Abstract
Saposin C deficiency, a rare variant form of Gaucher disease, is due to mutations in the prosaposin gene (PSAP) affecting saposin C expression and/or function. We previously reported that saposin C mutations affecting one cysteine residue result in autophagy dysfunction. We further demonstrated that the accumulation of autophagosomes, observed in saposin C-deficient fibroblasts, is due to an impairment of autolysosome degradation, partially caused by the reduced amount and enzymatic activity of CTSB (cathepsin B) and CTSD (cathepsin D). The restoration of both proteases in pathological fibroblasts results in almost completely recovery of autophagic flux and lysosome homeostasis.
Gaucher disease, the most prevalent lysosomal storage disorder, is caused by a deficit of glucosylceramidase or, rarely, of its activator saposin C (only six cases have been reported in the world so far). The disorder is characterized by the accumulation of glucosylceramide and other lipids in the lysosomes of monocyte/macrophage lineage. Stored substrates lead to enlargement of lysosomes and compromise their functionality. Mutations in glucosylceramidase or in saposin C result in hepatosplenomegaly, anemia, bone crisis, pulmonary problems and in some cases, neurological involvement. Clinically, Gaucher disease is differentiated into type 1, non- neuronopathic variant, and types 2 and 3, acute and chronic neuronopathic variants, respectively. Of the six diagnosed saposin C-deficiency cases, we analyzed biological properties of fibroblasts from four recently described patients. Two of these cell lines carry a mutation involving one of the six cysteine residues. One patient, homozygous for a deletion that corresponds to seven amino acid residues, does not present neurological manifestations to date; the other one, carrying a missense mutation, displays a clear type 3 phenotype. The substitution or the deletion of a cysteine residue leads to a disruption of one of three disulfide bridges resulting in almost complete lack of saposin C and affecting the autophagic pathway. Autophagy is a highly regulated homeostatic and degradative process in which cells destroy and recycle their own components via the lysosomal machinery. This pathway involves the formation of a closed double-membrane structure, called an autophagosome, and its subsequent fusion with endosomes or lysosomes to form an autolysosome, where the sequestered materials are degraded to generate nutrients; finally, lysosomes are rederived from digested autolysosomes. Alteration of any of these steps can impair autophagy.
We provided experimental evidence of autophagy activation in saposin C-deficient fibroblasts utilizing microscopy-based green fluorescent protein (GFP)-LC3 puncta formation assay, western blot analysis of LC3-I and LC3-II forms, and MTOR activation. Enhanced autophagy could result from different mechanisms: increased autophagosome formation, impairment of the autophagosome-lysosome fusion, and defective degradation of the autolysosome. The first possibility was excluded because we did not observe upregulation of some proteins (BECN1, ATG5 or ATG7) involved in upstream steps of autophagosome formation. On the contrary, an increased level of BECN1 is observed in some lysosomal storage disorders, such as Niemann-Pick type C, GM1 gangliosidosis and neuronal ceroid-lipofuscinosis. Another possible explanation of autophagy dysfunction could be the blockade of autophagosome fusion with lysosomes, which occurs in multiple sulphatase deficency, mucopolysaccharidosis type III A, and Pompe disease. This was verified by exposing the cells to leupeptin, an inhibitor of lysosomal proteases. Leupeptin treatment increases dramatically the LC3-II level and dot numbers of GFP-LC3 in pathological fibroblasts compared with control fibroblasts, indicating the proper fusion of autophagosomes with lysosomes. Possible dysregulated lysosomal clearance was investigated by starving the cells and then incubating them in complete medium for short periods. Nutrient deprivation increased the LC3-II level, especially in saposin C-deficient fibroblasts, and inhibited the MTOR signaling pathway. Amino acid replenishment promotes the total disappearance of LC3-II and the reactivation of MTOR in control cells, but not in pathological fibroblasts. These results indicate that the degradation of autophagosomal content is impaired in saposin C-deficient fibroblasts leading to accumulation of enlarged and long-lasting autolysosomes. As a consequence, lysosomal reformation, regulated by MTOR reactivation, is delayed. We provided evidence that defective lysosomal clearance results from reduced levels and enzymatic activities of CTSB and CTSD. At the same time, we noted a difference in the behavior of glucosylceramidase-deficient fibroblasts. These cell lines do not show either autophagic dysfunction or reduction of both proteases, but even CTSB is almost doubled compared with control cells. Our findings support the idea that the complete lack of saposin C is responsible for altered autophagy. This could be associated with several functions of saposin C that activates and stabilizes glucosylceramidase, but also extracts and transports lipids from lysosomes to other compartments. Last, to determine whether dysregulation of autophagy is correlated with the low amount of the above mentioned proteases, we transiently overexpressed them and observed that lysosomal proteolysis and consequent reformation of lysosomes is almost completely restored.
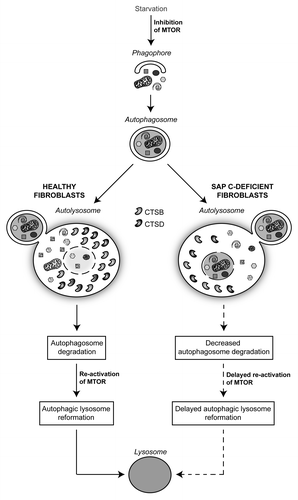
To summarize our results we propose a model in which the complete lack of saposin C triggers defective levels and enzymatic activities of CTSB and CTSD. The resulting reduction of autolysosome degradation impairs MTOR reactivation and delays lysosomal reformation (). We report for the first time that increased autophagy in a lysosomal storage disease is associated with a defective expression and activity of two lysosomal proteases.
Figure 1. Model of the autophagic flux in healthy and saposin C-deficient fibroblasts. In healthy cells, the content of autolysosomes, derived from autophagosome and lysosome fusion, is degraded by lysosomal proteases, leading to reactivation of MTOR and reformation of lysosomes. In pathological cells the complete lack of saposin C results in dysfunctional autophagy due to reduced level and enzymatic activity of two proteases (CTSB and CTSD). Consequently, delayed degradation of autolysosomes triggers inhibition of MTOR and defective lysosome reformation.
Our data are strengthened by a recent published study about autophagic dysfunction in Niemann-Pick type C disease. Although aberrant autophagy in this disorder has been associated with an increased level of BECN1, it has been newly demonstrated that decreased lysosomal cathepsin (B and L) activities, resulting from cholesterol accumulation, lead to an impaired clearance of autophagosomes. In our study we report not only a defective enzymatic activity of CTSB and CTSD, but also a decrease of their amount in the saposin C-deficient lysosomes. Further studies will be necessary to elucidate the mechanism(s) underlying the downregulation of these proteases in saposin C-deficient models.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.