Abstract
In a recent study, characterization of novel mechanisms that regulate lethal autophagy revealed that C18-ceramide stress mediates LC3B-II-ceramide binding on mitochondrial membranes to target autophagosomes for mitophagy induction and tumor suppression.
Autophagy promotes cell survival during starvation or it can progress to cell death (lethal autophagy). However, the mechanisms by which autophagy promotes or inhibits cell survival are unclear. Ceramide is a bioactive sphingolipid that mediates cell death. Recently, mammalian ceramide synthases 1–6 (CERS1 to CERS6), which regulate the de novo generation of ceramides with specific fatty acid chain lengths, have been discovered. For example, CERS1 and CERS6 preferentially generate C18- and C16-ceramide, respectively. Ceramides generated by CERS1 to CER6 have distinct biological roles. CERS1 and C18-ceramide have emerged as tumor suppressors in preclinical and clinical studies. However, mechanisms of ceramide signaling in the regulation of lethal autophagy have been enigmatic.
In a recent study, the roles and mechanisms involved in the regulation of lethal autophagy mediated in response to ceramide stress were investigated. The data suggest that C18-pyridinium ceramide (C18-Pyr-Cer), a ceramide analog containing a pyridinium tether in the 14C-sphingosine backbone, D-erythro-14C18-pyridinium ceramide bromide, which accumulates in mitochondria because of the positive charge localized within the pyridinium ring or endogenous C18-ceramide generation by CERS1 expression selectively mediate lethal autophagy, which is independent of BAX and BAK1 as well as caspase, by inducing mitophagy. CERS1 expression, generating mainly C18-ceramide-mediated localization of ceramide on the outer mitochondrial membrane, leads to mitophagy, whereas CERS6 expression, which mainly generates C16-ceramide, has no effect on mitochondrial localization of ceramide or mitophagy. However, C16-Pyr-Cer, which localizes to mitochondria, induces mitophagy. These data suggest that different subcellular localization of endogenous ceramides rather than their fatty acid chain lengths has a key role in mediating their distinct biological functions. How CERS1-mediated C18-ceramide is transported to the outer mitochondria membrane is still unknown.
Moreover, CERS1 and C18-ceramide or C18-Pyr-Cer induce LC3B-phosphotidylethanolamine (PE) lipidation, forming LC3B-II, which then binds ceramide on the mitochondrial membrane upon DNM1L/DRP1-mediated mitochondrial fission, targeting autolysosomes to mitochondria and leading to inhibition of mitochondrial function and oxygen consumption. Accordingly, expression of mutant LC3B with impaired ceramide binding, as predicted by molecular modeling, prevents CERS1-mediated mitochondrial targeting, recovering oxygen consumption (). The ceramide–LC3B-II interaction seems to involve the central hydrophobic domain of the protein; the Ile35 and Phe52 residues localized within this hydrophobic domain of LC3B were identified as important for ceramide binding. Notably, data also indicate that PE conjugation (lipidation) of LC3B also influences the ceramide–LC3B-II interaction, because binding of LC3BG120A (the nonlipidated mutant) to ceramide is reduced compared with that of the wild-type protein, whereas, LC3B mutants that lack ceramide binding are lipidated, but not localized to mitochondria in response to ceramide stress. These data suggest that ceramide-LC3B-II interaction targets autophagosomes to mitochondria without affecting LC3B-lipidation. However, upstream mechanisms by which ceramide stress induces LC3B lipidation in HNSCC cells remain unknown.
Figure 1. Mechanism by which C18-ceramide induces lethal autophagy involves, at least in part, the lipidation of LC3B, forming LC3B-II, and the interaction of LC3B-II and ceramide on mitochondrial membranes upon DNM1L-mediated mitochondrial fission, which targets autolysosomes to mitochondria for lethal mitophagy.
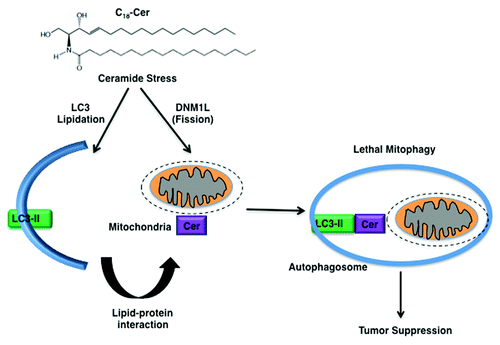
Thus, these data suggest that ceramide-mediated mitophagy, which requires LC3B-II formation and ceramide-LC3B-II binding on mitochondrial membranes, is regulated downstream of DNM1L-mediated mitochondrial fission. DNM1L-mediated mitochondrial fission induces LC3B lipidation and mitophagy, which requires PARK2/Parkin and PINK1 or BNIP3 function. Also, it is still unknown if PARK2 and PINK1 are also involved in the regulation of ceramide-mediated lethal mitophagy. However, the involvement of ceramide as one of the factors that targets LC3B-II–containing autolysosomes to mitochondria for induction of lethal mitophagy via the ceramide–LC3B-II interaction upon DNM1L-induced fission is new and distinct from starvation-induced autophagy, which is generally protective for survival.
In addition, knockdown of CERS1 abrogates sodium selenite–induced mitophagy, and stable LC3B knockdown protects against CERS1- and C18-ceramide-dependent mitophagy and block tumor suppression in vivo. This is one of the first mechanistic links to explain the autophagy paradox, defining how lethal autophagy is regulated, at least in part, by ceramide stress.
Overall, these data define at least one key component of the autophagy paradox: LC3B-II autophagosomes targeting to mitochondrial membranes via C18-ceramide–LC3B-II interaction regulate lethal mitophagy, leading to tumor suppression. These data should help clarify the role of ceramide as a possible receptor for mitochondrial targeting of LC3B-II and autophagosomes, which is linked to HNSCC tumor suppression in response to induction of CERS1 and C18-ceramide. Defining at least one specific mechanism of how the tumor suppressor function of CERS1 and C18-ceramide is regulated via lethal mitophagy might have an impact on the development of therapeutics against various human cancers.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.