Abstract
Hepatitis C virus (HCV) is able to induce autophagy via endoplasmic reticulum (ER) stress, but the exact molecular signaling pathway is not well understood. We found that the activity of the mechanistic target of rapamycin complex 1 (MTORC1) was inhibited in Huh7 cells either harboring HCV-N (genotype 1b) full-genomic replicon or infected with JFH1 (genotype 2a) virus, which led to the activation of UNC-51-like kinase 1 (ULK1) and thus to autophagy. We then analyzed activity upstream of MTORC1, and found that both protein kinase, AMP-activated, α (PRKAA, including PRKAA1 and PRKAA2, also known as AMP-activated protein kinase, AMPKα) and AKT (refers to pan AKT, including three isoforms of AKT1-3, also known as protein kinase B, PKB) were inhibited by HCV infection. The inhibition of the AKT-TSC-MTORC1 pathway contributed to upregulating autophagy, but inhibition of PRKAA downregulated autophagy. The net effect on autophagy was from AKT, which overrode the inhibition effect from PRKAA. It was further found that HCV-induced ER stress was responsible for the inhibition of the AKT pathway. Metformin, a PRKAA agonist, inhibited HCV replication not only by activating PRKAA as previously reported, but also by activating AKT independently of the autophagy pathway. Taken together, our data suggested HCV inhibited the AKT-TSC-MTORC1 pathway via ER stress, resulting in autophagy, which may contribute to the establishment of the HCV-induced autophagy.
Introduction
Hepatitis C virus (HCV) is a major cause of chronic liver disease. It has infected more than 170 million people worldwide, approximately 3% of the world’s population.Citation1 In up to 80% of infected patients, a chronic infection persists, often leading to liver cirrhosis and hepatocellular carcinoma.Citation2,Citation3 HCV infection can induce nonhepatic diseases such as type 2 diabetes and lymphoma.Citation4,Citation5 Current therapy is performed with peg-interferon plus ribavirin, but only a portion of patients can eliminate the virus. The outcome of antiviral treatment is dependent on the viral genotype. No effective vaccine against HCV is currently available.Citation6
HCV is an enveloped, single-stranded, positive-sense RNA virus belonging to the family of Flaviviridae. The RNA genome is about 9.6 kb, encoding four structural proteins (core, E1, E2 and p7) and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A and NS5B).Citation7 The structural proteins, core, E1 and E2, are the major components of the viral particle. The nonstructural proteins participate in genome replication. HCV replication involves the formation of a membrane-associated replication complex. Various structural and nonstructural proteins localize inside the ER lumen or integrate into the ER membrane.Citation8,Citation9 It is conceivable that HCV is able to induce ER stress, a cellular response when the capacity of the ER to fold and modify proteins is exceeded.Citation10 The activation of ER stress can activate signaling pathways involved in apoptosis and autophagy.Citation11,Citation12
Autophagy is a cellular catabolic process by which cells degrade their own damaged organelles and long-lived proteins inside a double-membraned vesicle.Citation13,Citation14 Autophagy can be activated under stresses such as nutrient starvation, organelle damage, deprivation of growth factors and ER stress.Citation12,Citation15 Autophagy is induced during bacterial or viral infections, such as HCV infection.Citation16-Citation20 Autophagy is initiated by the UNC-51-like kinase 1/2 complex (ULK1 and ULK2, the mammalian orthologs of autophagy-related 1, Atg1).Citation21,Citation22 The ULK1/2 complex is regulated by the mechanistic target of rapamycin complex 1 (MTORC1), which is a key regulator involved in autophagy.Citation23-Citation26 Under basal conditions, MTORC1 phosphorylates a serine (S757) of ULK1, which inhibits ULK1/2’s kinase activity to inhibit autophagy.Citation27 The ULK1/2 complex is also regulated by protein kinase, AMP-activated, α (PRKAA, including PRKAA1 and PRKAA2, also known as AMP-activated protein kinase, AMPKα) which phosphorylates several other serine residues (S555, S777, etc.) of ULK1 to activate it under starvation conditions.Citation27-Citation29 The activity of MTORC1 is negatively regulated by both PRKAA and tuberous sclerosis 1/2 (TSC1/TSC2) complex.Citation30,Citation31 TSC1/TSC2 complex can be positively regulated by PRKAA or negatively regulated by AKT (refers to pan AKT, including three isoforms of AKT1-3).Citation32-Citation36 Therefore, PRKAA and AKT are important regulators of autophagy.
Autophagy can be upregulated by transfection with the subgenomic replicon or full-length mRNA of HCV, or direct viral infection.Citation16-Citation20 HCV induces the unfolded protein response (UPR), a cytoprotective signaling cascade induced by ER stress, which in turn activates the autophagic pathway.Citation17,Citation19,Citation20 HCV exploits the UPR-autophagy pathway to escape the innate immune response, allowing further HCV RNA replication.Citation20 Upon HCV infection, three sensors located on the ER membrane respond to ER stress and initiate the UPR via three different pathways, eukaryotic translation initiation factor 2-α kinase 3 (EIF2AK3, also known as PERK, RNA-dependent protein kinase [PRK]-like ER kinase), activating transcription factor-6 (ATF6) and ER to nucleus signal-1 (ERN1, also known as IRE1, inositol-requiring ER to nucleus signal-1). The suppression of any of the three pathways inhibits HCV-induced autophagy. The UPR induces DNA-damage-inducible transcript 3 (DDIT3/CHOP) expression which facilitates autophagosome formation (reviewed in ref. Citation37). However, the mechanism by which ER stress initiates and regulates autophagy during HCV infection has not been established. In this study, we analyzed the activity of the ULK1/2 complex, MTORC1 and upstream regulators to reveal the signaling pathway involved in HCV-induced autophagy.
Results
HCV infection induced autophagy
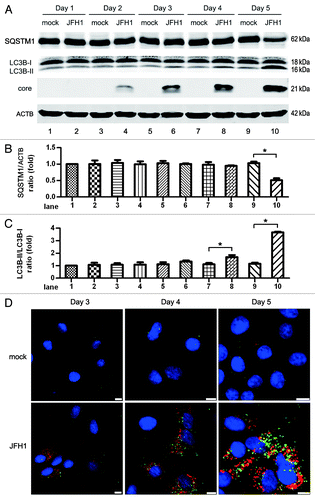
To mimic natural viral infection, we used cell culture-propagated HCV JFH1 (genotype 2a) to infect Huh7 cells and determined the induction of autophagy at different time points. The formation of the phosphatidylethanolamine-conjugated form of microtubule-associated protein 1 light chain 3 β (MAP1LC3B-II, abbreviated to LC3B-II) was highly activated and the level of sequestosome 1 (SQSTM1, also known as p62) was significantly decreased in JFH1-infected Huh7 cells at 5 d postinfection, as compared with mock groups infected with supernatants from naïve Huh7 cells (). To establish the connection between intracellular replication of JFH1 and autophagy, we used ultra-violet (UV)-inactivated JFH1 to infect Huh7 cells. As shown in Figure S1A, UV-inactivated virus did not increase the formation of LC3B-II and did not decrease the level of SQSTM1. A dose effect was seen with JFH1 virus in infected Huh7 cells (Fig. S1B). Ten multiplicity of infection (MOI) of JFH1 significantly induced autophagy and 1 MOI of JFH1 only moderately promoted the induction of autophagy on day 5. Moreover, 0.1 MOI virus had no influence on LC3B-II transformation and SQSTM1 expression compared with mock treatment. The puncta formation of endogenous LC3B was examined using confocal microscopy on days 3, 4 and 5 postinfection (). The fixed cells were permeabilized with saponin before staining to remove nonaggregated LC3, leaving autophagosome/autolysosome associated LC3.Citation38 From day 3 to day 5, increasing numbers of infected cells could be observed (, bottom row). On day 3, a few LC3B puncta formed in HCV-infected cells, whereas remarkable puncta formation was found in HCV-infected cells on days 4 and 5. In contrast, LC3B-II puncta could hardly be found in the cells not infected with HCV. These results show that the induction of autophagy was concomitant with HCV infection.
