Abstract
Autophagy is mediated by a unique organelle, the autophagosome, which encloses a portion of the cytoplasm for delivery to the lysosome. Phosphatidylinositol 3-phosphate (PtdIns3P) produced by the class III phosphatidylinositol 3-kinase (PtdIns3K) complex is essential for canonical autophagosome formation. RAB5A, a small GTPase localized to early endosomes, has been shown to associate with the class III PtdIns3K complex, regulate its activity and promote autophagosome formation. However, little is known about how endosome-localized RAB5A functions with the class III PtdIns3K complex. Here we identified a novel endoplasmic reticulum (ER)-localized transmembrane protein, ER membrane protein complex subunit 6 (EMC6), which interacted with both RAB5A and BECN1/Beclin 1 and colocalized with the omegasome marker ZFYVE1/DFCP1. It was shown to regulate autophagosome formation, and its deficiency caused the accumulation of autophagosomal precursor structures and impaired autophagy. Our study showed for the first time that EMC6 is a novel regulator involved in autophagy.
Keywords: :
Introduction
Autophagy is a catabolic process involving the degradation of cytoplasmic materials (e.g., proteins, lipids, glycogens and organelles). During this process, damaged organelles and long-lived proteins are sequestered in a double-membrane vesicle, known as the autophagosome or autophagic vacuole (AV), and delivered to lysosomes for degradation.Citation1,Citation2
Several signaling complexes and pathways are involved in the development of the autophagic response, including the Atg1/ULK1 complex, the Vps34/PIK3C3/class III PtdIns3K complex and two ubiquitin-like conjugation systems, involving ATG12–ATG5 and Atg8/LC3.Citation3-Citation6 In yeast, the Atg1 and Vps34 complexes are required for initiation and nucleation of a cup-shaped membrane sac, termed the phagophore.Citation1 The localized production of PtdIns3P by Vps34 can recruit proteins containing FYVE and PX domains, such as the WD40 repeat PtdIns3P effector Atg18 to the phagophore assembly site (PAS), the yeast-specific site of autophagosome formation.Citation7 The ubiquitin-like conjugation systems are required for expansion and completion of autophagosomes. Ubiquitin-like Atg12 is activated by the E1-like activating enzyme Atg7, transferred to the E2-like enzyme Atg10 and eventually conjugated to the target protein Atg5, which interacts with a small coiled-coil protein, Atg16. Atg12–Atg5-Atg16 then forms a multimeric complex via homo-oligomerization of Atg16.Citation6 The Atg8 C terminus is proteolytically removed by the cysteine protease Atg4, activated by Atg7, transferred to the E2-like enzyme Atg3 and eventually conjugated to PE.Citation6 The Atg12–Atg5 conjugate apparently has an E3-like activity for Atg8 lipidation.Citation8
In mammalian cells, the role of ATG proteins in autophagosome formation is largely unknown. The class III PtdIns3K complex, consisting of at least PIK3C3/Vps34, PIK3R4/Vps15 and BECN1/Vps30/Atg6, is essential for the formation of autophagosomes.Citation2 The endoplasmic reticulum (ER)-localized ATG14 is responsible for recruiting the class III PtdIns3K complex to the ER and generating a PtdIns3P-rich ER subdomain, termed the omegasome.Citation4,Citation5,Citation9 PtdIns3P generated by the class III PtdIns3K complex recruits the zinc finger FYVE domain-containing protein 1 (ZFYVE1) and mammalian Atg18 homologs, WIPI1 and WIPI2, to the omegasome, which provides a platform for the accumulation of other ATG proteins.Citation10
PIK3C3 is also required for several other vesicular trafficking processes, including sorting of hydrolytic enzymes to the lysosome/vacuole and early steps in the endocytic pathway. PtdIns3P produced by PIK3C3 results in the endosomal recruitment of FYVE domain-containing proteins, including EEA1, which is necessary for endosomal trafficking.Citation11 RAB5A, a small GTPase localized to early endosomes, regulates the fusion between endocytic vesicles and early endosomes, as well as the homotypic fusion between early endosomes.Citation12 It has been reported that both PIK3C3 and PIK3R4 are RAB5A effectors.Citation13 RAB5A binds directly with PIK3R4 and regulates the localization of the PIK3C3-PIK3R4 complex.Citation13 RAB5A also associates with the BECN1-PIK3C3 complex and promotes autophagosome formation.Citation14 This result is consistent with the findings of a later study indicating that RAB5A and PIK3C3 are involved in NS4B-induced autophagy.Citation15
In our human genomics project, we have cloned hundreds of functionally unknown human open-reading frames (ORFs) by searching the human Refseq and expressed sequence tag (EST) databases in GenBank. Using a cell-based functional screening platform based on an automated fluorescence microscopy system, which enables acquiring and quantitatively analyzing images of LC3 signals in cotransfected cells,Citation16 we identified several novel genes associated with cell autophagy, including EMC6 (also known as transmembrane protein 93, TMEM93). Here, we report the identification and characterization of EMC6 as a novel ER-localized protein, which interacts with RAB5A and BECN1, facilitates RAB5A ER localization, and modulates autophagosome formation.
Results
Bioinformatic analysis and expression profile of human EMC6
Transcription of the human EMC6 gene,which is located on chromosome 17p13.2, produces two different mRNAs (EMC6-v1 and EMC6-v2), both of which encompass two exons and one intron and share the same ORF (). The full-length EMC6-v1 and EMC6-v2 are 805 and 693 base pairs long, respectively. The ORF encodes a predicted 12.2 kDa protein of 110 amino acids with an isoelectric point of 10.08. The full-length cDNA and predicted amino acids are shown in . EMC6 is conserved in cow, mouse, chicken, zebrafish and Xenopus (). Transmembrane (TM) analysis (www.cbs.dtu.dk/services/TMHMM-2.0/) suggests that there are two conserved TM domains near the C-terminal end of the protein (, dashed lines). A functional domain (www.ebi.ac.uk/Tools/InterProScan/) search suggests that EMC6 is a potential member of the RAB5A interacting protein (RAB5AIP) superfamily. To our knowledge, no functional studies have been performed on this hypothetical gene.
Figure 1. Identification, sequence analysis and expression profile of EMC6. (A) Schematic of gene and cDNA structures of EMC6. There are two validated transcript variants of EMC6, and EMC v2 was cloned by RT-PCR. The boxes show the exons of these two variants with their relative sizes and positions in the EMC6 gene. (B) Nucleotide sequence and predicted amino acid sequences of human EMC6. The sequence eliminated in EMC6-v2 is boxed. The start and stop codons are underlined. Sequences of the two peptides used for generating the polyclonal antibody against EMC6 are highlighted in gray. The putative TM domains are indicated with dashed lines. (C) Phylogenetic analysis of EMC6. (D) Schematic representation of EMC6 and constructs used in this study. (E) EMC6 mRNA expression was analyzed by RT-PCR in human normal tissues and cell lines (F). GAPDH expression was amplified as an internal control.

The presence of the EMC6 mRNA was confirmed by RT-PCR analysis in a variety of normal human tissues, including pancreas, kidney, heart, liver, skeletal muscle, spleen, thymus, small intestine, colon, prostate, testis, peripheral blood lymphocytes (PBL), lung, placenta and ovary (). EMC6 mRNA was also detected in various cell lines by RT-PCR (). For subsequent experiments, a rabbit anti-EMC6 specific antibody was prepared by using chemically synthesized EMC6 peptides (, gray highlighted sequence) and validated by western blot and immunofluorescence (Fig. S1A and S1B).
