/ABL in mice through its ability to activate the kinase activity of ABL. The GRB2-binding motif in the BCR portion of BCR/ABL GRB2 binds the scaffold adapter GRB2-associated binding protein2 (GAB2) and a guanine-nucleotide RAS exchanger. Formation of this complex can activate RAS and promote the recruitment of other signaling molecules ( and reviewed in detail elsewhereCitation48-51).
Although a large number of signaling proteins have been found to interact with BCR/ABL through its many motifs (reviewed recently by Cilloni and SaglioCitation52), which of these are required downstream targets for leukemogenesis has been difficult to assess. One way to investigate the role of the different signaling proteins/pathways in the pathophysiology of CML is to test the ability of BCR/ABL to induce disease in murine bone marrow deficient for specific signaling proteins. For example, the demonstration that BCR/ABL still induced a CML-like MPN in Stat5a/b double knockout mice suggests that the STAT5 proteins are not necessary for BCR/ABL-induced disease.Citation53 The caveat with such conclusions is that retroviral expression of BCR/ABL has certain limitations (see below). Since the levels or BCR/ABL are probably much higher than in the “normal” CML situation due to the presence of multiple copies of the oncogene and expression from a very active retroviral promoter, adaptation of the cells to STAT5 deficiency to generate a CML-like MPN in the absence of 1 or 2 of the many signals that emerge from BCR/ABL is perhaps not unexpected but may not reflect clinical reality.
BCR/ABL Alone is Not Sufficient for CML Induction
It is likely that additional mutations or epigenetic changes in the genome are required together with the BCR/ABL mutation for the development of CML. Indeed, most neoplasms, solid or liquid, are thought to require more than one oncogenic mutation for disease initiation. Although CML has in general been thought to be an exception to this rule, individuals who have BCR/ABL in their bone marrow but do not have disease are reported to be common,Citation6-10 suggesting that multiple abnormalities are necessary for CML development. If this is the case, selected expansion of only those cells with the second mutation would be expected and may occur in a small fraction of BCR/ABL-positive but disease-free individuals. Because most of these BCR/ABL-positive healthy individuals have not been systematically observed, whether they are at risk for disease is not clear. Recently, a large cohort of healthy individuals with the t(14;18) BCL2 translocation were found to develop follicular lymphoma at a low frequency; however, it is not known whether these lymphomas carried new second or third mutations.Citation14 Another interesting point to consider is that the presence of additional mutations that are required for BCR/ABL to induce CML could contribute to LIC persistence because such mutations would probably not be directly targeted by ABL-specific TKIs. Hence, although the expression of BCR/ABL is necessary for full disease, as evidenced by TKI sensitivity, maintenance of the LICs might not require BCR/ABL but instead be due to an additional mutation.
Recently, we developed a conditional knockin allele of BCR/ABL and found that no CML-like MPN developed in mice expressing BCR/ABL in hematopoietic stem cells (HSCs) and their progeny.Citation54 There are many other examples of situations where more realistic models of oncogene-induced diseases, such as the AML-associated Flt3-itd knockinCitation55,56 or a sporadic T-cell acute lymphoblastic leukemia (T-ALL)-associated MYC activation allele,Citation57 do not develop disease. These situations contrast with models in which massive oncogene expression in large numbers of hematopoietic cells readily induces leukemias (AML or T-ALL). Our BCR/ABL knockin allele expressed the same human BCR/ABL cDNA as that used in standard retroviral models that do induce a CML-like MPN in mice.Citation13,32,34 However, the human BCR/ABL cDNA was knocked into the endogenous mouse Bcr locus so that it could be conditionally expressed with different tissue-specific Cre transgenes under the added control of the native Bcr regulatory elements. This might explain the limited phenotype of these mice compared to that observed in standard retroviral and transgenic models. All of these models come with several caveats; for example, the standard retroviral and transgenic models express multiple copies of the BCR/ABL oncogene from artificial promoters and the constructs induce integration site mutations,Citation32,58 and there is only one knockin model available for study. Although retroviral or transgenic overexpression is sufficient to cause MPN, the limitations of non-physiologic expression levels, random insertion-site mutations, selection during culture (in the retroviral model), and non-specificity of expression timing and locale could artificially select for disease development. Levels of BCR/ABL obtained from the endogenous locus in the knockin model are comparable to levels of mouse Bcr expressed from the other allele in the mice but are clearly not sufficient to cause CML during the lifetime of the mouse.Citation54 In some human cases the latency of the disease may be longer than the lifespan, possibly explaining why the BCR/ABL mutation is sometimes observed in humans who are free from disease.Citation6-10