Figure 1. HCV infection induces significant autophagy at 5 d postinfection at an MOI of 10. (A) Huh7 cells were infected with 10 MOI of HCV, and then harvested at different times for western blot analysis. The expression of SQSTM1, LC3B and HCV core protein was analyzed. ACTB was used as sample loading control. (B and C) The SQSTM1/ACTB ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. *p < 0.05 was considered significant. (D) The puncta formation of endogenous LC3B in JFH1-infected Huh7 cells was analyzed on day 3, 4 and 5. Green: LC3B, Red: HCV core protein, Blue: nucleus. Scale bars: 10 μm (Because the cell confluence was different among the time points, the magnification was adjusted to have a proper number of cells captured, resulting in the length of scale bars being different among the time points).
To exclude the possibility that autophagy was induced by cytopathic effects induced by long-term HCV infection, we determined the cell viability. As shown in Figure S2A, cytopathic effects were not observed in HCV-infected cells. To determine if HCV infection induced apoptosis, JFH1-infected cells were stained with Annexin-V and propidium iodide (PI) and analyzed using flow cytometry. Annexin-V can bind to apoptotic cells and PI stains dead cells. Cells that are considered viable are Annexin-V negative and PI negative. Cells that are in early apoptosis are Annexin-V positive and PI negative. Cells that are in late apoptosis or already dead are Annexin-V and PI positive. Cells that are Annexin-V negative and PI positive are dead. High concentrations of DMSO can induce apoptosisCitation39 and was used as a positive control. As shown in Figure S2B, the rate of cell death or apoptosis was similar in the HCV-infected cells and the mock group. High concentration of DMSO did induce cell death and apoptosis in Huh7 cells. Thus, HCV-induced autophagy was not caused by cytopathic effects.
HCV infection-induced autophagy was a complete process
To determine if HCV-induced autophagy is an incomplete or complete process, we treated JFH1-infected cells with an acidification inhibitor of lysosome degradation, chloroquine (CQ), and then measured the accumulation of LC3B-II and the degradation of SQSTM1, compared with mock-infected cells (). In the presence of CQ, LC3B-II was significantly increased and SQSTM1 degradation was significantly inhibited in JFH1-infected cells compared with that in mock-infected cells. We measured the gray scale of SQSTM1, LC3B-I, -II and ACTB (actin, β) in three independent experiments and found that in the presence of CQ, SQSTM1/ACTB only increased by 4.3% in the mock group, however, a 58.5% increase was observed in JFH1-infected cells (). Similarly, in the presence of CQ, LC3B-II/LC3B-I increased by 27.1% in mock-infected cells, but a 70.1% increase was observed in JFH1-infected cells (). These results show that HCV induced complete autophagy.
Figure 2. HCV infection induces complete autophagy at 5 d postinfection at an MOI of 10. (A) Huh7 cells were infected with 10 MOI of HCV for 5 d, and then treated with 50 μM chloroquine (CQ) for 6 h. The expression of SQSTM1, LC3B and HCV core protein was analyzed by western-blot. ACTB was used as sample loading control. (B and C) The SQSTM1/ACTB ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown.a Percentage increased by CQ treatment. *p < 0.05 was considered significant. (D) HCV-infected cells were transfected with mTagRFP-mWasabi-LC3B on day 3 postinfection and maintained for 2 d. Then, the cells were treated with 50 μM CQ and analyzed for the localization of indicated proteins. Blue: HCV core protein. Scale bars: 10 μm.
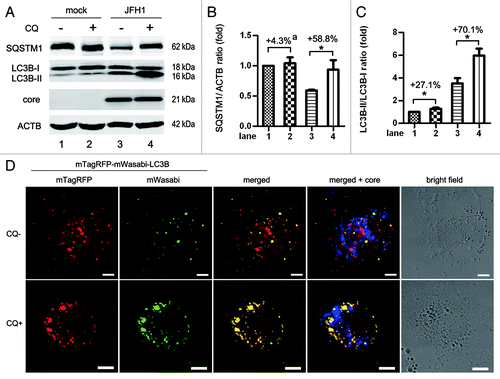
To confirm this, we used a tandem fluorescence-labeled LC3B construct, mTagRFP-mWasabi-LC3B,Citation40 to identify the formation of autolysosomes. The green fluorescence signal (mWasabi) was lost upon autophagosome fusion with acidic lysosome, while the red signal remains detectable. In the absence of CQ, frequent red puncta were observed in JFH1-infected Huh7 (, top row). In contrast, treatment with CQ greatly restored the green signals and resulted in increased yellow signals in HCV-infected cells (, bottom row). These findings suggest HCV infection induces complete autophagy.
It was a concern that CQ treatment only slightly induced LC3B-II accumulation in mock-infected Huh7 cells (). We analyzed the LC3B-II level in Huh7 cells at different time points post-CQ treatment and found CQ did not induce significant LC3B-II accumulation until 12 h post-treatment (Fig. S3). LC3B-II accumulation in normal cells might be not as susceptible to CQ-treatment as in HCV-infected cells where autophagy was highly upregulated.
Mizui et al. and Ke et al. both reported that HCV replication was inhibited by CQ treatment, which inhibited autophagosome-lysosome fusion and autophagic protein degradation.Citation20,Citation41 However, the core protein expression was not changed in our system (). We analyzed the HCV RNA abundance in HCV-infected cells by real-time PCR after CQ treatment. Concordant with the results of core protein expression, HCV RNA was not changed after CQ treatment (Fig. S4A). These results show that perturbation of the autophagy process with CQ had no impact on HCV replication in our system.
HCV infection inhibited MTORC1 and activated ULK1 to induce autophagy
As a key regulator in autophagy, the activity of MTORC1 was analyzed in HCV-N genome harboring Huh7 cells and JFH1-infected Huh7 cells. After HCV infection, the phosphorylation of Ser2448 (an activation site of MTORC1) and of ribosomal protein S6 kinases (RPS6Ks, substrates of MTORC1) were both decreased (, lanes 1–3). ULK1 S757 phosphorylation, which inhibits ULK1 function, was decreased (, lanes 1–3). The ULK1 complex was activated and initiated autophagy, as indicated by the increase of LC3B-II and the decrease of SQSTM1 (, lanes 1–3).
Figure 3. HCV inhibits the activity of MTORC1 and induces autophagy. (A) Mock-infected Huh7 (mock), Huh7 harboring HCV-N full-genomic replicon (HCV-N) together with Huh7 infected with 10 MOI JFH1 virus (JFH1) were harvested 5 d postinfection and phosphorylation of MTOR, RPS6K and ULK1, SQSTM1, LC3B, HCV core protein were analyzed by western blot. To activate MTORC1, Huh7 cells were treated with 50 ng/ml platelet-derived growth factor-BB (PDGF-BB, which stimulates the PI3K-AKT pathway to activate MTOR) for 6 h before harvest. ACTB was used as sample loading control. (B–E) The MTOR p-S2448/total MTOR ratio, the ULK1 p-S757/total ULK1 ratio, the SQSTM1/ACTB ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. *p < 0.05 was considered significant.