EMC6 is localized in the ER
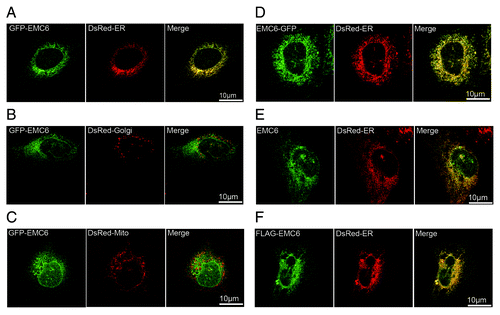
To examine the subcellular localization of EMC6, U2OS cells were cotransfected with GFP-EMC6 and DsRed-ER, DsRed-Golgi or DsRed-Mito plasmids and analyzed by confocal microscopy. GFP-EMC6 primarily colocalized with DsRed-ER (), but not with DsRed-Golgi () or DsRed-Mito (). The C-terminal GFP-fused EMC6 (EMC6-GFP) also colocalized with DsRed-ER (). Using indirect immunofluorescence, we confirmed that overexpressed EMC6 and N-terminal FLAG-tagged EMC6 (FLAG-EMC6) colocalized with DsRed-ER (). Together, these results indicate that EMC6 is localized in the ER.
Figure 2. EMC6 is localized in the ER. Confocal microscopy images are shown of U2OS cells: cotransfected with GFP-EMC6 and DsRed-ER (A), DsRed-Golgi (B) or DsRed-Mito (C); cotransfected with EMC6-GFP and DsRed-ER (D); cotransfected with EMC6 and DsRed-ER and then immunostained with an anti-EMC6 antibody after 24 h (E); cotransfected with FLAG-EMC6 and DsRed-ER and then immunostained with an anti-FLAG antibody after 24 h (F).
EMC6 regulates autophagy
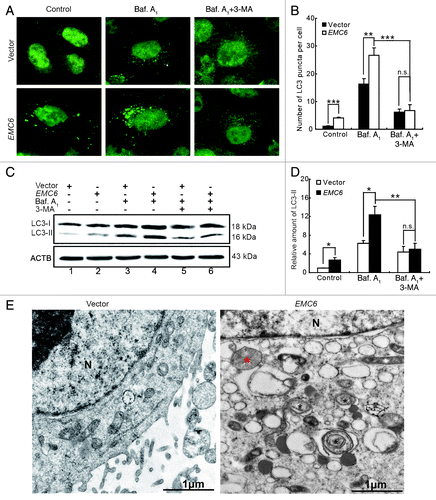
EMC6 was originally identified as an autophagy-related gene through a high-throughput, cell-based functional screening.Citation16 We then confirmed that overexpression of EMC6 resulted in punctate distribution of GFP-LC3 by fluorescence microscopy analysis (Fig. S2A and S2B). Western blot analysis revealed that overexpression of EMC6 also increased the level of GFP-LC3-II and free GFP in GFP-LC3 transfected cells (Fig. S2C). It is proposed that free GFP fragments result from the degradation of GFP-LC3 within autolysosomes; thus, an increased level of free GFP fragments indicated that EMC6 may promote autophagy flux. To further study the role of EMC6 in cell autophagy, bafilomycin A1, a lysosome inhibitor, was employed to monitor the autophagy flux in our studies. 3-MA, a PIK3C3 inhibitor was employed to indicate whether EMC6 acts upstream or downstream of the class III PtdIns3K complex. It was confirmed that the overexpression of EMC6 increased dot distribution of endogenous LC3 by immunofluorescence staining and fluorescence microscopy analysis, in contrast to the diffuse pattern in control cells (, left). Bafilomycin A1 treatment caused accumulation of the LC3 dots in both EMC6 and vector-transfected cells, but the abundance of LC3 dots in EMC6-transfected cells was much greater than that in vector-transfected cells (, middle). 3-MA treatment decreased the accumulation of LC3 dots in both EMC6 and vector-transfected cells in the presence of bafilomycin A1 (, right). We further analyzed the level of the membrane-bound LC3-phospholipid conjugate LC3-II by western blot. The LC3-II band was weak in both the EMC6 and vector groups, while it seemed more distinct in the EMC6 group (, lane 2 vs. lane 1). Bafilomycin A1 treatment caused accumulation of LC3-II in both EMC6 and vector-transfected cells, and the LC3-II band of the EMC6 group was much stronger than that of the vector group (, lane 4 vs. lane 3). 3-MA treatment decreased the accumulation of LC3-II in both EMC6 and vector-transfected cells treated with bafilomycin A1 (, lanes 5 and 6). Transmission electron microscopy (TEM) analysis revealed that EMC6 overexpression resulted in the appearance of autophagosome-like structures, unlike in the vector-transfected cells (). These results indicated that EMC6 overexpression could induce autophagy, which may involve the activation of the class III PtdIns3K complex.
Figure 3. EMC6 overexpression promotes cell autophagy. (A) Representative fluorescence microscopy images of the dot distribution of endogenous LC3 obtained from U2OS cells transfected with vector or EMC6 for 18 h, which were treated with 10 nM bafilomycin A1 (Baf. A1) for the last 4 h and/or 10 mM 3-MA for the last 6 h. (B) Quantification of LC3 dots in control or EMC6-overexpressing cells treated with reagents as indicated in (A). Data are means ± SD of at least 100 cells scored (**p < 0.01, ***p < 0.001, N.S: not significant). (C) Western blot analysis of endogenous LC3-II levels in U2OS cells treated as in (A). (D) Quantification of the amounts of LC3-II relative to ACTB treated as in (C). The average value in the vector-transfected cells without Baf. A1 treatment was normalized as 1. Data are the means ± SD of results from three experiments (*p < 0.05, **p < 0.01, N.S: not significant). (E) TEM analysis of U2OS cells transfected with vector or EMC6. Note the autophagosome-like structure (black star) and autolysosome-like structure (red star) in EMC6-overexpressing cells.
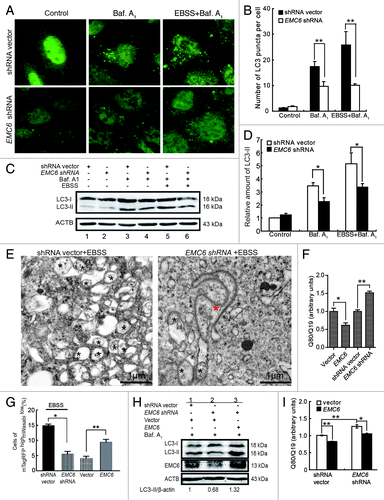
To confirm the essential role of EMC6 in autophagy, further analysis was performed in EMC6-silenced U2OS cells. The shRNA against EMC6 (EMC6 shRNA) was demonstrated to be effective by both RT-PCR and western blot (Fig. S3). We then examined the dot distribution of endogenous LC3 in EMC6-silenced cells. It was found that the accumulation of LC3 dots caused by either bafilomycin A1 alone or together with EBSS was hindered in EMC6-silenced cells compared with control cells (, middle and right). Western blot analysis also revealed that the accumulation of LC3-II caused by bafilomycin A1 alone or together with EBSS was decreased in EMC6-silenced cells compared with control cells (, lane 4 vs. lane 3 and lane 6 vs. lane 5). We further performed TEM analysis to confirm the impact of EMC6 knockdown on autophagy. The level of autophagosome induction by EBSS treatment in the EMC6-silenced cells was significantly lower than that in the control cells, most EMC6-silenced cells displayed anomalous double-membrane structures that looked like enlarged autophagosomal precursors still connected to the ER (, red star). These results implied that the knockdown of EMC6 may block autophagosome formation and then cause accumulation of autophagosomal precursors.
