Abstract
Type I interferons (IFNs) are induced during most viral infections and are considered to be the primary and universal means of innate viral control. However, several other innate mechanisms, including autophagy, have recently been shown to play an important role in antiviral defense. In our recent study, we utilized a herpes simplex virus 1 (HSV-1) infection model to investigate the relationship between cell type and innate antiviral immune mechanisms. Our study demonstrates that dorsal root ganglion (DRG) neurons undergo an innate antiviral response to HSV-1 that differs from the antiviral program induced in mitotic cells in three distinct ways. First, DRG neurons produce less type I IFN and undergo a less effective IFN antiviral program vs. mitotic cells in response to HSV-1 infection. Second, the type I IFN program initiated in DRG neurons induces less cell death than in mitotic cells. Third, in the absence of a robust type I IFN response, DRG neurons, but not mitotic cells, repy on autophagy in HSV-1 defense. Our findings reveal a cell type-specific requirement for autophagy in defense against HSV-1, and offer insight into the cell-appropriate antiviral defense mechanism employed by neurons.
Type I IFNs induce an antiviral state through the induction of numerous genes downstream of IFNAR [interferon (α,β and ω) receptor], whose known antiviral functions can be broadly classified into four functional categories: those that block viral infection and replication, induce cell death of infected cells, recruit or activate hematopoietic cells, and prime antiviral adaptive immune responses. Although the IFN pathway is remarkably effective at limiting viral replication, the lytic consequences of a robust type I IFN response may have severe pathological consequences in tissues predominated by nonrenewable cell types such as neurons. In contrast, autophagy is an evolutionarily ancient, prosurvival intracellular degradation pathway that has recently been shown to play a key role in antiviral defense against several viral pathogens including HSV-1. HSV-1 primarily replicates in two cell types in vivo, mucosal epithelial cells at the site of virus entry that are continuously replaced by local stem cells, and nonrenewable DRG neurons that innervate the primary site of infection. Both type I IFN and autophagy have previously been shown to be important in HSV-1 defense in vivo; thus, the HSV-1 model provided us an ideal system to interrogate the cell type dependency of innate antiviral defense mechanisms.
In our study, we first evaluated the contribution of type I IFNs in antiviral defense in mitotic cells and primary DRG neurons. We observed that mitotic cells including mouse embryonic fibroblasts (MEFs) and keratinocytes rely on type I IFN responses, while DRG neurons utilize type I IFNs to a much lesser extent to control HSV-1 infection. Thus, DRG neurons treated with recombinant IFNs lead to reduced induction of several key interferon-stimulated genes (ISGs) and result in significantly less IFN-dependent cell death. While DRG neurons are less protected from HSV-1 infection in response to exogenous IFN treatment, they are no more susceptible to HSV-1 infection in the absence of IFNs compared with MEFs. This led us to evaluate whether DRG neurons utilize an alternative mechanism(s) of antiviral defense, namely, autophagy. To test this hypothesis, we used a combination of mutant host and mutant viruses that are defective in autophagy or in evasion from autophagy. Specifically, we used selective Atg5 conditional knockout mice that lack Atg5 in keratinocytes or neurons to study the role of autophagy in anti-HSV-1 defense. We also used an HSV-1 mutant incapable of blocking autophagy in infected host cells. Using these approaches, we found that DRG neurons, but not mitotic cells, require autophagy for protection against HSV-1. As both type I IFN and autophagy play a key role in HSV-1 antiviral defense in DRG neurons, we further investigated the relationship between type I IFN and autophagy-dependent control of HSV-1 replication. Notably, we found that type I IFN and autophagy both operate independently in neurons to control HSV-1 infection by nonlytic mechanisms, whereas a robust, lytic type I IFN response alone is sufficient to control HSV-1 replication in mitotic cells. Thus, our findings imply a fundamental difference in the innate HSV-1 antiviral strategies employed by neurons vs. mitotic cells that are suited to the physiology of the cell types.
A key question that arises from our study is the mechanism by which autophagy controls HSV-1 replication in neurons. Current data suggest that control is at least in part due to innate cell intrinsic virus restriction, as we and others have observed autophagy-dependent control of HSV-1 replication at early time points in vivo and in infected neurons ex vivo. Innate control of HSV-1 through autophagy may be mediated directly or indirectly. In direct autophagy-dependent restriction, HSV-1 is controlled by degradation of whole virions and/or viral components in autophagosomes, through a process known as xenophagy. Previous studies have demonstrated HSV-1 virions inside the autophagosomes of infected cells. Several possible explanations can be contemplated for a neuron-specific mechanism of xenophagy in anti-HSV-1 defense. First, the efficiency of autophagy within DRG neurons may be greater than in mitotic cells. This could be due to increased induction of autophagy, increased efficiency of targeting of HSV-1 virions/viral components to autophagosomes, or increased autophagosome turnover (flux) in neurons. Second, the unique spatiotemporal features of the HSV-1 neuronal replication life cycle may be suited for xenophagic degradation of HSV-1. HSV-1 viral entry, viral replication, and viral egress all have unique characteristics in neurons and could provide unique opportunities for autophagosomal capture and degradation. Third, the lack of a robust lytic innate response against HSV-1 in neurons may reveal the ubiquitous contribution of autophagy to HSV-1 antiviral defense. This hypothesis predicts that robust type I IFN and other lytic pathways supersede autophagy in mitotic cells, and attenuation of these pathways in these cell types would unveil an antiviral role of autophagy. It is also possible that HSV-1 replication is controlled within neurons via indirect autophagy mechanisms, such as the role of autophagy in maintaining general cellular homeostasis and thereby contributing to the ability to withstand viral insult. However, the observations that (1) genetic deletion of autophagy in neurons renders them more susceptible to HSV-1 infection, and (2) infection with a mutant virus incapable of blocking host autophagy in wild-type neurons is impaired, point to the role of inducible autophagy in antiviral protection.
We predict that the findings of this study may apply in general to controlling intracellular pathogens in post-mitotic nonrenewable cell types, including neurons of the central nervous system and cardiomyocytes. Another pertinent question is whether autophagy restricts HSV-1 from reactivation. Further studies are necessary to determine if autophagy may be manipulated to prevent or treat chronic neuronal infections in humans ().
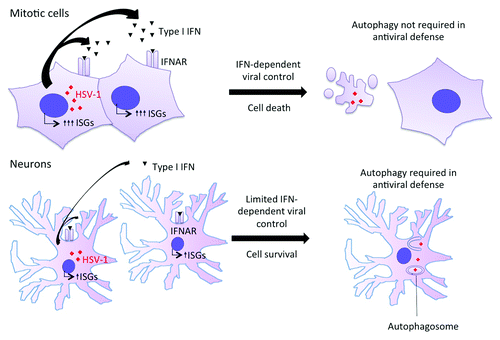
Figure 1. Mitotic cells and DRG neurons rely on different innate strategies to control HSV-1 replication. In mitotic cells such as mouse embryonic fibroblasts and keratinocytes, HSV-1 infection induces a robust type I IFN response. Type I IFN induces vigorous upregulation of ISGs, which provide protection against HSV-1 infection in part by inducing death of infected cells. Autophagy is not required in this context. In neurons, HSV-1 infection results in limited type I IFN production. Moreover, exposure to exogenous type I IFN induces a limited upregulation of certain ISGs. This altered type I IFN response is not sufficient to induce either cell death or complete protection against HSV-1 infection. Within neurons, both type I IFN and autophagy contribute to innate control of HSV-1.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.