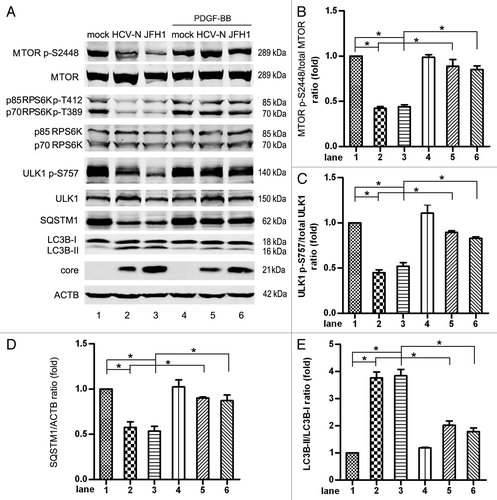
Mock- and HCV-infected cells were treated with 50 ng/ml platelet-derived growth factor-BB (PDGF-BB). PDGF-BB activates MTOR to control cell proliferation and cell survival through the PIK3CA (also known as PI3K)-AKT pathway.Citation42,Citation43 We found that the level of phosphorylated MTOR S2448 was not changed in mock-infected Huh7 cells, probably because MTOR is highly phosphorylated under basal conditions. The inhibited phosphorylation of MTOR S2448 and RPS6K was rescued in HCV-infected cells compared mock-infected cells ( lanes 4–6, and ). The activation of MTORC1 by PDGF-BB increased the phosphorylated form of ULK1 S757 (, lanes 4–6), decreased the expression of LC3B-II, and increased the expression of SQSTM1 in HCV-infected cells (, lanes 4–6). The expression of LC3B-II and SQSTM1 did not return to the basal levels found in mock cells, suggesting another mechanism is involved in HCV-induced autophagy. In conclusion, HCV infection inhibited the activation of MTORC1 and led to the activation of the ULK1, which initiated autophagy.
Several groups have reported that using RNA interference (RNAi) to knock down the expression of genes important in the formation of autophagic vacuoles could suppress HCV replication.Citation17,Citation41,Citation44-Citation48 As the HCV replication in Huh7 harboring HCV-N replicon may depend on the cell cycle,Citation49 we only analyzed the intracellular HCV RNA in JFH1 virus-infected cells. In contrast to previous studies, the core protein expression was not changed after PDGF-BB treatment (). Accordant with the core protein expression, HCV RNA level was not changed either in JFH1-infected cells after PDGF-BB treatment (Fig. S4B). These results showed that the inhibition of autophagy with PDGF-BB did not influence HCV replication.
HCV infection inhibited both PRKAA and AKT
PRKAA and AKT both regulate MTORC1 in autophagy. The activities of PRKAA and AKT were therefore determined in HCV-infected cells. The phosphorylation of T172 of PRKAA was inhibited by HCV-N and JFH1 compared to mock-infected Huh7 cells, but the phosphorylation of S485 was not significantly changed (). As both T172 and S485 sites are thought to be essential for the activity of PRKAA, the decreased phosphorylation of T172 might inhibit PRKAA’s activity (reviewed in ref. Citation30). This is supported by the decreased level of phosphorylation of acetyl-CoA carboxylase (ACACA, a substrate of PRKAA) S79, indicating inhibition of PRKAA in HCV-infected cells ().
Figure 4. HCV inhibits the activity of PRKAA and AKT. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were harvested 5 d postinfection and phosphorylation of PRKAA and ACACA was analyzed by western blot. (B) The PRKAA p-T172/total PRKAA ratio from at least three independent experiments of (A) was shown. (C) Phosphorylation of AKT and the level of phospho-AKT substrates were analyzed by western-blot [the AKT antibodies recognize all three isoforms of AKT(AKT1-3)]. Nontreated and AKT Inhibitor VIII (AKT in, a pleckstrin homology (PH) domain-dependent AKT inhibitor)-treated 293 cells were used as control to indicate the band of AKT. (D) The AKT p-S473/total AKT ratio from at least three independent experiments of (C) was shown. *p < 0.05 was considered significant.
![Figure 4. HCV inhibits the activity of PRKAA and AKT. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were harvested 5 d postinfection and phosphorylation of PRKAA and ACACA was analyzed by western blot. (B) The PRKAA p-T172/total PRKAA ratio from at least three independent experiments of (A) was shown. (C) Phosphorylation of AKT and the level of phospho-AKT substrates were analyzed by western-blot [the AKT antibodies recognize all three isoforms of AKT(AKT1-3)]. Nontreated and AKT Inhibitor VIII (AKT in, a pleckstrin homology (PH) domain-dependent AKT inhibitor)-treated 293 cells were used as control to indicate the band of AKT. (D) The AKT p-S473/total AKT ratio from at least three independent experiments of (C) was shown. *p < 0.05 was considered significant.](/cms/asset/be72c980-e2c6-41d8-b279-1f25fd7cb5e7/kaup_a_10922791_f0004.gif)
We analyzed the phosphorylation of AKT S473 and T308, two activation sites of AKT (reviewed in ref. Citation50). Under basal conditions, the AKT S473 site was moderately phosphorylated, but a phosphorylated T308 signal was not detected (). During HCV infection, phospho-AKT S473 decreased as shown by the western blot assay (, lanes 2 and 3). Nontreated 293 cells and AKT Inhibitor VIII (abbreviated as AKT in, a pleckstrin homology (PH) domain-dependent AKT inhibitor)-treated 293 cells were used as a control to indicate the phosphorylated bands of AKT S473 and T308. We proceeded to see if the substrates of AKT were changed. Phospho-AKT substrate (RXXS*/T*) antibody was utilized and western blot was performed as an AKT substrate assay. AKT belongs to the Arg-directed kinases or AGC-family kinases, including cAMP-dependent protein kinase (PKA), cGMP-dependent protein kinase (PKG) and protein kinase C.Citation51,Citation52 These kinases share a substrate specificity characterized by Arg at position −3 relative to the phosphorylated Ser or Thr. Thus, when the activity of AKT changes, the bands detected by this antibody may shift. As shown in , 5 bands decreased after HCV infection (arrows). Similar results were obtained from AKT-specific inhibitor-treated 293T cells. Together, these results suggest that HCV infection also inhibited AKT.