Figure 4. Knockdown of EMC6 impairs cell autophagy. (A) Representative fluorescence microscopy images of the dot distribution of endogenous LC3 obtained from U2OS cells transfected with shRNA vector or EMC6 shRNA, which were cultured for 30 h and treated with 10 nM bafilomycin A1 (Baf. A1) and/or EBSS for the last 4 h. (B) Quantification of LC3 dots in control or EMC6-silenced cells treated with reagents as indicated in (A). Results are means ± SD of at least 100 cells scored (**p < 0.01). (C) Western blot analysis of LC3-II levels in U2OS cells treated as in (A). (D) Quantification of the amounts of LC3-II relative to ACTB treated as in (C). The average value in the shRNA vector-transfected cells without Baf. A1 treatment was normalized as 1. Data are the means ± SD of results from three experiments (*p < 0.05). (E) TEM analysis of U2OS cells transfected with shRNA vector or EMC6 shRNA for 24 h and then incubated in EBSS for 2 h. The autophagosome-like structures are shown with a black star, and an anomalous double-membrane structure is shown with a red star. (F) U2OS cells were cotransfected with polyQ80-luciferase (or polyQ19-luciferase) and the indicated plasmids for 24 h; firefly luciferase activity was measured using the Dual Luciferase Reporter System (means ± SD; *p < 0.05, **p < 0.01). (G) HCT116 cells were cotransfected with mTagRFP-mWasabi-LC3 and the indicated plasmids for 24 h, and then incubated with or without EBSS for 4 h. The percentages of mTagRFPhighmWasabilow cells were detected using flow cytometry (means ± SD; *p < 0.05, **p < 0.01). (H) HCT116 cells were cotransfected with the indicated plasmids for 24 h, and then treated with Baf. A1 for 4 h. LC3-II and EMC6 levels were analyzed by western blot. (I) HCT116 cells were cotransfected with polyQ80-luciferase (or control polyQ19-luciferase), vector (or EMC6) and shRNA vector (or EMC6 shRNA) as indicated for 24 h, and then firefly luciferase activaty was measured using the Dual-Luciferase Reporter System (means ± SD; *p < 0.05, **p < 0.01).
To further investigate the regulation of autophagy by EMC6, we examined degradation of polyQ aggregates formed by a stretch of 80 glutamine residues (polyQ80) known to be an autophagy substrate.Citation17-Citation19 Degradation of polyQ19 was also monitored and served as an internal control. The results showed that degradation of polyQ was impaired in EMC6-silenced cells and enhanced in EMC6-overexpressing cells (), indicating that EMC6 is required for autophagic degradation of cellular components. We further employed mTagRFP-mWasabi-tandemly tagged LC3 (mTagRFP-mWasabi-LC3) to monitor autophagosome-lysosome fusion.Citation20 mWasabi is more sensitive to acidic pH than mTagRFP, which retains fluorescence even at acidic pH. Therefore, colocalization of mWasabi and mTagRFP fluorescent signals indicates a compartment that has not fused with a lysosome, such as the phagophore or an autophagosome. In contrast, a strong mTagRFP signal with absent or low mWasabi corresponds to an amphisome or autolysosome. In this study, the cells were cotransfected with mTagRFP-mWasabi-LC3 and shRNA vector or EMC6 shRNA, then treated with EBSS for 4 h and analyzed using flow cytometry. As shown in , the percentage of mTagRFPHighmWasabiLow cells was significantly decreased in cells transfected with EMC6 shRNA, compared with control cells (p < 0.05). The cells cotransfected with mTagRFP-mWasabi-LC3 and vector or EMC6 expression plasmid were also analyzed; it was shown that the percentage of mTagRFPHighmWasabi-LC3Low cells was significantly increased in EMC6 overexpressing cells (). These observations indicated that autophagic flux was impaired in EMC6-silenced cells and promoted in EMC6-overexpressing cells.
We also performed a rescue experiment by cotransfection of the EMC6 expression plasmid with EMC6 shRNA. HCT116 cells were transfected as indicated in and then treated with bafilomycin A1. Western blot analysis showed that the level of endogenous LC3-II was significantly decreased (, lane2) in cells transfected with EMC6 shRNA. Simultaneously, those cells overexpressing EMC6 exhibited increased accumulation of endogenous LC3-II (, lane3). The polyQ80 degradation assay further demonstrated that EMC6 rescued the EMC6 shRNA-induced inactivation of autophagy and disabled degradation of polyQ80 (), indicating that endogenous EMC6 is required for autophagy.
Knockdown of EMC6 impairs autophagosome formation
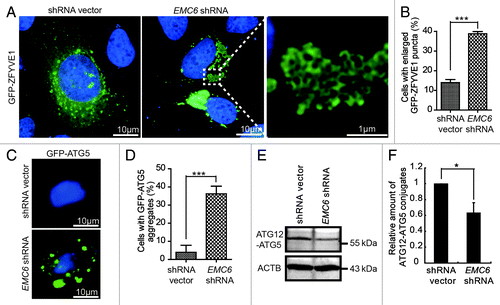
To further investigate the step at which the autophagy process is interrupted in EMC6-silenced cells, we examined the formation of omegasomes, which can be specifically labeled by the ER-associated PtdIns3P-binding protein ZFYVE1. U2OS cells were cotransfected with GFP-ZFYVE1 and EMC6 shRNA or shRNA vectors and observed under a fluorescence microscope. Compared with shRNA vector transfected cells, EMC6 shRNA-transfected cells displayed abnormally enlarged GFP-ZFYVE1-labeled structures, which seemed to result from the aggregation of small GFP-ZFYVE1-labeled vesicles (, right). The percentage of cells with enlarged GFP-ZFYVE1 structures in EMC6 shRNA-transfected cells was significantly higher than that in control cells (, ***p < 0.001). Furthermore, those enlarged GFP-ZFYVE1-labeled structures were partially colocalized with the ER (Fig. S4), suggesting that they may be caused by accumulation of omegasomes. We then detected the formation of ATG5-labeled membrane structures, which were also considered as autophagosomal precursors. As shown in , a larger number of membrane-binding GFP-ATG5 structures could be observed in cells transfected with EMC6 shRNA but not in control cells. The percentage of cells with GFP-ATG5 aggregates in EMC6 shRNA transfected cells was significantly higher than that in control cells (, ***p < 0.001). Subsequently, we analyzed the level of ATG12–ATG5 conjugation in U2OS cells transfected with EMC6 shRNA or shRNA vector. As shown in , compared with control cells, the levels of conjugated ATG12–ATG5 were decreased in EMC6 shRNA-transfected cells, indicating that the ATG12–ATG5 conjugation essential for autophagosome formation was impaired in EMC6-silenced cells. Taken together, these results indicate that EMC6 may regulate autophagosome formation.