It is safe to say that, by itself, expression of BCR/ABL in the knockin model is not sufficient for immediate leukemogenesis even though we observed expression of the constitutively active BCR/ABL tyrosine kinase in HSCs and in other hematopoietic progenitors. Instead, cooperating mutations, epigenetic changes, or other strategies appear to be required to overcome the barriers to BCR/ABL-induced transformation in both mice and humans. In support of the idea that additional genetic changes are necessary, we found that BCR/ABL and AML1/ETO compound mutant knockin mice do develop MPN.Citation54 Furthermore, although human CML occurs at any age, the frequency increases with age and radiation exposure increases the risk of CML after a long latency. Finally, mathematical models predict that 2 or more mutations in the HSC may be needed for CML to develop.Citation59,60 It is also possible that the number of leukemic cells that are needed for phenotypic disease is so high that the development of disease from a single transformed HSC requires a significant length of time. In this case, healthy mice and humans with the oncogene might progress to disease only if they live long enough.
Despite the caveats about the various disease models mentioned above, there are data suggesting that different phenotypes in the different models may be attributable to distinct levels of BCR/ABL. Levels of BCR/ABL in the hematopoietic tissues (blood or bone marrow) of patients previously diagnosed with CML are reported to correlate inversely with the rare chance of being “cured” using TKIs alone.Citation20,21 These reports have been challenged by others who argue that saying that the patients with low BCR/ABL levels are those who can stop treatment is circular, as only treated patients with low oncogene levels are offered the opportunity to withdraw TKI therapy. Furthermore, there is debate about what defines a low-level positive qRT-PCR result for BCR/ABL. Variations in the assay between different laboratories make it difficult to standardize the definition of undetectable minimal residual disease. Moreover, it is also unclear what a low-level positive finding for BCR/ABL actually represents: is it fewer transcripts in many cells or many transcripts from few cells? To circumvent this issue, detection of BCR/ABL DNA has been used to provide evidence of minimal residual disease. Consistent with the need for more than the BCR/ABL mutation for disease to develop, the presence of BCR/ABL DNA in patients in a treatment-free remission does not predict relapse risk.
It has been reported that 40% of the 10% of patients (4% overall) who have a sustained deep molecular response (very low levels of residual BCR/ABL expression) while on TKIs can stop taking the TKI without relapsing. For example, in the STIMCitation20 and TWISTERCitation21 clinical studies of TKI discontinuation in patients with a deep molecular remission, BCR/ABL DNA was present in all of the patients in treatment-free remission (or cure) for several years, indicating that eradication of BCR/ABL-containing cells was not responsible for the continued remission. It is possible that cells with BCR/ABL expression were not the source of the original CML clone. Other possibilities include the presence of residual pre-neoplastic single-mutant BCR/ABL-expressing cells, or an added layer of complex immunologic suppression of CML clones.
The presence of the BCR/ABL mutation in the hematopoietic system for several years (e.g., the STIM and TWISTER studiesCitation20,21) supports the hypothesis that normal stem cells with the BCR/ABL mutation survive, but on balance do not perpetuate the disease. CML patients who are in treatment-free remission after imatinib withdrawal yet express the BCR/ABL oncogene in their hematopoietic systemCitation20,21 are phenotypically similar to healthy BCR/ABL-carrying “previvors”.Citation6-10 Data from BCR/ABL-expressing cell lines indicate that very low levels of BCR/ABL do not transform cells to factor-independent growth in hematopoietic cell lines.Citation27 Uncovering the reasons why expression of BCR/ABL from the Bcr locus, at least at low levels, is not sufficient for development of CML will lead to ideas for new strategies to cure CML.