HCV promoted autophagy induction via inhibiting AKT activation
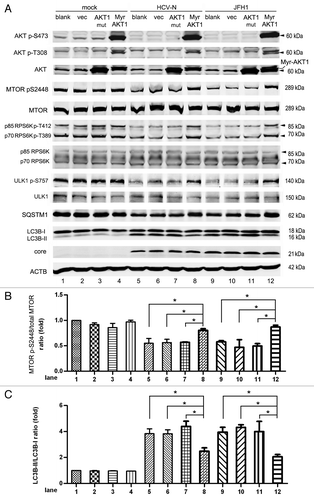
As PRKAA is generally considered a positive regulator of autophagy and AKT a negative regulator, we speculated that the inhibition of AKT-inhibited MTORC1 would result in autophagy. To verify this, we expressed activated AKT in HCV-infected Huh7 cells. Because all three isoforms of AKT (AKT1-3) share significant homology,Citation53,Citation54 we measured only one isoform, AKT1, in this study. An inactive AKT1 (AKT1 mut, mutated AKT1 with T308A and S473A), a myristoylated AKT1 (Myr-AKT1, a myristoylation signal fused to the N-terminal of AKT1, which consistently activates AKT1), and an empty vector (vec) were transduced into mock-infected and HCV-infected Huh7 cells using lentiviruses. Nontransduced cells were used as a control (blank) (). Exogenously expressed Myr-AKT1 showed highly phosphorylated S473 and T308 compared with the blank, vector and AKT1 mut groups. Western blots of mock-infected Huh7 cells, blank, vector, AKT1 mut and Myr-AKT1 groups showed no significant phosphorylation differences (, lanes 1–4). As MTOR S2448 is highly phosphorylated under basal conditions, activated AKT should not increase the phosphorylation of MTOR S2448. In HCV-N replicon cells and JFH1-infected cells (, lanes 5–12), the levels of phosphorylated MTOR, RPS6K and ULK1 increased in the Myr-AKT1 expressed group compared with the other three groups. The level of LC3B-II was decreased (, lanes 5–12) and the level of SQSTM1 was increased in the Myr-AKT1 expressed group. There was no difference among the blank, vector and AKT1 mut groups in LC3B-II and SQSTM1 expression. HCV inhibited AKT1’s activation and then inhibited the activity of MTORC1. Inhibition of MTORC1 activated ULK1 to induce autophagy. The expression of Myr-AKT1 in HCV-infected Huh7 did not depress LC3B-II expression to the basal level, suggesting that other mechanisms may induce HCV-mediated autophagy. Considering the high degree of homogeneity of the three isoforms of AKT, our results support HCV mediates autophagy induction through AKT pathway inhibition.
Figure 5. The inhibition of AKT induced by HCV infection leads to autophagy. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were transduced with mutant AKT (AKT mut, T308A, S473A), myristoylated AKT1 (Myr-AKT) with empty vector as control via lentivirus on day 3 postinfection and harvested at 5 d postinfection. Cell lysates were prepared and phosphorylation of AKT, MTOR, RPS6K and ULK1, SQSTM1, LC3B and HCV core protein were analyzed by western blot. ACTB was used as a sample loading control. (B and C) The MTOR p-S2448/total MTOR ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. *p < 0.05 was considered significant.
HCV inhibited the AKT-TSC-MTORC1 pathway to enhance autophagy
It is clear that AKT is able to regulate MTOR via the TSC1/2 complex,Citation35 so we proceeded to determine if HCV induces autophagy through this pathway. TSC2 gene knockdown was performed using lentiviral based shRNA in Huh7 cells and a nontarget shRNA was used as a negative control (NTC). Knockdown of TSC2 had no influence on AKT’s activity, which indicates that TSC2 worked downstream of AKT (, lanes 1 and 2). The knockdown of TSC2 had no influence on the phosphorylation of MTOR in mock-infected Huh7 cells, but increased the phosphorylation of MTOR S2448 in HCV-infected cells (). The activation of MTOR reversed the increase of LC3B-II and the decrease of SQSTM1 seen in HCV-N and JFH1-infected Huh7 cells (). These results suggest that HCV infection inhibits the AKT-TSC-MTORC1 pathway, enhancing autophagy. The expression of LC3B-II in HCV-infected, TSC2 knockdown cells was not back to the basal level found in nontreated mock cells. This suggests that nonTSC2 pathways participate in HCV-induced autophagy. Transduction of lentiviral TSC2 shRNA slightly increased the conversion of LC3B-II in mock-infected Huh7 cells (, lanes 1 and 2), probably because VSV-G protein from lentiviral particles can induce autophagy.Citation55
Figure 6. HCV inhibits the AKT-TSC2-MTORC1 pathway to induce autophagy. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were transduced with TSC2 shRNA or nontarget shRNA (NTC) via lentivirus on day 3 postinfection and harvested at 5 d postinfection. Cell lysates were prepared and phosphorylation of AKT, MTOR and ULK1, SQSTM1, LC3B and HCV core protein were analyzed by western blot. ACTB was used as a sample loading control. (B and C) The MTOR p-S2448/total MTOR ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. *p < 0.05 was considered significant.
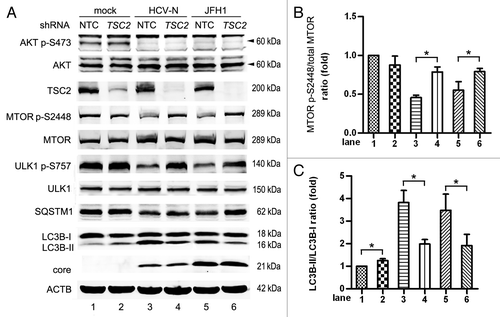
It should be noted that the HCV core protein level was not changed after AKT-TSC-MTORC1 pathway was activated using Myr-AKT1 or suppression of TSC2 expression using shRNA. Transduction of the empty vector, mutated AKT1 and activated AKT1 did not change HCV RNA levels as compared with blank control (Fig. S4C). JFH1-infected cells with TSC2 knockdown did not have different viral RNA levels from controls without TSC2 knockdown (Fig. S4D). These results demonstrated inhibition of autophagy by interference with the AKT-TSC2-MTOR pathway had no impact on HCV replication.
HCV-induced ER stress resulted in the inhibition of AKT
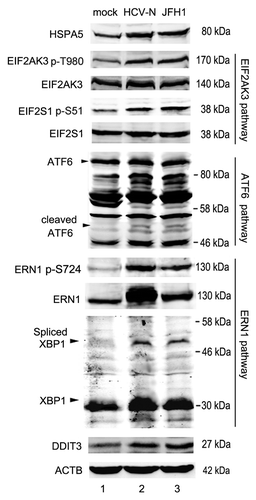
HCV has been reported to induce autophagy via ER stress.Citation16,Citation17,Citation19,Citation20 ER stress can be induced by three different pathways. Briefly, under ER stress conditions, the ER chaperone protein, HSPA5/GRP78/Bip, binds to unfolded proteins and thereby renders each ER stress transducers including EIF2AK3 (PERK), ATF6 and ERN1 (IRE1) to be activated. Activated EIF2AK3 phosphorylates EIF2S1 (eIF2α), which activates the expression of DDIT3, and leads to cell cycle arrest or apoptosis. The second sensor, ATF6 (90 kDa), is transported to the Golgi apparatus and cleaved by proteolysis. The released N-terminal cytosolic domain (50 kDa) of ATF6 is transported into the nucleus, and also leads to the activation of DDIT3. The third sensor, ERN1, cleaves x-boxing-protein 1 (XBP1) mRNA precursor (encodes a 31 kDa protein) to mature XBP1 mRNA (encodes a 54-kDa protein) by unique splicing. This pathway also induces DDIT3 transcription.Citation56 Experiments were performed to determine if these three UPR pathways were activated in HCV-infected cells. As shown in , HCV infection upregulated the expression of HSPA5 and increased the phosphorylation of EIF2AK3, which in turn increased the phosphorylation of EIF2S1. HCV infection also activated the cleavage of ATF6, resulting in the expression of cleaved ATF6 (50 kDa). Both total ERN1 and phosphorylated ERN1 were increased in HCV-infected cells, which induced the expression of spliced XBP1 (54 kDa). The activation of the three arms of UPR increased the expression of DDIT3 (). These results suggest that HCV infection induced ER stress by activating all of the three UPR pathways.
Figure 7. HCV induced ER stress. Mock-infected Huh7 (Huh7), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were harvested 5 d postinfection. The expression of HSPA5, ATF6, XBP1 and DDIT3, and the phosphorylation of EIF2AK3, EIF2S1 and ERN1 were analyzed by western blot. ACTB was used as a sample loading control.