Figure 5. Knockdown of EMC6 impairs autophagosome formation. (A) U2OS cells cotransfected with GFP-ZFYVE1 and shRNA vectors or EMC6 shRNA for 24 h were observed under a fluorescence microscope. Nuclei were stained with H33342. (B) Percentages of cells with enlarged GFP-ZFYVE1 structures of the total GFP-ZFYVE1 cells were quantified (means ± SD, ***p < 0.001). (C) U2OS cells cotransfected with a plasmid expressing GFP-ATG5 and shRNA vectors or EMC6 shRNA for 24 h were extracted using 0.02% saponin and fixed with 4% paraformaldehyde. Nuclei were stained with H33342. Cells were observed under a fluorescence microscope. (D) Percentage of cells with accumulated GFP-ATG5 structures was quantified by scoring at least five random fields (means ± SD, ***p < 0.001). (E) Western blot analysis of ATG12–ATG5 conjugates in U2OS cells transfected with shRNA vectors or EMC6 shRNA for 24 h. (F) Quantification of the amounts of ATG12–ATG5 conjugates relative to ACTB. The average value in the shRNA vector-transfected cells was normalized as 1. Data are the means ± SD of results from three experiments (*p < 0.05).
EMC6 is associated with RAB5A and BECN1 in the autophagosomal membrane
To determine whether EMC6 is a component of the autophagosomal membrane, we analyzed the colocalization of overexpressed EMC6 with GFP-ZFYVE1 () and FLAG-EMC6 with GFP-BECN1 () by confocal microscopy. In addition, the N-terminal GFP-BECN1 could be pulled down with glutathione S-transferase (GST)-EMC6 from whole cell lysates (, lane 3). These results suggest that EMC6 may associate with the BECN1 complex localized on the autophagosomal membrane.
Figure 6. EMC6 is associated with the RAB5A-BECN1 complex. (A, B, D, E and H) Direct or indirect immunofluorescence analysis by confocal microscopy was performed on cells, and nuclei were stained with H33342 as indicated. (A) U2OS cells were cotransfected with EMC6- and GFP-ZFYVE1-expressing plasmids for 24 h and then stained with an anti-EMC6 antibody. (B) U2OS cells were cotransfected with plasmids expressing FLAG-EMC6 and GFP-BECN1 for 24 h and stained with an anti-FLAG antibody. (C) GST-EMC6 fusion protein and the GST protein immobilized on Glutathione-Sepharose beads were incubated with GFP-BECN1-transfected HCT116 cell lysates at 4°C for 4 h. GFP and GST were detected in the washed beads by western blot. (D) U2OS cells were cotransfected with plasmids expressing FLAG-EMC6 and GFP-RAB5A for 24 h and then stained with an anti-FLAG antibody. (E) U2OS cells were cotransfected with plasmids expressing EMC6-GFP and DsRed-RAB5A for 24 h. (F) HCT116 cells were transfected with a plasmid expressing FLAG-EMC6 for 24 h. Total cell extracts were subjected to IP using either an anti-FLAG or a nonspecific control mIgG, as indicated. RAB5A was detected in the IP proteins by western blot. (G) GST-EMC6 fusion protein and the GST protein immobilized on Glutathione-Sepharose beads were incubated with GFP-RAB5A-transfected HCT116 cell lysates at 4°C for 4 h. GFP and GST were detected in the washed beads by western blot. (H) The GST-EMC6 fusion protein or the GST protein immobilized on Glutathione-Sepharose beads were incubated with His-RAB5A or His-RAB5A loaded with GDP or GTPγS. His-RAB5A and GST were detected in the washed beads by western blot. (I) U2OS cells were transfected with FLAG-EMC6 for 24 h and then stained with anti-EMC6 or anti-EEA1 antibody.
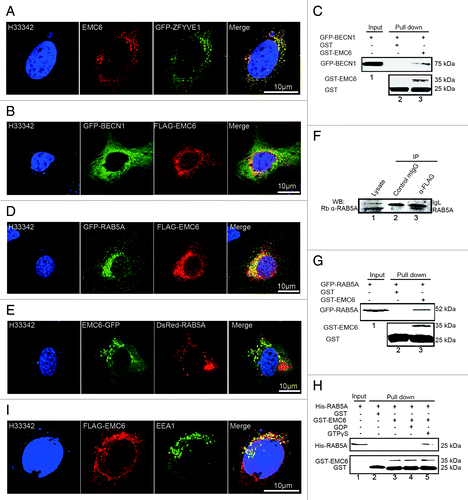
EMC6 was predicted to interact with RAB5A by bioinformatic analysis. To verify this association, we also observed the colocalization of FLAG-EMC6 with N-terminal GFP-fused RAB5A (GFP-RAB5A) () and C-terminal GFP-fused EMC6 (EMC6-GFP) with DsRed-RAB5A () by confocal microscopy. Co-immunoprecipitation (Co-IP) and GST-pull-down analysis were also applied to confirm the association of EMC6 and RAB5A in mammalian cells. U2OS cells were transfected with FLAG-EMC6, lysed and then subjected to immunoprecipitation (IP) with an anti-FLAG antibody. Western blot analysis revealed that endogenous RAB5A coprecipitated with FLAG-EMC6 (, lane 3). This interaction was considered specific, since the control IgG failed to bind RAB5A (, lane 2). The interaction between EMC6 and RAB5A could also be observed by using the pull-down approach (). In vitro binding study was also performed with recombinant proteins, and it was shown that GST-EMC6 could bind His-RAB5A loaded with GTPγS but not that loaded with GDP or His-RAB5A alone (), indicating that EMC6 interacts directly with activated RAB5A. We also captured the colocalization of FLAG-EMC6 with another early endosome-localized protein EEA1 (), suggesting that EMC6 may interact with early endosome-localized RAB5A.
EMC6 modulates the ER localization of RAB5A and EEA1
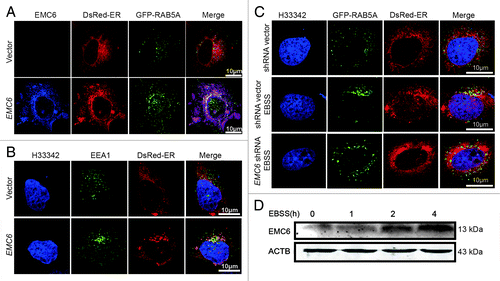
Although RAB5A is an important regulator of the endocytic pathway, it was reported to modulate aggregation and toxicity of a polyglutamine expansion mutant through macroautophagy, as well as suggested to modulate the formation of autophagosomes via regulating PIK3C3 activity.Citation14 However, it is widely thought that ER localization of the class III PtdIns3K complex is essential for autophagy. This idea is supported by the ER localization of ATG14, an essential regulator of autophagosome formation, which interacts with BECN1.Citation4,Citation9 Thus, to exert its positive role in autophagy, RAB5A may need to be in close proximity to the ER in order to activate PIK3C3. In this study, we demonstrated that EMC6 regulates autophagy by localizing to the ER and associating with RAB5A and BECN1. Hence, it is reasonable to assume that EMC6 may be involved in recruiting RAB5A to the ER.