Uncovering Molecular Barriers to Transformation by BCR/ABL
It is believed that the BCR/ABL tyrosine kinase is not required for LIC persistence in the presence of TKIs, i.e., that LICs are not “addicted” to BCR/ABL.Citation22 LICs that persist in TKI-treated CML are poorly understood cells that probably originate from the HSC population. Because LICs, like HSCs, self-renew to later produce several proliferating and non-proliferating progenitors they are at any one time a minor fraction of the neoplastic population, making them difficult to study (). As mentioned earlier, the mechanism(s) underlying the refractoriness of LICs to imatinib treatment are a key clinical mystery. There is currently a large gap in our knowledge about the environmental, genetic, or cellular mechanisms that contribute to LIC persistence that, when filled by analysis of human samples and the most realistic models, will contribute to curing CML.
Understanding the mechanisms of LIC persistence will provide key information about not only the molecular changes that enhance BCR/ABL-mediated transformation, but also those that block it. Ultimately, such understanding will lead novel therapies for CML. Recent clinical data from the STIMCitation20 and TWISTERCitation21 studies, as well as those from healthy individuals with sustained BCR/ABL in their bone marrow,Citation7-9 suggest there are molecular barriers to transformation by BCR/ABL.
First, the inherent properties of BCR/ABL may present a molecular barrier to transformation. BCR/ABL is in fact a relatively weak oncogene compared to others. For example, expression of the AML-associated fusion oncogene MOZ–TIF2 in common myeloid progenitors (CMPs) () and granulocyte monocyte progenitors (GMPs) induces leukemia in the BTT model.Citation61 In contrast, BCR/ABL fails to induce disease when it is expressed in HSC progeny such as CMPs and GMPs using the same system. However, this difference between BCR/ABL and other, stronger, oncogenes may be due to the induction of simultaneous and opposing transformative and tumor suppressive signals by BCR/ABL. Hence, a greater ability to transform certain cell types in vivo may reflect either stronger oncogenic properties or weaker barriers to transformation in those cells.
Because the BCR/ABL knockin mice did not develop disease, we posit that physiologic BCR/ABL expression may in fact be insufficient for development of CML because the balance of tumorigenic and tumor suppressive pathways favors tumor suppression. It is possible that adaptive changes in gene expression or secondary mutations arise when BCR/ABL is expressed from the endogenous locus in mice to prevent the development of disease. A corollary is that the MPN induced in retroviral or transgenic models may be due, at least in part, to insertion mutations or very high levels of BCR/ABL expression that overcome the BCR/ABL-induced tumor suppressive pathways. In support of the idea that tumor suppressive forces put brakes on the promotion of disease induction or progression by BCR/ABL, Sherr and colleagues discovered that a deficiency of Arf in mice leads to induction of a more aggressive acute lymphoblastic leukemia following retroviral expression of either p210 or p185 BCR/ABL.Citation62,63 Further in-depth non-biased analyses of transformation-protective changes in gene expression or mutations induced by BCR/ABL in the disease-free BCR/ABL knockin mice and in HSCs from humans who have the BCR/ABL mutation but do not have disease will provide clues to how the leukemogenic tendencies of BCR/ABL are suppressed.
Future Steps Toward a Cure
A deeper understanding of the role of BCR/ABL in the pathophysiology of CML is a necessary step to overcome the persistence of LICs even after years of continuous TKI therapy.
