
Abstract
Knowledge of the mechanisms underlying chemoresistance is important in the development of novel targeted treatments for ovarian cancer. We recently reported that targeting endothelin A receptor/β-arrestin-1, a binding partner of Wnt/β-catenin, is sufficient to sensitize ovarian cancer to chemotherapy. This result highlights endothelin-1 receptor antagonists as potential anticancer therapeutics.
Abbreviations
| APC | = | adenomatous polyposis coli |
| AXIN1 | = | axis inhibition 1 |
| CCND1 | = | cyclin D1 |
| ETAR | = | endothelin A receptor |
| ETBR | = | endothelin B receptor |
| EMT | = | epithelial to mesenchymal transition |
| EOC | = | epithelial ovarian cancer |
| ET-1 | = | endothelin-1 |
| GPCR | = | G-protein coupled receptor |
| GSK3 | = | glycogen synthetase kinase 3 |
| MMP-2 | = | matrix metalloproteinase 2 |
| TCF4 | = | T-cell-specific transcription factor-4 |
| β-TrCP | = | β-transducin repeat containing protein |
| VEGFR-2 | = | vascular endothelial growth factor receptor-2 |
Epithelial ovarian cancer (EOC) is a disease plagued by recurrences and progressive chemoresistance. EOC cells assure their growth advantage and resistance to chemotherapy through the appropriation of key pathways, such as those controlled by G-protein coupled receptors (GPCRs). Accumulating molecular and in vivo evidence demonstrates that the activation of autocrine and paracrine signaling by the binding of endothelin-1 (ET-1, EDN1) to its GPCRs, endothelin A receptor (ETAR, EDNRA) and endothelin B receptor (ETBR, EDNRB), elicits pleiotropic effects in tumor cells and in the host microenvironment, modulating the epithelial to mesenchymal transition (EMT), chemoresistance, and the expansion of vascular networks.Citation1 Of particular interest, ETAR was shown to be aberrantly activated in EOC and its expression has been correlated with platinum resistance and EMT marker expression.Citation2 This discovery was followed by analysis of EOC samples from The Cancer Genome Atlas (TCGA) that showed evidence for worse survival in EOC patients with ETAR overexpression.Citation3-4 In addition to ETAR overexpression in chemoresistant EOC cells, ETBR also appears to have protumorigenic activity by promoting angiogenesis and lymphangiogenesis and evasion of the immune response.Citation5 Hence, ETAR and ETBR, which are heterogeneously expressed in EOC cells, have emerged as key targets for cancer therapy.Citation6-7 Emerging evidence demonstrates that acquisition of chemoresistance is highly dependent on contextual cues such as interactions with the tumor microenvironment and crosstalk with other signaling pathways. Noting that crosstalk between ET-1 signaling and other growth factor pathways drives tumor progression via the scaffold protein β-arrestin-1 (ARRB1) that serves as a co-pilot to organize complex signaling networksCitation8–9, we hypothesized that an β-arrestin-1–mediated mechanism in ET-1 signaling may play a particularly important role in evasion of the drug response.
To investigate the mechanism underlying this resistance, we used resistant EOC cell lines generated by prolonged treatment with cisplatinum or taxol. Upon ETAR activation of these resistant cells, β-arrestin-1 formed a nuclear complex with β-catenin and p300, resulting in histone acetylation that led to chromatin reorganization and enhanced transcription of genes such as ET-1 that are responsible for regulating the rate limiting step of the drug responseCitation10. Similarly, 13 of 13 platinum-resistant patients had increased recruitment of β-arrestin-1 and β-catenin on the ET-1 promoter, providing further evidence that ETAR/β-arrestin-1 cooperates with Wnt signaling to acquire a chemoresistant phenotype through amplification of the ET-1 autocrine loop. This work expands what was previously known about the chemoresistance-associated functions of ETAR, outlining a model in which ET-1 co-opts Wnt components for its own agenda, thus sustaining EMT, stemness features, cell invasion, and metastasis ().