To confirm the above assumption, we monitored the localization of RAB5A and EEA1 by confocal microscopy. In cells cotransfected with the vector, GFP-RAB5A and DsRed-ER, no obvious colocalization of GFP-RAB5A and DsRed-ER was observed (, upper panel). In cells cotransfected with the EMC6-expressing plasmid, GFP-RAB5A and DsRed-ER, most cells displayed apparent colocalization of GFP-RAB5A, EMC6 and DsRed-ER near the nucleus (, lower panel). We also found that unlike the control cells (, upper panel), most EMC6 overxpressing cells exhibited obvious colocalization of EEA1 with DsRed-ER (, lower panel). These results suggest that EMC6 may regulate the recruitment of RAB5A and EEA1 to the ER or bridge the contacts between the ER and early endosomes. The colocalization of GFP-RAB5A and DsRed-ER was also observed in cells under starvation conditions (, middle panel), but not in control cells (, upper panel), coincident with the upregulation of EMC6 protein in starved cells (; Fig. S5A and S5B). Furthermore, starvation-induced colocalization of GFP-RAB5A and DsRed-ER was not observed in EMC6-silenced cells (, lower panel), suggesting that the defective autophagy in EMC6-deficient cells may be caused by the absence of ER-localized RAB5A. We also observed that EMC6 protein was upregulated in the presence of rapamycin (Fig. S5C and S5D). It was noted that the upregulation of EMC6 induced by both starvation and rapamycin was not sensitive to wortmannin (Fig. S5), indicating that it is upstream of the PtdIns3P requirement. This conclusion is coincident with the observation that EMC6 overexpression-induced autophagy is sensitive to 3-MA.
Figure 7. EMC6 modulates the ER localization of RAB5A and EEA1. (A–C) Indirect immunofluorescence analysis by confocal microscopy was performed on cells, and nuclei were stained with H33342 as indicated. (A) U2OS cells were cotransfected with vector (or EMC6), and plasmids expressing GFP-RAB5A and DsRed-ER at a ratio of 3:1:1 for 24 h, stained with anti-EMC6 antibody and APC-conjugated anti-rabbit secondary antibodies. (B) U2OS cells were cotransfected with vector (or EMC6) and a plasmid expressing DsRed-ER at a ratio of 3:1 for 24 h, stained with anti-EEA1 antibody and FITC-conjugated anti-rabbit secondary antibodies. (C) U2OS cells were cotransfected with EMC6 shRNA (or shRNA vector), and plasmids expressing GFP-RAB5A and DsRed-ER at a ratio of 3:1:1 for 24 h and then incubated with or without EBSS for 2 h. (D) U2OS cells were incubated in EBSS for the indicated time, and EMC6 was detected by western blot.
EMC6ΔTM suppresses cell autophagy
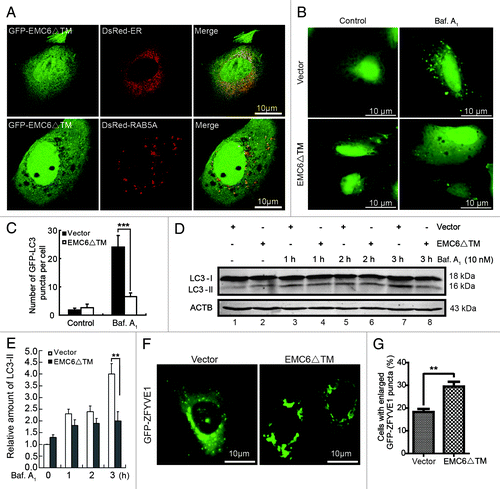
As mentioned above, EMC6 is predicted to be a membrane protein with two TM regions at the C terminus. Thus, the C-terminal region of EMC6 may be essential for its ER localization, and the N terminus may play a role in binding RAB5A. We constructed a truncated mutant of EMC6 (EMC6ΔTM) in which 65 C-terminal amino acids were deleted () and then investigated its location and function. U2OS cells were cotransfected with GFP-EMC6ΔTM and DsRed-ER or DsRed-RAB5A. Instead of localizing to the ER, GFP-EMC6ΔTM displayed a diffuse cytoplasmic expression pattern in the confocal images (, upper panel), indicating that the C-terminal TM domain of EMC6 is required for its specific localization. However, GFP-EMC6ΔTM distinctly colocalized with DsRed-RAB5A (, lower panel), suggesting that the N terminus of EMC6 may play a role in binding RAB5A. We also observed that the accumulation of LC3 dots caused by bafilomycin A1 was hindered in EMC6ΔTM-expressing cells compared with control cells (). Western blot analysis revealed that the rate of LC3-II accumulation slowed in EMC6ΔTM-overexpressing cells (, lane 4 vs. lane 3 and lane 6 vs. lane 5) in the presence of bafilomycin A1. After 3 h of bafilomycin A1 treatment, EMC6ΔTM overexpressing cells showed a notable downregulation in LC3-II accumulation compared with control cells (, lane 8 vs. lane 7). Furthermore, we cotransfected GFP-ZFYVE1 and EMC6ΔTM or vector plasmids into U2OS cells and analyzed the distribution of GFP-ZFYVE1. As shown in , unlike the control vector group, cotransfection of EMC6ΔTM resulted in enlarged ZFYVE1 structures similar to those observed in EMC6-silenced cells. The percentage of cells with enlarged GFP-ZFYVE1 structures was significantly higher in EMC6ΔTM transfected cells than that in control cells (). These results indicate that the putative C-terminal TM domain of EMC6 is necessary for the localization and biological function of EMC6, and EMC6ΔTM may function as an antagonist of EMC6 to inhibit autophagy activity.
Figure 8. EMC6ΔTM suppresses cell autophagy. (A) U2OS cells were cotransfected with plasmids expressing GFP-EMC6ΔTM and DsRed-ER or DsRed-RAB5A for 24 h and observed by confocal microscopy. (B) Representative fluorescence microscopy images obtained from U2OS cells cotransfected with GFP-LC3 and vector or EMC6ΔTM for 30 h and treated with or without 10 nM bafilomycin A1 (Baf. A1) for the last 4 h. (C) Quantification of GFP-LC3 dots in control or EMC6ΔTM-expressing cells treated with reagents as indicated in (B). Results are means ± SD of at least 100 cells scored (***p < 0.001). (D) U2OS cells were transfected with vector or EMC6ΔTM for 24 h, and incubated with or without 10 nM Baf. A1 for the indicated times. The levels of endogenous LC3-I and LC3-II were analyzed by western blot. (E) Quantification of the amounts of LC3-II relative to ACTB treated as in (D). The average value in the vector-transfected cells without Baf. A1 treatment was normalized as 1. Data are the means ± SD of results from three experiments (**p < 0.01). (F) U2OS cells were cotransfected with GFP-ZFYVE1 and EMC6ΔTM or vector for 24 h and observed by fluorescence microscopy. (G) Numbers of cells with enlarged GFP-ZFYVE1 structures were quantified (means ± SD) by scoring at least five random fields. **p < 0.01.
Discussion
Macroautophagy is initiated by the formation of phagophores. Although it has been studied for several decades, the origin of the autophagic membrane remains unclear. Several investigations have indicated that the phagophore is unique, with an autophagosomal membrane that is relatively protein poor with a lack of known organelle markers.Citation21-Citation23 Meanwhile, contradictory data from several other laboratories support the notion that the phagophore, while being unique, may arise from the ER or Golgi complex.Citation24 Gratifyingly, the subcellular localization of several Atg proteins provides us with more information about the source of the autophagic membrane. Atg9 is the only membrane-associated Atg protein described thus far that is absolutely required for autophagosome formation, and it has not been well studied. However, Atg9 knockout mice are unable to survive after birth,Citation25 and the identical phenotype has been found in mice lacking Atg5, Atg7 and Atg3, suggesting a crucial role of ATG9 in autophagy. In yeast, Atg9 cycles between the PAS and a peripheral pool consisting of clusters of vesicles and tubules.Citation26 Mammalian ATG9 transits between the endosome and Golgi compartment in normal cultured cells, while it delocalizes to a peripheral compartment after starvation.Citation27 These results support a role of the Golgi and/or endosomal compartment in phagophore formation.