There is a need for more realistic in vivo models to provide a better understanding of why current therapies fail to eradicate LICs. The use of a murine stem-cell retroviral vector to express BCR/ABL in hematopoietic stem/progenitor cells has resulted in improved model systems in which MPN similar to CML develops consistently.Citation64 The disease induced in this model includes increased numbers of peripheral-blood myeloid cells, hepatosplenomegaly with extramedullary hematopoiesis, and granulocyte infiltration elsewhere. In addition, the retrovirally induced disease is oligoclonal and transplantable into secondary recipient mice. The ability of BCR/ABL to induce a CML-like MPN in mice more efficiently and reproducibly using a murine stem-cell vector is probably the result of successful expression of BCR/ABL in the cell type that can evolve into a MPN. Nonetheless, this model expresses massive levels of the oncogene, can result in random integration site mutations, and uses cell culture prior to transplantation, which could lead to clonal selection of resultant mutant cells in an artificial system.
Generation of knockin mouse models of CML has been problematic and is an area for continued future work. Early germline knockin mice produced through expression of BCR/ABL from the Bcr promoter were reported to exhibit embryonic lethality that was presumed at the time to be due to the toxicity of the constitutively active tyrosine kinase during embryonic development.Citation65 However, because conditional expression of BCR/ABL in embryonic and adult HSCs (using the Vav-Cre transgene to drive expression) or germ line knockin of BCR/ABL did not display embryonic lethality in our studies, these earlier models probably had additional lethal mutations that were not detected at the time.Citation54 However, because this new BCR/ABL knockin model expresses an intronless cDNA and both alleles of Abl are intact it does not perfectly mimic the human mutation.
Prospective serial screening of a large series of healthy individuals to determine whether the presence of the BCR/ABL oncogene in the blood predicts future development of CML would be of interest. This notion is supported by the fact that BCR/ABL knockin miceCitation54 do not develop disease, together with the existence of healthy CML-free humans with BCR/ABL in their hematopoietic system.Citation6-10 However, because CML occurs at a frequency of 1 to 2 cases per 100,000 per year, a very large cohort would be needed coupled with analysis of an equal number of individuals without BCR/ABL transcripts or DNA in their peripheral blood or bone marrow. This screening would be similar to recent studies showing that the t(14;18) translocation is found in healthy individuals and could be considered the best-known predictive marker for the development of follicular lymphoma.Citation14 Together with data on chronic lymphocytic leukemia/monoclonal B-cell lymphocytosis and multiple myeloma/monoclonal gammopathy of undetermined significance, these findings on follicular lymphoma suggest that the t(14;18) translocation is another premalignant state that has the potential to progress into a full-blown hematopoietic neoplasia.
The patient material used in the study of t(14;18) as a putative marker of follicular lymphoma may be useful for CML studies. The t(14;18) study used a large European cohort of more than 500,000 healthy individuals who were followed for cancer for more than 15 years. The researchers identified 100 patients who were diagnosed with follicular lymphoma during follow up and found that they had the t(14;18) translocation in their original blood samples. We propose that these same DNA samples could be used to search for individuals in this cohort with BCR/ABL mutations who have, or have not, gone on to develop CML. As the incidence of CML is estimated to be half that of follicular lymphoma we predict that there might be 50 patients who were diagnosed with CML over the 15-year observation span of this cohort. If this study group contained a fraction of healthy individuals harboring the t(9:22) translocation that ultimately went on to develop CML years later, BCR/ABL positivity could be considered a premalignant state that has the potential to progress to a hematopoietic neoplasia.
Finally, the expression patterns of stem cell progenitors in healthy individuals with BCR/ABL in their bone marrow compared with patients with chronic phase CML have yet to be studied. Additional clinical data and more realistic mouse models will provide new insights into the in vivo mechanism of tyrosine kinase-associated leukemogenesis, including the additional mutations necessary for disease development. Improvements in our ability to study the basic science of tyrosine kinase transformation in vivo will improve our ability to positively influence patient outcome.