Figure 1. Interplay between ETAR/β–arrestin-1 and Wnt/β-catenin drives chemoresistance in ovarian cancer. In chemoresistant ovarian cancer cells, binding of endothelin-1 (ET-1, EDN1) to its receptors leads to recruitment of β-arrestin-1 (ARRB1). β-arrestin inhibits the destruction complex composed of glycogen synthetase kinase 3 (GSK3), axis inhibition 1 (AXIN1), β-transducin repeat containing protein (β-TrCP), and adenomatous polyposis coli (APC), and thus promotes the accumulation of a non-Ser/Thr phosphorylated active form of β-catenin. β-arrestin-1 shuttles with β-catenin into the nucleus, where it interacts with p300 histone acetyltransferase to enhance β-catenin/T-cell-specific transcription factor-4 (TCF4)-transactivation, thus promoting the transcription of genes such as EDN1, matrix metalloproteinase 2 (MMP-2), or cyclin D1 (CCND1) and leading to enhanced chemoresistance, epithelial to mesenchymal transition (EMT), stemness, cell plasticity, invasion, and metastasis. As a signal transducer, endothelin A receptor (ETAR, EDNRA)/β-arrestin-1 initiates transactivation of the vascular endothelial growth factor receptor-2 (VEGFR-2) through SRC. In parallel, paracrine production of ET-1 activates endothelin B receptor (ETBR, EDNRB) expressed on endothelial cells, promoting expansion of vascular networks. The dual ETAR and ETBR antagonist macitentan targets not only cancer cells (which express ETAR and ETBR) but also tumor-associated stromal elements (which express ETBR).
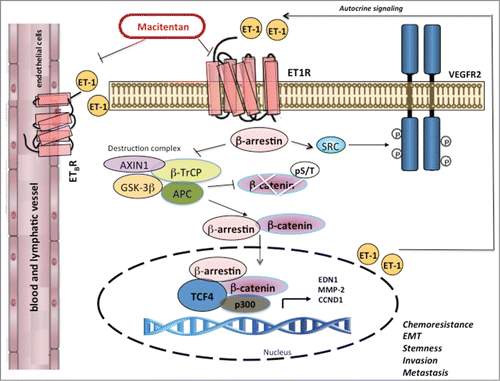
The challenge that lies ahead is integrating our improved understanding of the interconnected molecular mechanisms promoted by the ET-1 axis in chemoresistant EOC cells with novel therapeutic options to improve patient outcomes. Can ET-1 receptors be directly targeted to resensitize EOC cells to cisplatinum or taxol? Treatment of resistant cells with the approved small molecule macitentan, a dual ETAR/ETBR antagonist that prevents formation of the β-arrestin-1/β-catenin/p300 complex on the target, caused a strong reduction in growth and invasiveness. In vivo, macitentan significantly inhibited tumor growth, neovascularization, intravasation, and peritoneal dissemination in chemoresistant EOC xenografts by interfering with ETAR expressed on EOC cells and ETBR expressed on endothelial cells. One striking observation is that the combination of macitentan and chemotherapy restored sensitivity to cisplatinum and taxol. Consistent with the in vitro results, analysis in human platinum-resistant EOC tissues showed that ETAR overexpression is significantly associated with chemoresistance and poor prognosis, emphasizing ETAR as a potential predictive marker of drug response. Taken together, the results of our study provide novel mechanistic insights into how Wnt/β-catenin signaling is wired to ETAR/β-arrestin-1 to enable EOC cells to become unresponsive to chemotherapeutics. In the nucleus, the β-arrestin-1–β-catenin connection represents the initial scaffold on which transcriptional regulatory complexes could be built to regulate epigenetic modifications. Furthermore, in the cytosol, distinct β-arrestin-1 complexes can recruit factors that activate crosstalk with tyrosine kinase receptor, such as vascular endothelial growth factor receptor-2 (VEGFR-2), indicating that β-arrestin-1 might control an intriguing spectrum of pathway interactions that regulate drug sensitivity.