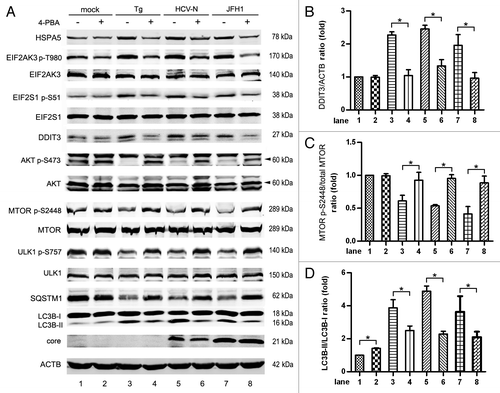
It was reported that ER stress negatively regulates the AKT-TSC-MTORC1 pathway, resulting in the enhancement of autophagy.Citation57 We examined whether the inhibition of AKT by HCV infection was mediated by ER stress. 4-phenylbutyric acid (4-PBA) acts as a chemical chaperone in the ER to prevent the activation of UPR and inhibits ER stress.Citation58 In mock-infected Huh7, 4-PBA did not change the level of phosphorylated AKT, MTOR and ULK1, but slightly enhanced LC3B-II expression, and slightly decreased SQSTM1 expression (, lanes 1 and 2). Thapsigargin (Tg) is a commonly used agent known to rapidly elicit ER stress by inhibiting the ER Ca2+ pump. As shown in , lanes 3–8, 4-PBA rescued Tg HCV induced UPR, as indicated by decreased phosphorylation of EIF2AK3 and EIF2S1, HSPA5 and DDIT3 (). The inhibition of ER stress by 4-PBA restored the phosphorylation of AKT S473, which increased the phosphorylation of MTOR S2448 and ULK1 S757 in Tg-treated and HCV-infected cells (). 4-PBA also decreased LC3B-II expression and increased SQSTM1 expression slightly in HCV- infected cells, but the level of LC3B-II was not reversed to the basal condition ().
Figure 8. 4-phenylbutyric acid (4-PBA) inhibited HCV-induced ER stress and autophagy. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were treated with 5mM 4-PBA (acts as a chemical chaperone in the ER to prevent the activation of UPR) for 12 h before harvesting at 5 d postinfection. Huh7 cells treated with 300 nM thapsigargin (Tg, an agent known to rapidly elicit ER stress by inhibiting the ER Ca2+ pump) for 12 h were used as positive control. Phosphorylation of EIF2AK3, EIF2S1, AKT, MTOR and ULK1, HSPA5, DDIT3, SQSTM1, LC3B and HCV core protein were analyzed by western blot. ACTB was used as a sample loading control. (B–D) The DDIT3/ACTB ratio, the MTOR p-S2448/total MTOR ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. *p < 0.05 was considered significant.
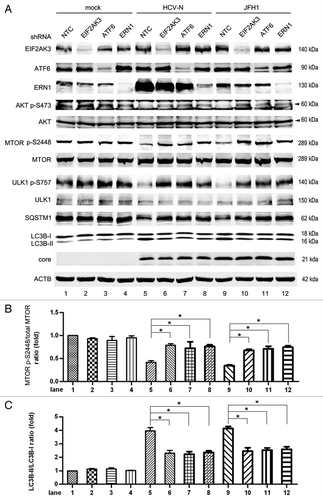
The three pathways of UPR (EIF2AK3, ATF6 and ERN1) were studied using knockdown with lentiviral-based shRNA in Huh7 cells. In mock-infected Huh7, knockdown of EIF2AK3, ATF6 and ERN1 had no significant influence on the phosphorylation of AKT, MTOR and ULK1, or the protein expression of LC3B-II and SQSTM1 (, lanes 1–4). In HCV-infected cells, knockdown of each of the three pathways partly restored the phosphorylation of AKT S473, which subsequently restored the phosphorylation of MTOR S2448 (, lanes 5–12). The restoration of MTOR activity inhibited the activity of ULK1, and finally inhibited HCV-induced autophagy. This mechanism of action is supported by the increase of LC3B-II () and the decrease of SQSTM1 (). These results verified that all of the three UPR pathways are involved in inhibiting the AKT-autophagy pathway.
Figure 9. Knockdown of the ER stress pathway inhibited HCV-induced autophagy. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were transduced with EIF2AK3, ATF6, ERN1 shRNA or non-target shRNA (NTC) via lentivirus on day 3 postinfection and harvested 5 d postinfection. Cell lysates were prepared and phosphorylation of AKT, MTOR and ULK1, SQSTM1, LC3B and HCV core protein were analyzed by western blot. ACTB was used as a sample loading control. (B and C) The MTOR p-S2448/total MTOR ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. *p < 0.05 was considered significant.
In the same way, we also determined HCV core protein expression and HCV replication after inhibition of UPR pathways using 4-PBA or shRNA. In contrast to the studies of Sir et al. and Ke and Chen,Citation17,Citation20 both the expression level of core protein ( and ) and of intracellular HCV RNA (Fig. S4E and S4F) were not changed after 4-PBA treatment or suppression of UPR pathways using shRNAs. This suggests that inhibition of autophagy has no impact on HCV replication.
Activation of PRKAA by AICAR enhanced HCV induced-autophagy
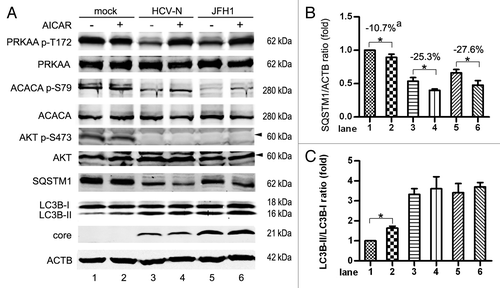
It is generally accepted that the activation of PRKAA promotes the induction of autophagy. Based on the aforementioned data we obtained that the activity of PRKAA was inhibited during HCV replication (), AICAR,Citation59 a PRKAA activator, was then utilized to determine if HCV induced inhibition of PRKAA activation would restrict HCV-induced autophagy.
As shown in lanes 3 to 6 of , AICAR rescued the inhibition of PRKAA activation induced by HCV. The phosphorylation of ACACA was enhanced by AICAR in HCV-infected cells (, lanes 3 to 6), confirming that AICAR activated PRKAA. Treatment of AICAR decreased SQSTM1 expression in HCV-infected cells. AICAR had no impact on the phosphorylation of AKT S473 in HCV-infected cells. This verifies the finding of activation of PRKAA by AICAR enhanced HCV-induced autophagy and suggests that HCV downregulates autophagy through inhibition of PRKAA activation. Taken together with the finding HCV could induce autophagy via inhibiting AKT phosphorylation, these results implied the positive-regulation effect by HCV-induced inhibition of AKT activation was much stronger than the negative-regulation role from inhibition of PRKAA activation, which ultimately led to autophagy induction.
Figure 10. Activation of PRKAA enhanced HCV-induced autophagy. (A) Mock-infected Huh7 (mock), HCV-N cells and Huh7 infected with 10 MOI JFH1 virus (JFH1) were treated with 1mM AICAR (a specific PRKAA activator) for 12 h before harvesting 5 d postinfection. Phosphorylation of PRKAA, ACACA and AKT, SQSTM1, LC3B and HCV core protein were analyzed by western blot. ACTB was used as a sample loading control. (B and C) The SQSTM1/ACTB ratio and the LC3B-II/LC3B-I ratio from at least three independent experiments of (A) were shown. aPercentage decreased by AICAR. *p < 0.05 was considered significant.