PtdIns3P production is essential for autophagosome formation.Citation28 Upon starvation, PtdIns3P production is induced in cellular compartments dynamically connected to the ER, which is thought not to normally have high levels of PtdIns3P.Citation29 ATG14, a component of the class III PtdIns3K complex is essential for PtdIns3P synthesis and autophagosome formation, as ATG14 knockdown results in disappearance of the omegasome.Citation10 It has been shown that ATG14 localizes to the ER as well as the phagophore, and that the ER localization of ATG14 is essential for its function in regulating autophagosome formation,Citation10,Citation30 indicating that the ER may be the site of phagophore formation. This idea is further supported by the localization of a non-ATG protein, ZFYVE1, which binds PtdIns3P through its FYVE domains.Citation29 ZFYVE1 is associated with the Golgi complex in normal cultured cells and translocates to an ER-associated membrane (omegasome) upon starvation. The ATG5 complex, LC3, ATG14 and WIPI2, have all been observed to be recruited to the omegasome, suggesting that the omegasome may function as a platform for autophagosome formation.Citation29
In contrast to the idea that the phagophore originates from the ER, a recent study shows that ATG5 and LC3 colocalize with the mitochondria and that a protein anchored in the outer leaflet of mitochondria can be found on forming autophagosomes.Citation31 Thus, the true origin of the autophogic membrane remains to be determined.
Here we have demonstrated that EMC6, a newly identified ER-localized transmembrane protein, was essential for autophagy progression, which supports the idea that the ER may provide a platform for phagophore formation. We also found that a mutated EMC6 was unable to localize to the ER and did not induce autophagosomes but instead hindered autophagosome formation, indicating that ER localization of EMC6 was essential for its ability to regulate autophagy and EMC6ΔTM may function as an antagonist of EMC6. In this study, we observed the accumulation of LC3-labeled structures, ATG5 structures, unconventional ZFYVE1 structures and anomalous double-membrane structures that looked like enlarged autophagosomal precursors in EMC6-silenced cells. The accumulation of ATG5 structures was also observed in RAB5A-inhibited and PIK3C3-defective cells.Citation14 EMC6 seemed to function upstream of the class III PtdIns3K complex, as EMC6 overexpression-induced autophagy was sensitive to the PIK3C3 inhibitor 3-MA, while EBSS-and rapamycin-induced upregulation of EMC6 was insensitive to the PIK3C3 inhibitor wortmannin. Thus, a defect of EMC6 function may result in inactivation of the class III PtdIns3K complex and then impaired autophagosome formation.
RAB5A is a small GTPase localized to early endosomes, which regulates endocytic traffic. It was also shown to associate with the BECN1-PIK3C3 complex and promote autophagosome formation.Citation14 This observation is consistent with the findings of an earlier study reporting that RAB5A interacts with PIK3C3 and modulates its activity.Citation13 Even so, the question remains as to how RAB5A interacts with the ER-localized BECN1-PIK3C3 complex.
The ER is a large, singular, membrane-bound organelle, which performs a variety of functions in eukaryotic cells. It is in contact with almost every membrane-bound organelle, including mitochondria, endosomes, Golgi and peroxisomes.Citation32 Recent work has illustrated the direct contacts between ER and early endosomes. It was shown that the ER-localized protein tyrosine phosphatase PTP1B interacts directly with the endocytic cargo EGFR at the ER-endosome contact sites.Citation33 However, the factors mediating ER-endosome contact and the functions of these contacts are still unknown. Here we demonstrated that EMC6, a newly identified ER-localized transmembrane protein, could interact with RAB5A and modulate the ER localization of RAB5A and EEA1. We also found that EMC6 colocalized with BECN1 and played an essential role in the autophagy pathway. EMC6 controls the degradation of polyQ80, which is known as an autophagy substrate. EMC6 silencing also resulted in the accumulation of ATG5 structures and unconventional enlarged ZFYVE1 structures. All these results were consistent with the phenotype observed in RAB5A-inhibited and PIK3C3-defective cells. There are two possible roles of EMC6 in autophagy regulation: on the one hand, EMC6 may regulate the recruitment of RAB5A to the ER and then facilitate the interaction between RAB5A and PIK3C3, thereupon promoting autophagosome formation; the recruitment of EEA1 to the ER may be mediated by the PtdIns3P produced in the omegasome membrane. On the other hand, the interaction between EMC6 and RAB5A may bridge contacts between the ER and early endosomes, therefore leading to the recruitment of the PIK3C3-PIK3R4 complex originally located on the early endosome membrane; such a complex may further interact with BECN1 and ATG14 located on the ER membrane and form another stable complex contributing to autophagosome formation. Altogether, our findings provide new insights into the regulation of autophagy.
Materials and Methods
Antibodies and reagents
The specific polyclonal antibody against EMC6 was prepared by immunizing rabbits with chemically synthesized EMC6 peptides (, gray highlighted sequences), purified by peptide affinity chromatography via CNBr-activated SepharoseTM 4 Fast Flow (GE Healthcare, 17-0981-01) according to the manufacturer’s instructions. Other antibodies used in this study were: anti-LC3B (Sigma Aldrich, L7543), anti-EEA1 (Cell Signaling Technology, 3288), anti-ATG5 (Cell Signaling Technology, 8540), anti-FLAG (Sigma Aldrich, F3165), anti-β-actin/ACTB (Sigma Aldrich, A5316), anti-RAB5A (Santa Cruz, sc-309) and anti-GST (Santa Cruz, sc-459) antibodies; IRDye 800-conjugated anti-GFP antibody (Rockland, 600-432-215), DyLight 800/DyLight 680-conjugated secondary antibodies against mouse (Rockland, 610-145-002/610-144-002) or rabbit (Rockland, 611-145-002/611-144-002) IgG; FITC/RBITC/APC-conjugated secondary antibodies against mouse (Bioss Inc., bs-0296G-FITC/bs-0296G-RBITC/bs-0296G-APC) or rabbit (Bioss Inc., bs-0295G-FITC/bs-0295G-RBITC/bs-0295G-APC) IgG. Other reagents used in this study were: cDNA libraries of human normal adult tissues (Clontech, 636742), The Dual-Luciferase® Reporter (DLR™) Assay System (Promega, E1910), guanosine gamma thio-phosphate (GTPγS, Sigma, G8634), bafilomycin A1 (Sigma Aldrich, B1793), wortmannin (Sigma Aldrich, W1628), Hoechst 33342 (Sigma Aldrich, 14533), saponin (Sigma Aldrich, S7900) and 3-methyladenine (3-MA, Sigma Aldrich, M9281).