Combining current basic science and clinical knowledge to understand why some patients with the BCR/ABL fusion do not have disease and why BCR/ABL knockin mice are disease-free will provide groundwork for the next steps toward new therapies. To bridge the knowledge gaps, it will be necessary to identify cooperating mutations in mice and/or sequence CML cells from patients with chronic-phase disease. Pressing questions include whether there are BCR/ABL cooperators that are required for CML to develop, whether individuals with BCR/ABL without disease later progress to CML, and whether the treatment-free “cured” state is protective against further disease. Clearly, there is more to learn about the chronic phase of CML, which appears to be caused by more factors than simply the presence of a single BCR/ABL mutation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to members of the Ross lab for continuous and critical review of the data. TSR holds the Jeanne Ann Plitt Professorship in Breast Cancer Research and the H. Ben and Isabelle T. Decherd Chair in Internal Medicine at the University of Texas Southwestern Medical Center.
Funding
TSR was a Leukemia and Lymphoma Society Scholar when the experiments in this manuscript were initiated. This work was also funded in part by the Burroughs Wellcome Fund (Clinical Scientist Award; TSR) and the National Cancer Institute.
References
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243:290-3; PMID:4126434; http://dx.doi.org/10.1038/243290a0
- Heisterkamp N, Groffen J. Molecular insights into the Philadelphia translocation. Hematol Pathol 1991; 5:1-10; PMID:2050600
- Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984; 36:93-9; PMID:6319012; http://dx.doi.org/10.1016/0092-8674(84)90077-1
- Hernandez SE, Krishnaswami M, Miller AL, Koleske AJ. How do Abl family kinases regulate cell shape and movement? Trends Cell Biol 2004; 14:36-44; PMID:14729179; http://dx.doi.org/10.1016/j.tcb.2003.11.003
- Pasternak G, Hochhaus A, Schultheis B, Hehlmann R. Chronic myelogenous leukemia: molecular and cellular aspects. J Cancer Res Clin Oncol 1998; 124:643-60; PMID:9879825; http://dx.doi.org/10.1007/s004320050228
- Boquett JA, Alves JR, de Oliveira CE. Analysis of BCRABL transcripts in healthy individuals. Genet Mol Res 2013; 12:4967-71; PMID:24301757; http://dx.doi.org/10.4238/2013.October.24.8
- Bayraktar S, Goodman M. Detection of BCR-ABL positive cells in an asymptomatic patient: a case report and literature review. Case Report Med 2010; 2010:939706
- Biernaux C, Loos M, Sels A, Huez G, Stryckmans P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995; 86:3118-22; PMID:7579406
- Bose S, Deininger M, Gora-Tybor J, Goldman JM, Melo JV. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: biologic significance and implications for the assessment of minimal residual disease. Blood 1998; 92:3362-7; PMID:9787174
- Ismail SI, Naffa RG, Yousef AM, Ghanim MT. Incidence of bcrabl fusion transcripts in healthy individuals. Mol Med Rep 2014; 9:1271-6; PMID:24535287
- Limpens J, Stad R, Vos C, de Vlaam C, de Jong D, van Ommen GJ, Schuuring E, Kluin PM. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood 1995; 85:2528-36; PMID:7727781
- McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 1989; 57:79-88; PMID:2649247; http://dx.doi.org/10.1016/0092-8674(89)90174-8
- Schuler F, Dolken L, Hirt C, Kiefer T, Berg T, Fusch G, Weitmann K, Hoffmann W, Fusch C, Janz S, et al. Prevalence and frequency of circulating t(14;18)-MBR translocation carrying cells in healthy individuals. Int J Cancer 2009; 124:958-63; PMID:19030176; http://dx.doi.org/10.1002/ijc.23958