AICAR did not increase the conversion of LC3B-II in HCV-infected cells. This might be because LC3B-II was degraded in the autolysosome. Our previous data showing HCV induced complete autophagy may support this viewpoint.
We noticed that AICAR did not significantly enhance the phosphorylation of PRKAA T172 in mock-infected Huh7 cells (probably because T172 is highly phosphorylated under basal conditions), but slightly increased the level of LC3B-II and decreased the level of SQSTM1 (, lanes 1 and 2). The analysis of three independent experiments showed that SQSTM1/ACTB decreased by 10.7% in mock-infected cells after AICAR treatment. A more significant decrease was observed in HCV-N (25.3%) and JFH1 (27.6%)-infected cells (). These results suggest that AICAR might induce autophagy independent of PRKAA in the mock group at basal condition, but when infected by HCV, AICAR might promote HCV induced autophagy by activating PRKAA.
We evaluated the impact of AICAR enhanced autophagy on HCV replication. The core protein () and the intracellular HCV RNA expression (Fig. S4G) in JFH1-infected cells did not change after AICAR treatment, indicating the enhancement of autophagy by AICAR had no impact on HCV replication.
Metformin had a minimal effect on HCV-induced autophagy due to the activation of both PRKAA and AKT
Besides AICAR, we used another activator of PRKAA, metformin (a widely used drug for treatment of type 2 diabetes as a PRKAA activator),Citation60 to analyze the function of PRKAA in HCV-induced autophagy. Because metformin was reported to inhibit HCV replication,Citation61,Citation62 we examined the impact of metformin on HCV replication. Considering that HCV replication in Huh7 harboring HCV-N replicon may depend on the cell cycle as mentioned above,Citation49 we used the JFH1 virus infection system in this experiment.
As shown in lanes 4 and 5 of , metformin activated phosphorylation of PRKAA T172 and ACACA S79. Like AICAR, metformin should enhance HCV-induced autophagy by the activation of PRKAA. Interestingly, we found that metformin had no significant impact on the expression of SQSTM1 and LC3B-II (, lanes 4 and 5), suggesting that metformin had minimal effect on HCV-induced autophagy. Our data revealed that metformin could not only activate PRKAA, but also activated AKT in Huh7 cells (; Fig. S5). As mentioned above, PRKAA and AKT have opposite effects on autophagy induction. The positive regulation of autophagy by activation of PRKAA might be counteracted by the activation of AKT, resulting in unchanged induction of autophagy. This speculation was further verified by the additional treatment with AKT inhibitor VIII. In the presence of AKT inhibitor VIII, the enhanced phosphorylation of AKT S473 induced by metformin was reversed (, lanes 5 and 6). Similar to AICAR, metformin plus AKT inhibitor only activated PRKAA, resulting in the enhancement of autophagy as indicated by the degradation of SQSTM1 (, lanes 5 and 6). We analyzed the activation of MTORC1 by determining the phosphorylation of RPS6K. As shown in , in HCV-infected cells, metformin did not change the phosphorylation of RPS6K as compared with control. This might be due to the opposing effects of PRKAA and AKT on MTORC1. Metformin and AKT inhibitor treatment slightly inhibited the phosphorylation of RPS6K in HCV-infected cells compared with the metformin-treated group, because the inhibition of AKT activated MTORC1. These results confirmed HCV-induced autophagy is the result of the interaction between the inhibition of PRKAA and the inhibition of AKT. Metformin and AKT inhibitor cotreatment did not increase LC3B-II (, lane 6), in accordance with the results we obtained for AICAR with the same underlying mechanism.
Figure 11. Metformin inhibits HCV replication by activating PRKAA and AKT. (A) Mock- infected Huh7 (mock) and Huh7 infected with 10 MOI JFH1 virus (JFH1) were treated with 1mM metformin (activates PRKAA, a widely used drug for treatment of type 2 diabetes) without or with 0.25 μM AKT Inhibitor VIII (a pleckstrin homology (PH) domain-dependent AKT inhibitor) for 3 d before harvesting 5 d postinfection. Phosphorylation of PRKAA, ACACA, AKT and RPS6K, SQSTM1, LC3B and HCV core protein were analyzed by western blot. ACTB was used as a sample loading control. (B) JFH1-infected Huh7 cells (MOI = 10) were treated with 1 mM metformin (met) without or with 0.25 μM AKT Inhibitor VIII (AKT in) 24 h postinfection. Intracellular HCV RNA was analyzed by real-time PCR at 24, 48 and 72 h post-treatment. The amount of intracellular HCV RNA was normalized to the control group at 24 h. The control groups were supplied with DMSO. *p < 0.05 was considered significant. (C) Cell viability was analyzed by MTT assay in groups mentioned in (B). The value of each group was normalized to control group at each time-point respectively.
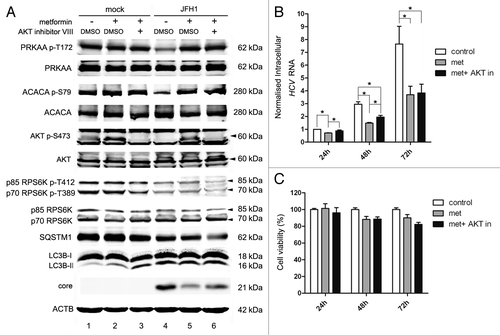
In mock-infected Huh7 cells, metformin did not significantly enhance the phosphorylation of PRKAA T172 and ACACA S79, but significantly enhanced the phosphorylation of AKT S473 (, lanes 1 and 2). As mentioned above, the activation of AKT inhibits autophagy, thus, metformin should theoretically inhibit autophagy in mock-infected cells by activating AKT, but metformin slightly enhanced basal autophagy as indicated by the increase of LC3B-II (, lanes 1 and 2), suggesting that metformin might also induce autophagy through other mechanisms independent of PRKAA and AKT activation.
Metformin inhibited HCV replication by activating both PRKAA and AKT
It has been reported that metformin can activate PRKAA and subsequently inhibit HCV replication.Citation61,Citation62 Thus, we tested the ability of metformin to inhibit HCV replication in our system. As shown in lane 5 of , treatment with metformin inhibited the expression of core protein in HCV-infected cells at 72 h post-treatment. HCV RNA was also depressed at 24, 48 and 72 h post-treatment with metformin as compared with control group (). These findings are in agreement with Mankouri et al. and Nakashima et al.,Citation61,Citation62 who reported that activation of PRKAA by metformin inhibits HCV replication.
As AKT was also activated by metformin in our study, we examined if AKT also participated in the inhibition of HCV replication. As shown in lane 6 of , treatment with metformin and AKT inhibitor slightly increased core protein expression at 72 h and HCV RNA at 24 and 48 h post-treatment comparing with metformin alone, which suggested that activation of AKT by metformin also contributed to the inhibition of HCV replication. However, the intracellular HCV RNA level did not show significant differences between metformin and AKT inhibitor treated and control group at 24 h and between metformin mono- and co-treated groups at 72 h. These results suggest that AKT acted more rapidly but not persistently on the inhibition of HCV RNA. In contrast, PRKAA acted more slowly than AKT, but persistently.
The two treatment groups had no significant effect on cell viability compared with controls ().