Plasmid construction
The EMC6 cDNA was amplified from a human kidney cDNA library (Clontech, 637204) by PCR with the forward primer P1 (5′-CGTCTGAGGGAACGCTAAGT-3′) and reverse primer P2 (5′- GAATGAGGAGCAGGGAGAG -3′). The insert was released by EcoRI and subcloned into the EcoRI site of pcDNA.3.1/myc-His (-) B (Invitrogen, V85520) to construct the pcDB-EMC6 plasmid, abbreviated EMC6 in this study. Based on this plasmid, we constructed the following plasmids in succession: FLAG-EMC6, GFP-C3-EMC6 (abbreviated GFP-EMC6), EMC6-N1-GFP (abbreviated EMC6-GFP), GST-EMC6, EMC6ΔTM and GFP-EMC6ΔTM (see for details). GFP-BECN1, GFP-RAB5A, DsRed-RAB5A, GFP-ZFYVE1 and FLAG-ATG12 plasmids were also constructed in our laboratory. All plasmids were confirmed by DNA sequencing.
DsRed-ER, DsRed-Golgi and DsRed-Mito plasmids were kindly provided by Quan Chen (Chinese Academy of Sciences, China). The GFP-ATG5 plasmid was a gift from Weiguo Zhu (Peking University, China). The mTagRFP-mWasabi-LC3 plasmid was kindly provided by Jian Lin (Peking University, China). The PolyQ80-luciferase and polyQ19-luciferase plasmids were kindly provided by Conrad C. Weihl (Washington University School of Medicine, USA). Specific shRNA mediating EMC6 gene knockdown with the targeting sequence, 5′-GCCTCTTCACCTACGTCCTGTTCTGGACG-3′, and nonsilencing shRNA vector were constructed by ORIGEN Corporation. The shRNA vector sequence with no sequence homology to any known human gene was used as the control.
Cell culture, transfections and treatments
U2OS and HCT116 cell lines were maintained in DMEM (Invitrogen, 12800-017) supplemented with 10% fetal bovine serum (FBS). Other cell lines used for RT-PCR were cultured routinely in our laboratory. Cells were transfected using MegaTran 1.0 Transfection Reagent (ORIGEN, TT200004) according to the manufacturer’s instruction. All experiments were performed on logarithmically growing cells. Cell autophagy was induced by nutrient deprivation, through incubation in Earle’s balanced salt solution (EBSS, Sigma Aldrich, E7510). Autophagy inhibition was achieved by treating cells with 10 nM of bafilomycin A1, a vacuolar-type H+-ATPase inhibitor, which can block the fusion of autophagosomes with lysosomes.
RT-PCR assay
Different cell lines were cultured to the exponential phase, and total RNA samples were extracted with the TRIzol reagent (Invitrogen, 15596-026). RT-PCR was performed using the ThermoScript RT-PCR System (Invitrogen, 11146-024). Primers used for amplifying EMC6 were 5′-CGTCTGAGGGAACGCTAAGT-3′ and 5′- GAATGAGGAGCAGGGAGAG -3′.
Poly Q degradation assay
U2OS cells were cotransfected with polyQ80-luciferase (or control polyQ19-luciferase) constructs and indicated plasmids using MegaTran 1.0 Transfection Reagent according to the manufacturer’s protocol. Twenty-four h later, firefly luciferase activity was measured using the Turner Biosystems Veritas Microplate Luminometer (Bio-Rad) by dispensing Luciferase Assay Buffer II (LAR II) from the Dual-Luciferase® Reporter Assay System according to the manufacturer’s protocol.
Immunofluorescence, fluorescence and confocal microscopy
U2OS cells were cultured in confocal dishes and treated as indicated, fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. The dishes were then incubated with FBS overnight and exposed to primary antibody for 1 h at 4°. After being washed three times with PBS, the dishes were immersed with FITC/RBITC/APC-conjugated secondary antibody solution. Nuclei were stained with Hoechst 33342 (H33342). Morphological alterations in the cells were observed and documented with an Olympus FV1000 confocal microscope (Olympus).
To observe the endogenous LC3 puncta, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, incubated with FBS overnight, exposed to LC3 antibody, stained with FITC-conjugated secondary antibody and then observed by fluorescence microscopy.
Cells transfected with GFP-LC3 or GFP-ZFYVE1 plasmids were observed by fluorescence microscopy. The number of GFP-LC3 puncta per cell or the percentage of enlarged GFP-ZFYVE1 structures was assessed in five non-overlapping fields, and statistical data were obtained from three independent experiments.
To observe the accumulated GFP-ATG5 structures in U2OS cells, transfected cells were treated with 0.02% saponin prepared using PHEM buffer (60 mM Na-PIPES, 25 mM Na-HEPES, 10 mM EGTA, 2 mM MgCl2, pH 6.9) supplemented with 0.19 M NaCl and fixed with 4% paraformaldehyde. Cells were then observed under a fluorescence microscope. The percentage of accumulated GFP-ATG5 structures was assessed in five non-overlapping fields, and statistical data were obtained from three independent experiments.
Transmission electron microscopy
Treated U2OS cells were initially fixed in 0.1 M sodium phosphate buffer containing 3% glutaraldehyde (pH 7.4) and fixed in 0.1 M sodium phosphate buffer containing 1% OsO4 (pH 7.2) for 2 h at 4°C. The cells were then dehydrated in a graded series of ethanol. Cells were embedded into Ultracut (LEICA ULTRACUT R) and sliced into 60-nm sections. Ultrathin sections were stained with uranyl acetate and lead citrate and observed under a JEM-1230 transmission electron microscope (JEOL).
Flow cytometry
HCT116 cells were cotransfected with a plasmid expressing mTagRFP-mWasabi-LC3 and shRNA vector, EMC6 shRNA, vector or EMC6 expression plasmid at a ratio of 1:3 for 24 h, followed by culture in EBSS for 4 h (vector and EMC6 expression plasmid-transfected cells were not treated with EBSS). The cells were then harvested and resuspended in ice-cold PBS. mTagRFP and mWasabi fluorescent signals were analyzed on a FACS Calibur flow cytometer (Becton Dickinson). Both fluorescences were excited at 488 nm, while the mWasabi signal was detected at 509 nm, and the mTagRFP signal was detected at 584 nm. The percentage of mTagRFPhighmWasabilow cells was assessed from three independent experiments.
IP and western blot analysis
For IP analysis, cells were collected and disrupted in lysis buffer (300 mM NaCl, 50 mM Tris pH 8.0, 0.4% NP-40, 10 mM MgCl2, and 2.5 mM CaCl2) containing protease inhibitors (Roche Diagnostics, 04693116001). Total cell extracts (1 mg per sample) were mixed with pre-cleared protein G sepharoseTM Fast Flow (GE Healthcare, 17-0618-01) and appropriate antibodies, and then incubated for 4 h at 4°C. The beads were collected by centrifugation, washed five times using washing buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.4% NP-40, and 5 mM MgCl2), resuspended in 2 × SDS loading buffer and then analyzed by western blot as described previously. The protein bands were visualized using DyLight 800/DyLight 680-conjugated secondary antibodies, and the infrared fluorescence image was obtained using an Odyssey infrared imaging system (LI-COR Biosciences).
GST pull-down assay
Recombinant GST or GST fusion proteins were expressed in Escherichia coli strain BL21 (DE3) and purified. Equal amounts of these proteins were mixed with whole cell lysates extracted from transfected cells and Glutathione-SepharoseTM4B (GE Healthcare, 17-0756-01) for 4 h at 4°C. After five washes, the beads were resuspended in 2 × SDS loading buffer and analyzed by western blot.