Although these findings reveal the interplay between ETAR and Wnt/β-catenin signaling, a number of questions remain. For example, does nuclear β-arrestin-1 have a common role in mediating β-catenin transcription in other ET-1-driven tumors? How does β-arrestin-1 translocate to the nucleus in response to ETAR activation and is the receptor part of the nuclear complex? What causes the relatively high level of ETAR in chemoresistant EOC? Does miRNA regulation play a role in promoting chemoresistance by upregulating ETAR? These and other issues, such as whole-genome ChIP-seq analysis to identify new β-arrestin-1 partners, are currently being addressed to understand the early events leading to the acquisition of chemoresistance in EOC and other ET-1-driven malignancies.
Targeting multiple networks using the dual ETAR and ETBR antagonist macitentan to overcome compensatory mechanisms of therapy escape provides the potential advantage of targeting not only cancer cells (which typically express ETAR) but also microenvironment-associated elements, such as vascular, lymphatic, and inflammatory cells and fibroblasts, which all express ETBR. The present study therefore has biological and clinical relevance for the development of new prognostic tools and novel treatments to overcome chemoresistance, and offers a strong rationale for translation of the experimental approach of combining macitentan with chemotherapy into immediate clinical evaluation in this disease setting.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Rosanò L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2013; 13:637–51; PMID:23884378; http://dx.doi.org/10.1038/nrc3546
- Rosanò L, Cianfrocca R, Spinella F, Di Castro V, Nicotra MR, Lucidi A, Ferrandina G, Natali PG, Bagnato A. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin Cancer Res 2011; 17:2350–60; PMID:21220476; http://dx.doi.org/10.1158/1078-0432.CCR-10-2325
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474: 609–615; PMID:21720365; http://dx.doi.org/10.1038/nature10166
- Teoh JP, Park KM, Wang Y, Hu Q, Kim S, Wu G, Huang S, Maihle N, Kim IM. Endothelin-1/Endothelin A receptor-mediated biased signaling is a new player in modulating human ovarian cancer cell tumorigenesis. Cell Signal 2014; 26:2885–95; PMID:25194819; http://dx.doi.org/10.1016/j.cellsig.2014.08.024
- Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, Katsaros D, O'Brien-Jenkins A, Gimotty PA, Coukos G. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med 2008; 14: 28–36; PMID:18157142; http://dx.doi.org/10.1038/nm1699
- Kim SJ, Kim JS, Kim SW, Brantley E, Yun SJ, He J, Maya M, Zhang F, Wu Q, Lehembre F, et al. Macitentan (ACT-064992), a tissue-targeting endothelin receptor antagonist, enhances therapeutic efficacy of paclitaxel by modulating survival pathways in orthototic models of metastatic human ovarian cancer. Neoplasia 2011; 13:167–79; PMID:21403842
- Coffman L, Mooney C, Lim J, Bai S, Silva I, Gong Y, Yang K, Buckanovich RJ. Endothelin receptor-A is required for the recruitment of antitumor T cells and modulates chemotherapy induction of cancer stem cells. Cancer Biol Ther 2013; 14:184–92; PMID:23192269; http://dx.doi.org/10.4161/cbt.22959
- Rosanò L, Cianfrocca R, Masi S, Spinella F, Di Castro V, Biroccio A, Salvati E, Nicotra MR, Natali PG, Bagnato A. Beta-arrestin links endothelin A receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc Natl Acad Sci U S A 2009; 106:2806–11; PMID:19202075; http://dx.doi.org/10.1073/pnas.0807158106
- Rosanò L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Spadaro F, Salvati E, Biroccio AM, Natali PG, Bagnato A. -arrestin-1 is a nuclear transcriptional regulator of endothelin-1-induced -catenin signaling. Oncogene 2013; 32:5066–77; PMID:23208497; http://dx.doi.org/10.1038/onc.2012.527
- Rosanò L, Cianfrocca R, Tocci P, Spinella F, Di Castro V, Caprara V, et al. Endothelin A receptor/-arrestin signaling to the Wnt pathway renders ovarian cancer cells resistant to chemotherapy. Cancer Res 2014; Nov 6. pii: canres.3133.2013. [Epub ahead of print