Discussion
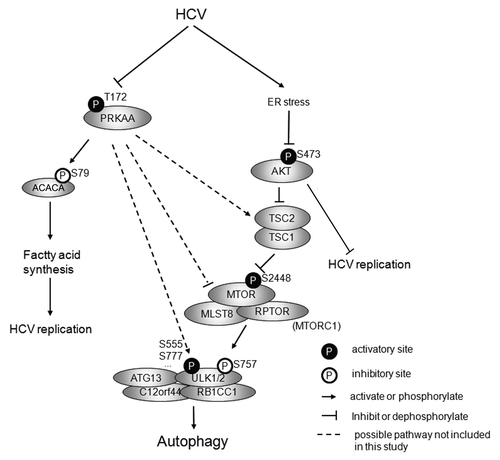
In this study we analyzed several proteins that are involved in the regulation of autophagy during HCV infection. We found that HCV-N full-length replicon and JFH1 virus both induced ER stress which inhibited the AKT-TSC-MTORC1 pathway and subsequently upregulated autophagy (summarized in ). We also found PRKAA was inhibited in HCV-infected cells. The activation of PRKAA by AICAR, an agonist of PRKAA, rescued HCV-induced inhibition of PRKAA activation and slightly enhanced HCV induced autophagy. In contrast, metformin, another PRKAA agonist, activated both PRKAA and AKT and decreased HCV replication.
Figure 12. Proposed model for HCV-induced autophagy. HCV infection induces ER stress, which inhibits AKT-TSC-MTORC1 pathway to inhibit the inhibitory site of ULK1 and subsequently upregulates autophagy. HCV also inhibits PRKAA activity to downregulate autophagy through different possible pathways. During HCV infection, the inhibition of AKT results in the upregulation of autophagy and overrides the inhibition of PRKAA-driven downregulation of autophagy, which finally leads to autophagy. The inhibition of PRKAA enhances HCV replication by inhibiting the activity of ACACA. The inhibition of AKT also promotes HCV replication, but the underlying mechanism is unknown.
Ke and ChenCitation20 used 10 MOI of HCV to infect Huh7 cells (the same cell line used in our study). UPR and the transformation from LC3B-I to LC3B-II were significantly activated on day 6 to 9 after infection. We used the same MOI to infect Huh7 cells and found a significant increase of LC3B-II did not appear until 5 d after infection, similar to this previous report. We examined the puncta formation of endogenous LC3. Significant puncta formation was found in HCV-infected cells, but not in uninfected cells. More significant puncta formation was present on day 5 compared with days 3 and 4. We concluded that the induction of autophagy reflected the state of HCV infection. Because the infection duration lasts for 5 d, Huh7 cells were seeded at very low density (approximately 5% confluence) before infection, to avoid overgrowth induced cellular apoptosis. We determined the cell viability on day 5 postinfection and found that although cellular proliferation was slightly inhibited, a cytopathic effect and apoptosis were not observed by microscopy. Since the Huh7 cell line is not as permissive for HCV infection as Huh7.5 and Huh7.5.1 (two derived cell lines of Huh7), the replication of HCV in Huh7 cells might not be very efficient and apoptosis may not follow infection. Taken together, the autophagy in our study was primarily induced by HCV infection, but not from cytopathic effects.
The treatment of JFH1-infected cells with CQ inhibited the degradation of SQSTM1 and resulted in the aggregation of LC3B-II, suggesting that complete autophagy was induced in our system. This is consistent with Ke and Chen’s report.Citation20 However, Sir et al. reported that transfection of JFH1 RNA into Huh7.5 cells induces an incomplete autophagy.Citation17 This controversy might be caused by the use of different experimental systems such as the use of different cell lines or transfection with viral RNA vs. infection with viral particles.
In contrast to our findings, a recent study by Krishnan’s group found that HCV activated MTORC1 to initiate host translation 3 d after Huh7.5 cells were infected with 1 MOI of JFH1 virus.Citation63 There is no information about the status of autophagy in Krishnan’s system, but it is clear that the activation of MTORC1 downregulates autophagy.Citation23-Citation26 In our study, we assayed two substrates of MTORC1, RPS6K and ULK1, and both showed inhibition of MTORC1. We also infected Huh7 cells with 1 MOI of JFH1 and determined the activity of MTORC1. We did not see activation of MTORC1 on day 3 (Fig. S7) and only a slight inhibition of MTORC1 was observed on day 5 with 1 MOI. The reason for this difference between our study and Krishnan’s might be that we used different cell lines and they came from disparate sources. Huh7.5 cells are derived from Huh7 cells and is highly permissive for HCV replication.Citation64 Results from different studies using different Huh7 cell stocks may differ from each other. Sainz et al. reported that Huh7 cell lines from different sources do vary in morphology, cell growth and permissiveness for HCV infection.Citation65 Therefore, the infection model used could exert an influence on the results obtained.
Activation of the MTORC1 pathway could not completely reverse HCV- induced autophagy, which suggests other mechanisms independent of MTOR could be at work. Shrivastava et al. found that JFH1 induced autophagy in immortalized human hepatocytes (IHH) through the upregulation of BECN1 independent of MTOR, which might explain our finding.Citation66 It was suggested that HCV-induced autophagy might act upstream of MTOR and activate MTORC1. The differences between Shrivastava’s study and ours might be due to the different infection systems we used. It seems that the regulation of autophagy by MTORC1 might be more important in our system.
Our studies revealed that the activity of AKT was inhibited by HCV infection in Huh7 cells. Similarly, the study by Banerjee et al. reveals that HCV impairs the AKT signaling pathway for insulin resistance in Huh7 and immortalized human hepatocytes.Citation67 However, four other studies reported that HCV activated the AKT pathway to enhance cell survival or increase proliferation.Citation68-Citation71 To determine if HCV activates AKT, we analyzed the status of AKT phospho-S473 at 24, 48 and 72 h post-HCV infection and did not see enhanced phosphorylation of AKT (data not shown). Despite the discrepancy in cell types, cell resources, HCV genotypes and HCV strains, the cell-culture conditions might also alter the outcome of HCV infection. Like PDGF-BB, activated AKT1 and knockdown of TSC2 could not completely reverse autophagy.
It was noted that AKT S473 and T308 were highly phosphorylated in 293T cells, but phospho-AKT substrates were more intense in Huh7 cells. Because the substrates recognized by this antibody could also be regulated by other Arg-directed kinases, the phosphorylation of AKT might not be consistent with the phosphorylation of AKT substrates in different cell lines.
Although it is known that ER stress induces autophagy,Citation12 the underlying mechanisms are not well understood. The possible signaling pathways involved in ER stress-induced autophagy were reviewed by Cheng and Yang.Citation72 Based on our findings that inhibition of the AKT-TSC-MTORC1 pathway led to autophagy, and HCV was able to induce acute and chronic ER stress,Citation73 we asked whether ER stress regulated the AKT pathway. ER stress was responsible for the inhibition of AKT during HCV infection. This is consistent with several reports that ER stress inhibits the AKT pathway.Citation57,Citation74 Qin et al. reported that ER stress negatively regulated the AKT-TSC-MTOR pathway to enhance autophagy.Citation57 It has been reported that the EIF2AK3-EIF2S1 pathway takes part in ployQ72-induced Atg12 upregulation and LC3 conversion.Citation75 Thus, other mechanisms independent of the AKT-MTOR pathway might be involved in ER stress-induced autophagy. This could explain why activating MTOR by PDGF-BB, expressing activated AKT, or knocking down TSC2 in HCV-infected cells cannot completely reverse autophagy. It is notable that using 4-PBA or suppressing the ER stress pathway by knocking down UPR-related molecules in HCV-infected cells could not completely reverse autophagy either. There might be a few reasons for this phenomenon: (a) chemicals had side effects; 4-PBA slightly enhanced basal autophagy in Huh7 cells as shown in ; (b) 4-PBA could not completely inhibit ER stress; (c) depressing one pathway of UPR could not completely reverse autophagy; and (d) some other mechanism independent of ER stress might be involved in HCV-induced autophagy. Although the ER stress-AKT pathway during HCV infection was not included in this study, it might be explained by a previous study. ER stress inhibited AKT signaling through inhibition of MTORC2, an activating kinase for AKT.Citation74
The expression of ERN1 was significantly higher in HCV-N cells than in JFH1-infected cells (). This discrepancy might have resulted from the different genotypes of HCV we used, as the HCV replicon used in our system was genotype 1b, whereas the JFH1 virus was genotype 2a. The duration of HCV infection might affect host protein expression,Citation76 as HCV-N was continuously replicated in the stable cell line and JFH1 virus only infected the cells for a few days (5 d maximal) before analysis.