- Roulland S, Kelly RS, Morgado E, Sungalee S, Solal-Celigny P, Colombat P, Jouve N, Palli D, Pala V, Tumino R, et al. t(14;18) Translocation: a predictive blood biomarker for follicular lymphoma. J Clin Oncol 2014; 32:1347-55; PMID:24687831
- Nucifora G, Birn DJ, Erickson P, Gao J, LeBeau MM, Drabkin HA, Rowley JD. Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood 1993; 81:883-8; PMID:8427996
- Erickson P, Gao J, Chang KS, Look T, Whisenant E, Raimondi S, Lasher R, Trujillo J, Rowley J, Drabkin H. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1ETO, with similarity to Drosophila segmentation gene, runt. Blood 1992; 80:1825-31; PMID:1391946
- Khan AM, Bixby DL. BCR-ABL inhibitors: Updates in the management of patients with chronic-phase chronic myeloid leukemia. Hematology 2013; 19; 249-58; PMID:24143950; http://dx.doi.org/10.1179/1607845413Y.0000000119
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355:2408-17; PMID:17151364; http://dx.doi.org/10.1056/NEJMoa062867
- Kavalerchik E, Goff D, Jamieson CH. Chronic myeloid leukemia stem cells. J Clin Oncol 2008; 26:2911-5; PMID:18539972; http://dx.doi.org/10.1200/JCO.2008.17.5745
- Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, Legros L, Charbonnier A, Guerci A, Varet B, et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 11:1029-35; PMID:20965785; http://dx.doi.org/10.1016/S1470-2045(10)70233-3
- Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, Dang P, Goyne JM, Slader C, Filshie RJ, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood 2013; 122:515-22; PMID:23704092; http://dx.doi.org/10.1182/blood-2013-02-483750
- Savona M, Talpaz M. Getting to the stem of chronic myeloid leukaemia. Nat Rev Cancer 2008; 8:341-50; PMID:18385684; http://dx.doi.org/10.1038/nrc2368
- Volpe G, Panuzzo C, Ulisciani S, Cilloni D. Imatinib resistance in CML. Cancer Lett 2009; 274:1-9; PMID:18653275; http://dx.doi.org/10.1016/j.canlet.2008.06.003
- Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene 2003; 22:7389-95; PMID:14576846; http://dx.doi.org/10.1038/sj.onc.1206942
- Kantarjian HM, Talpaz M, Giles F, O’Brien S, Cortes J. New insights into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann Intern Med 2006; 145:913-23; PMID:17179059; http://dx.doi.org/10.7326/0003-4819-145-12-200612190-00008
- Zhang X, Ren R. Bcr-Abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood 1998; 92:3829-40; PMID:9808576
- Barnes DJ, Schultheis B, Adedeji S, Melo JV. Dose-dependent effects of Bcr-Abl in cell line models of different stages of chronic myeloid leukemia. Oncogene 2005; 24:6432-40; PMID:16007188
- Gaiger A, Henn T, Horth E, Geissler K, Mitterbauer G, Maier-Dobersberger T, Greinix H, Mannhalter C, Haas OA, Lechner K, et al. Increase of bcr-abl chimeric mRNA expression in tumor cells of patients with chronic myeloid leukemia precedes disease progression. Blood 1995; 86:2371-8; PMID:7662984
- Hochhaus A, O’Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L, Goldman JM, Muller MC, Radich JP, Rudoltz M, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009; 23:1054-61; PMID:19282833; http://dx.doi.org/10.1038/leu.2009.38
- Kumari A, Brendel C, Hochhaus A, Neubauer A, Burchert A. Low BCR-ABL expression levels in hematopoietic precursor cells enable persistence of chronic myeloid leukemia under imatinib. Blood 2012; 119:530-9; PMID:22101898; http://dx.doi.org/10.1182/blood-2010-08-303495