In vitro binding studies with recombinant proteins
Recombinant His fused RAB5A was expressed in Escherichia coli strain BL21 (DE3) and purified. His-RAB5A (2 mM) was then used for GST-pull down assay directly or preincubated with 10 mM GDP or GTPγS in 20 mM HEPES, pH 7.2, 100 mM K-acetate, 0.5 mM MgCl2, 2 mM EDTA and 1 mM dithiothreitol for 30 min at 25°C. The mixture was then added to Glutathione-Sepharose beads containing 0.1 nM of GST or GST-EMC6 and left at 4°C for 60 min. Finally, the beads were washed three times with the same buffer containing 15 mM MgCl2 and 0.05% TX-100, and protein associated with the beads was detected by SDS-PAGE followed by immunoblotting with anti-RAB5A antibodies.
Statistical analysis
Data are presented as the mean ± SD. Differences between groups were analyzed using the Student’s t-test for continuous variables. Statistical significance in this study was set at p < 0.05. All reported p values are two-sided. All analyses were performed with GraphPad Prism 5.
| Abbreviations: | ||
| EMC6 | = | ER membrane protein complex subunit 6 |
| Atg | = | autophagy-related |
| GFP | = | green fluorescent protein |
| LC3 | = | microtubule-associated protein 1 light chain 3 (MAP1LC3) |
| 3-MA | = | 3-methyladenine |
| EBSS | = | Earle’s balanced salt solution |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| PIK3C3 | = | PtdIns3K class III |
| PIK3R4 | = | PtdIns3K regulatory subunit 4 |
| PtdIns3P | = | phosphatidylinositol 3-phosphate |
| BECN1 | = | Beclin 1 |
| TEM | = | transmission electron microscopy |
| ZFYVE1 | = | zinc finger FYVE domain-containing protein 1 |
| EEA1 | = | early endosome antigen 1 |
Additional material
Download Zip (7.9 MB)Acknowledgments
This work was supported by National Key Basic Research Program of China (973, 2011CB910103).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/22742
References
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458 - 67; http://dx.doi.org/10.1038/nrm2708; PMID: 19491929
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124 - 31; http://dx.doi.org/10.1016/j.ceb.2009.11.014; PMID: 20034776
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 2005; 16:2544 - 53; http://dx.doi.org/10.1091/mbc.E04-08-0669; PMID: 15743910
- Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008; 19:5360 - 72; http://dx.doi.org/10.1091/mbc.E08-01-0080; PMID: 18843052
- Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol 2009; 11:468 - 76; http://dx.doi.org/10.1038/ncb1854; PMID: 19270693
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859 - 64; http://dx.doi.org/10.1038/embor.2008.163; PMID: 18704115
- Obara K, Ohsumi Y. PtdIns 3-Kinase orchestrates autophagosome formation in yeast. J Lipids 2011; 2011:498768; http://dx.doi.org/10.1155/2011/498768; PMID: 21490802
- Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298 - 302; http://dx.doi.org/10.1074/jbc.C700195200; PMID: 17986448
- Obara K, Ohsumi Y. Atg14: a key player in orchestrating autophagy. Int J Cell Biol 2011; 2011:713435; http://dx.doi.org/10.1155/2011/713435; PMID: 22013444
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, et al. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 2010; 190:511 - 21; http://dx.doi.org/10.1083/jcb.200911141; PMID: 20713597
- Misra S, Miller GJ, Hurley JH. Recognizing phosphatidylinositol 3-phosphate. Cell 2001; 107:559 - 62; http://dx.doi.org/10.1016/S0092-8674(01)00594-3; PMID: 11733055
- Olkkonen VM, Stenmark H. Role of Rab GTPases in membrane traffic. Int Rev Cytol 1997; 176:1 - 85; http://dx.doi.org/10.1016/S0074-7696(08)61608-3; PMID: 9394917
- Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip SC, et al. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat Cell Biol 1999; 1:249 - 52; http://dx.doi.org/10.1038/12075; PMID: 10559924
- Ravikumar B, Imarisio S, Sarkar S, O’Kane CJ, Rubinsztein DC. Rab5 modulates aggregation and toxicity of mutant huntingtin through macroautophagy in cell and fly models of Huntington disease. J Cell Sci 2008; 121:1649 - 60; http://dx.doi.org/10.1242/jcs.025726; PMID: 18430781
- Su WC, Chao TC, Huang YL, Weng SC, Jeng KS, Lai MM. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol 2011; 85:10561 - 71; http://dx.doi.org/10.1128/JVI.00173-11; PMID: 21835792
- He P, Peng Z, Luo Y, Wang L, Yu P, Deng W, et al. High-throughput functional screening for autophagy-related genes and identification of TM9SF1 as an autophagosome-inducing gene. Autophagy 2009; 5:52 - 60; http://dx.doi.org/10.4161/auto.5.1.7247; PMID: 19029833
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11:1107 - 17; http://dx.doi.org/10.1093/hmg/11.9.1107; PMID: 11978769
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36:585 - 95; http://dx.doi.org/10.1038/ng1362; PMID: 15146184
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, Wei Y, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012; 8:1215 - 26; http://dx.doi.org/10.4161/auto.20284; PMID: 22647982
- Fengsrud M, Erichsen ES, Berg TO, Raiborg C, Seglen PO. Ultrastructural characterization of the delimiting membranes of isolated autophagosomes and amphisomes by freeze-fracture electron microscopy. Eur J Cell Biol 2000; 79:871 - 82; http://dx.doi.org/10.1078/0171-9335-00125; PMID: 11152279
- Réz G, Meldolesi J. Freeze-fracture of drug-induced autophagocytosis in the mouse exocrine pancreas. Lab Invest 1980; 43:269 - 77; PMID: 7401637
- Punnonen E-L, Pihakaski K, Mattila K, Lounatmaa K, Hirsimäki P. Intramembrane particles and filipin labelling on the membranes of autophagic vacuoles and lysosomes in mouse liver. Cell Tissue Res 1989; 258:269 - 76; http://dx.doi.org/10.1007/BF00239447; PMID: 2582478
- Orsi A, Polson HE, Tooze SA. Membrane trafficking events that partake in autophagy. Curr Opin Cell Biol 2010; 22:150 - 6; http://dx.doi.org/10.1016/j.ceb.2009.11.013; PMID: 20036114
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, et al. Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci U S A 2009; 106:20842 - 6; http://dx.doi.org/10.1073/pnas.0911267106; PMID: 19926846
- Mari M, Griffith J, Rieter E, Krishnappa L, Klionsky DJ, Reggiori F. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 2010; 190:1005 - 22; http://dx.doi.org/10.1083/jcb.200912089; PMID: 20855505
- Young ARJ, Chan EYW, Hu XW, Köchl R, Crawshaw SG, High S, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006; 119:3888 - 900; http://dx.doi.org/10.1242/jcs.03172; PMID: 16940348
- Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009; 186:773 - 82; http://dx.doi.org/10.1083/jcb.200907014; PMID: 19797076
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; http://dx.doi.org/10.1083/jcb.200803137; PMID: 18725538
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 2009; 11:385 - 96; http://dx.doi.org/10.1038/ncb1846; PMID: 19270696
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656 - 67; http://dx.doi.org/10.1016/j.cell.2010.04.009; PMID: 20478256
- English AR, Zurek N, Voeltz GK. Peripheral ER structure and function. Curr Opin Cell Biol 2009; 21:596 - 602; http://dx.doi.org/10.1016/j.ceb.2009.04.004; PMID: 19447593
- Eden ER, White IJ, Tsapara A, Futter CE. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol 2010; 12:267 - 72; PMID: 20118922