Abstract
The administration of rapamycin, an MTOR-dependent autophagy activator, for the treatment of neurodegenerative diseases has been tested in several animal models. Thus, whether autophagy activation would lead to the clearance of abnormal accumulation of aggregated proteins in neurodegenerative diseases is worthy of exploration. We have recently shown that rapamycin administration at the early pathological stage of a mouse model with frontotemporal lobar dementia (FTLD-U) characterized with cytoplasmic TARDBP/TDP-43(+)/ubiquitin(+) inclusions (UBIs) in the diseased neurons could rescue the learning/memory deficiency and the abnormal motor function disorder of the mice. This was accompanied by a decreased level of CASP3/caspase-3 and a reduction of the neuronal loss in the mouse forehead. Moreover, autophagy activation at a late pathological stage also could improve motor function, which was accompanied by a reduction of the TARDBP(+) UBIs. This study has set the principal for therapy of neurodegenerative diseases with the TARDBP protein, i.e., amyotrophic lateral sclerosis (ALS)-TDP and FTLD-TDP43, with the use of autophagy activators.
Keywords: :
A wide range of protein misfolding diseases, also called proteinopathies, is caused by the abnormal accumulation of aggregated forms of proteins in the cytoplasm of the diseased cells. Recent studies have shown that rapamycin or its analogs can upregulate MTOR-dependent autophagy in animal models of several proteinopathy-related neurodegenerative diseases including Alzheimer, Parkinson and Huntington diseases. This is somewhat expected since autophagy activators such as rapamycin induce early autophagosome formation and enhance late autolysosome degradation, therefore accelerating the removal of aggregation-prone proteins, which provides its neuroprotective effect. However, the failure of rapamycin as a therapeutic reagent for neurodegenerative disorders has also been reported, such as in the case of a Drosophila model of Alzheimer disease. Rapamycin also aggravates neuronal death in a mouse model of ALS. In these failure cases of rapamycin treatment, defective lysosome fusion or impaired autophagic flux at the late stage may have led to autophagosome accumulation, despite the induction of formation of the autophagosomes by rapamycin at the early stage. This indicates that different pathological mechanisms might have different responses to rapamycin treatment, and case-by-case studies are needed to clarify the potential of rapamycin and other autophagy activators to treat distinct proteinopathy-related neurodegenerative diseases.
TARDBP is a 43-kDa, ubiquitously expressed nuclear protein that has been implicated in a number of cellular functions, including transcriptional repression and regulation of alternative splicing. TARDBP proteinopathies have been reported to occur in several neurodegenerative diseases. For instance, many FTLD-U and ALS cases are characterized by cytoplasmic TARDBP(+) UBIs containing the full-length TARDBP, polyubiquitinated-TARDBP, and phosphorylated TARDBP as well as carboxyl 25-kDa/35-kDa fragments of TARDBP. Previously, we have generated a mouse model with neurological and pathological phenotypes mimicking the FTLD-U syndrome by calcium/camodulin-dependent protein kinase II α (CAMK2A/CaMKII-α) promotor-driven overexpression of TARDBP in the mouse forebrains including the cortex and the hippocampal region. The transgenic mice show impaired learning/memory ability, progressive motor dysfunction, neuronal apoptosis and brain atrophy. This mouse model appeared to be an ideal research tool for pathological/clinical analysis and therapeutic/drug development for FTLD-U, and TARDBP proteinopathies in general.
To investigate whether rapamycin could be applied as a useful therapeutic drug, we started the treatment in the FTLD-U transgenic (Tg) mice at 2 months of ages, right after the symptoms of learning/memory deficiency. After 1 month of administration, the impaired learning/memory capabilities are partially rescued. Upon continued treatment of the mice with rapamycin to the age of 6 months, the Tg mice exhibit improvement of performance in the rotarod test, indicating alleviation of the age-dependent progressive motor dysfunction by rapamycin treatment. In addition, rapamycin treatment lowers the CASP3-dependent neuronal apoptosis and the level of the aggregated form of TARDBP, suggesting that the ability of autophagic clearance induced by rapamycin effectively removes the insoluble cytosolic TARDBP aggregates and thus provides a neuron protection outcome.
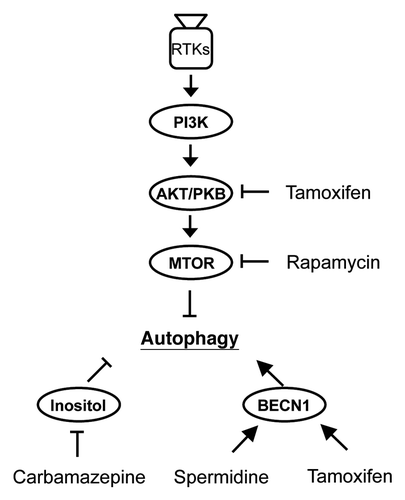
To investigate whether autophagy activation in general could provide therapeutic benefit at the late stage of disease progression, we have also tested the protective effect of rapamycin and three other autophagy activators, spermidine, carbamazepine and tamoxifen, in 6 month-old Tg mice. Of the four chemicals, rapamycin and tamoxifen activate autophagy through an MTOR-dependent pathway, whereas spermidine and carbamazepine could activate autophagy through an MTOR-independent pathway (). Administration of these four chemical drugs to 6 month-old Tg mice for 1 month effectively ameliorates the motor dysfunction, and this is accompanied by the reduction of the amount of cytosolic TARDBP(+) UBIs as well as enhancement of neuronal survival in the forebrain of the FTLD-U Tg mice. These results are highly suggestive that overall activation of the autophagic clearance would benefit patients with FTLD-TDP43.
Figure 1. Sites of actions by four different therapeutic reagents along the MTOR-dependent and MTOR-independent autophagy pathways. Autophagy is negatively regulated by MTOR, a downstream target of PI3K and AKT/PKB. Rapamycin functions as an MTOR inhibitor and triggers MTOR-dependent autophagy induction. Tamoxifen also functions as a MTOR-dependent autophagy inducer through AKT/PKB inhibition, and as with another autophagy chemical inducer, spermidine, they can both induce dissociation of the autophagy proteins BECN1 and BCL2, thus enhancing the MTOR-independent autophagy pathway simultaneously. Carbamazepine enhances MTOR-independent autophagy through reducing the intracellular level of inositol. RTKs, receptor tyrosine kinases.
The pathological and cellular mechanisms of downregulation of autophagy in the FTLD-U Tg mice, and in TARDBP proteinopathies in general, are still under investigation. One scenario could be that the cytosolic TARDBP(+) UBIs tackle and trap the ubiquitin-binding protein SQSTM1/p62, a functional component of autophagy, thus reducing the capability of the cells to remove the abnormal aggregates through autophagic degradation. Another possibility is that the early steps of autophagosome formation are disrupted due to the loss-of-function of TARDBP. TARDBP is required for the maintenance of autophagy by regulation of the stability of Atg7 mRNA. Thus, the disruption of autophagy happening in the FTLD-U mouse model could also result from a combined improper metabolism and dysfunction of TARDBP, such as the cleavage of the full-length TARDBP by CASP3 and its trapping in the UBIs.
In summary, we have provided evidence that autophagy activation is an effective therapeutic route for the treatment of a TARDBP Tg mouse model with FTLD-U phenotypes, thus giving one more way to attempt clinical treatment of FTLD-TDP43 and other neurodegenerative diseases with TARDBP proteinopathies.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.