- Ramaraj P, Singh H, Niu N, Chu S, Holtz M, Yee JK, Bhatia R. Effect of mutational inactivation of tyrosine kinase activity on BCRABL-induced abnormalities in cell growth and adhesion in human hematopoietic progenitors. Cancer Res 2004; 64:5322-31; PMID:15289338; http://dx.doi.org/10.1158/0008-5472.CAN-03-3656
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcrabl gene of the Philadelphia chromosome. Science 1990; 247:824-30; PMID:2406902; http://dx.doi.org/10.1126/science.2406902
- Elefanty AG, Hariharan IK, Cory S. bcr-abl, the hallmark of chronic myeloid leukaemia in man, induces multiple haemopoietic neoplasms in mice. EMBO J 1990; 9:1069-78; PMID:1691092
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcrabl-transduced bone marrow. Blood 1998; 92:3780-92; PMID:9808572
- Wong S, Witte ON. Modeling Philadelphia chromosome positive leukemias. Oncogene 2001; 20:5644-59; PMID:11607816; http://dx.doi.org/10.1038/sj.onc.1204638
- Kelliher MA, McLaughlin J, Witte ON, Rosenberg N. Induction of a chronic myelogenous leukemia-like syndrome in mice with v-abl and BCRABL. Proc Natl Acad Sci U S A 1990; 87:6649-53; PMID:2204061; http://dx.doi.org/10.1073/pnas.87.17.6649
- Honda H, Hirai H. Model mice for BCRABL-positive leukemias. Blood Cells Mol Dis 2001; 27:265-78; PMID:11358387; http://dx.doi.org/10.1006/bcmd.2000.0374
- Honda H, Oda H, Suzuki T, Takahashi T, Witte ON, Ozawa K, Ishikawa T, Yazaki Y, Hirai H. Development of acute lymphoblastic leukemia and myeloproliferative disorder in transgenic mice expressing p210bcrabl: a novel transgenic model for human Ph1-positive leukemias. Blood 1998; 91:2067-75; PMID:9490692
- Huettner CS, Koschmieder S, Iwasaki H, Iwasaki-Arai J, Radomska HS, Akashi K, Tenen DG. Inducible expression of BCRABL using human CD34 regulatory elements results in a megakaryocytic myeloproliferative syndrome. Blood 2003; 102:3363-70; PMID:12855552; http://dx.doi.org/10.1182/blood-2003-03-0768
- Jaiswal S, Traver D, Miyamoto T, Akashi K, Lagasse E, Weissman IL. Expression of BCRABL and BCL-2 in myeloid progenitors leads to myeloid leukemias. Proc Natl Acad Sci USA 2003; 100:10002-7; PMID:12890867; http://dx.doi.org/10.1073/pnas.1633833100
- Koschmieder S, Gottgens B, Zhang P, Iwasaki-Arai J, Akashi K, Kutok JL, Dayaram T, Geary K, Green AR, Tenen DG, et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 2005; 105:324-34; PMID:15331442; http://dx.doi.org/10.1182/blood-2003-12-4369
- Voncken JW, Kaartinen V, Pattengale PK, Germeraad WT, Groffen J, Heisterkamp N. BCRABL P210 and P190 cause distinct leukemia in transgenic mice. Blood 1995; 86:4603-11; PMID:8541551
- McWhirter JR, Galasso DL, Wang JY. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol 1993; 13:7587-95; PMID:8246975
- McWhirter JR, Wang JY. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. EMBO J 1993; 12:1533-46; PMID:8467803
- Zhang X, Subrahmanyam R, Wong R, Gross AW, Ren R. The NH(2)-terminal coiled-coil domain and tyrosine 177 play important roles in induction of a myeloproliferative disease in mice by Bcr-Abl. Mol Cell Biol 2001; 21:840-53; PMID:11154271; http://dx.doi.org/10.1128/MCB.21.3.840-853.2001
- He Y, Wertheim JA, Xu L, Miller JP, Karnell FG, Choi JK, Ren R, Pear WS. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcrabl. Blood 2002; 99:2957-68; PMID:11929787; http://dx.doi.org/10.1182/blood.V99.8.2957
- Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell 2003; 12:27-37; PMID:12887890; http://dx.doi.org/10.1016/S1097-2765(03)00274-0
- Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993; 75:175-85; PMID:8402896; http://dx.doi.org/10.1016/S0092-8674(05)80094-7
- Puil L, Liu J, Gish G, Mbamalu G, Bowtell D, Pelicci PG, Arlinghaus R, Pawson T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J 1994; 13:764-73; PMID:8112292
- Sattler M, Mohi MG, Pride YB, Quinnan LR, Malouf NA, Podar K, Gesbert F, Iwasaki H, Li S, Van Etten RA, et al. Critical role for Gab2 in transformation by BCRABL. Cancer Cell 2002; 1:479-92; PMID:12124177; http://dx.doi.org/10.1016/S1535-6108(02)00074-0
- Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer 2013; 13:559-71; PMID:23842646; http://dx.doi.org/10.1038/nrc3563
- Cilloni D, Saglio G. Molecular pathways: BCR-ABL. Clin Cancer Res 2012; 18:930-7; PMID:22156549; http://dx.doi.org/10.1158/1078-0432.CCR-10-1613
- Sexl V, Piekorz R, Moriggl R, Rohrer J, Brown MP, Bunting KD, Rothammer K, Roussel MF, Ihle JN. Stat5ab contribute to interleukin 7-induced B-cell precursor expansion, but abl- and bcrabl-induced transformation are independent of stat5. Blood 2000; 96:2277-83; PMID:10979977
- Foley SB, Hildenbrand Z, Soyombo AA, Wu Y, Magee J, Oravecz-Wilson K, Ross TS. Expression of BCRABL p210 from a knockin allele enhances bone marrow engraftment without inducing neoplasia. Cell Rep 2013; 5:51-60; PMID:24095735; http://dx.doi.org/10.1016/j.celrep.2013.08.037
- Lee BH, Tothova Z, Levine RL, Anderson K, Buza-Vidas N, Cullen DE, McDowell EP, Adelsperger J, Frohling S, Huntly BJ, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell 2007; 12:367-80; PMID:17936561; http://dx.doi.org/10.1016/j.ccr.2007.08.031
- Li L, Piloto O, Nguyen HB, Greenberg K, Takamiya K, Racke F, Huso D, Small D. Knock-in of an internal tandem duplication mutation into murine FLT3 confers myeloproliferative disease in a mouse model. Blood 2008; 111:3849-58; PMID:18245664; http://dx.doi.org/10.1182/blood-2007-08-109942
- Loosveld M, Bonnet M, Gon S, Montpellier B, Quilichini B, Navarro JM, Crouzet T, Goujart MA, Chasson L, Morgado E, et al. MYC fails to efficiently shape malignant transformation in T-cell acute lymphoblastic leukemia. Genes Chromosomes Cancer 2014; 53:52-66; PMID:24249258; http://dx.doi.org/10.1002/gcc.22117
- Daley GQ, Baltimore D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcrabl protein. Proc Natl Acad Sci USA 1988; 85:9312-6; PMID:3143116; http://dx.doi.org/10.1073/pnas.85.23.9312
- Vickers M. Estimation of the number of mutations necessary to cause chronic myeloid leukaemia from epidemiological data. Br J Haematol 1996; 94:1-4; PMID:8757501
- Michor F. Mathematical models of cancer stem cells. J Clin Oncol 2008; 26:2854-61; PMID:18539964; http://dx.doi.org/10.1200/JCO.2007.15.2421
- Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004; 6:587-96; PMID:15607963; http://dx.doi.org/10.1016/j.ccr.2004.10.015
- Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2006; 103:6688-93; PMID:16618932; http://dx.doi.org/10.1073/pnas.0602030103
- Mullighan CG, Williams RT, Downing JR, Sherr CJ. Failure of CDKN2AB (INK4AB-ARF)-mediated tumor suppression and resistance to targeted therapy in acute lymphoblastic leukemia induced by BCR-ABL. Genes Dev 2008; 22:1411-5; PMID:18519632; http://dx.doi.org/10.1101/gad.1673908
- Cherry SR, Biniszkiewicz D, van Parijs L, Baltimore D, Jaenisch R. Retroviral expression in embryonic stem cells and hematopoietic stem cells. Mol Cell Biol 2000; 20:7419-26; PMID:11003639; http://dx.doi.org/10.1128/MCB.20.20.7419-7426.2000
- Castellanos A, Pintado B, Weruaga E, Arevalo R, Lopez A, Orfao A, Sanchez-Garcia I. A BCR-ABL(p190) fusion gene made by homologous recombination causes B-cell acute lymphoblastic leukemias in chimeric mice with independence of the endogenous bcr product. Blood 1997; 90:2168-74; PMID:9310467