It is consistent with previous reports that HCV inhibited PRKAA to facilitate HCV genome replication.Citation61 The inhibition of PRKAA downregulates autophagy, but HCV needs the autophagy machinery to initiate replication and produce infectious virus particles.Citation41,Citation45,Citation46,Citation77 Therefore, other pathways are likely to upregulate autophagy. Our finding showed that the AKT-TSC-MTORC1 pathway needs to be inhibited to activate autophagy.
Theoretically, the activity of PRKAA should not be completely inhibited, because the activity of PRKAA is needed for autophagy.Citation27-Citation29,Citation78 Under basal conditions, MTORC1 phosphorylates ULK1 S757 and disrupts the interaction between PRKAA and ULK1, thus inhibiting autophagy.Citation27 HCV inhibits MTORC1 and dephosphorylates ULK1 S757, which may promote the phosphorylation of other ULK1 sites by PRKAA and subsequently induce autophagy. This could explain why the treatment of HCV-infected cells with AICAR activated PRKAA and enhanced autophagy further. It seems that the initiation of autophagy during HCV infection depends on the balance between these two pathways, but the positive-regulation effect of inhibited AKT is much stronger than the negative-regulation of inhibited PRKAA, and the tipping of this balance leads to autophagy. PRKAA directly affects ULK1 by phosphorylation, and indirectly regulates it via the TSC1/2 complex or MTORC1.Citation34,Citation79 The exact pathway by which PRKAA regulates HCV-induced autophagy is not included in this study. Possible pathways are summarized in .Citation17,Citation41,Citation44-Citation48
A major concern of our study was that regulating the autophagy pathways with chemicals or shRNA of host factors on the autophagy-related pathways had no influence on JFH1 propagation. Sir and colleagues found that knockdown of genes important for autophagosome formation suppressed HCV RNA replication.Citation17 Ferraris, et al. reported that HCV NS3 and NS5A proteins, HCV RNA, and LC3-II colocalize on autophagosome-like membrane vesicles.Citation80 A very recent study by Sir, et al. showed that HCV uses the membranes of autophagic vacuoles for RNA replication.Citation47 However, Dreux, et al. reported that autophagy is required for the initiation of HCV replication, but is not required once the infection is established.Citation45 The induction of autophagy pathways occurs relatively late (on day 5) after HCV infection, when JFH1 infection had been established. It therefore seems reasonable that the inhibition of autophagy had no impact on HCV replication in our study. The inability of autophagy-related genes knockdown to influence intracellular production of HCV proteins and RNA from Tanida’s study also supports this explanation.Citation46 The discrepancy between these studies may result from the difference between the HCV particle-infection system and the transfection of HCV RNA or HCV replicon system and the difference in cell lines and resources.
Shrivastava, et al. and Ke, et al. both reported that knockdown of key modulators of autophagy enhanced the innate immune response to inhibit HCV replication. In our study, the innate immune response might not be inhibited by autophagy or the partial inhibition of autophagy might be redundant in inhibiting the innate immune response. Furthermore, Mizui, et al. and Ke, et al. both reported that CQ treatment inhibits HCV replication.Citation20,Citation41 Mizui, et al. suggest CQ targets autophagic proteolysis and inhibits HCV replication independently of producing interferon (IFN), which processes strong anti-HCV effects. However, Ke, et al. found that complete autolysosome formation represses the innate immune response, thus promoting HCV replication. In contrast, CQ treatment had no impact on HCV core protein and RNA expression in our system. These controversies might be explained by the different cell stocks used in these studies. In summary, considering the use of different cell lines or stocks in these studies and the fact that autophagy was not totally abrogated by treatment, the inhibition of autophagy was not sufficient to inhibit the established infection in our system.
As a PRKAA agonist, metformin was capable of overriding the inhibition of PRKAA induced by HCV to inhibit HCV replication.Citation61,Citation62 Interestingly, we found that metformin was not PRKAA specific. It could also activate AKT and this activation also contributed to the inhibition of HCV replication. Our findings indicate that the AKT effect is rapid and temporary, but PRKAA works more slowly and persistently to inhibit HCV. Metformin has anti-HCV effects independent of PRKAA activationCitation62 and our study provided a possible explanation for this finding. It is known that PRKAA abrogates lipid accumulation in HCV-infected cells and inhibits HCV replication.Citation61 It should be noted that autophagy was enhanced in metformin and AKT inhibitor cotreated cells as compared with cells treated with metformin alone (). It seems reasonable that enhanced autophagy promoted HCV replication in the co-treated cells. As the autophagy was not important for viral replication in our system, AKT activation by metformin might act to inhibit HCV replication independent of autophagy. It has been reported that blockage of the PIK3CA-AKT-MTOR pathway increases HCV replication,Citation67,Citation81 but the mechanism is not well understood.Citation68,Citation81 It could be that phosphorylation of HCV viral proteins participating in replication is regulated by the AKT pathway. Activation of the AKT-MTOR pathway can induce NS5A phosphorylation, leading to reduction in HCV replication.Citation82 This mechanism may act more rapidly than PRKAA, explaining why we only observed the inhibitory effect from AKT during early stages. The expression of core protein in the metformin-treated group was slightly higher than the combination group at 72 h post-treatment, although RNA expression was not different (). Cotreatment inhibited HCV replication more slowly than metformin alone. We speculated that core protein might accumulate in cotreated cells, which might result in the level of intracellular core protein lagging behind the RNA response. To verify this speculation, we analyzed the core protein level at 96 h post-treatment and found there was no difference between the two groups (Fig. S6). In contrast to Mankouri’s finding that AICAR inhibits HCV replication by activating PRKAA,Citation61 AICAR had no impact on HCV core protein in our study. We treated HCV- infected cells with AICAR only for 12 h, but HCV proteins did not decrease until 24 h post-treatment in Mankouri’s study.
We noted that the expression of activated AKT1 did not inhibit core protein or HCV RNA expression (; Fig. S4C). AKT has three isoforms (AKT1-3) with great homology.Citation53,Citation54 Some AKT functions may be specific to one isoform, but not others. Santi, et al. reported that AKT1-3 is localized mainly in the cytoplasm, mitochondria and nucleus, respectively, in many cancer cells,Citation83 which suggests isoform-specific functions. In our study, metformin treatment might not just act on one AKT isoform. AKT inhibitor VIII can inhibit all three AKT isoforms, as suggested by the manufacturer. The overexpression of AKT1 might not inhibit HCV replication and metformin might also activate AKT2 or AKT3, which could play a role in metformin-triggered inhibition of HCV replication.
In conclusion, we found that HCV infection inhibited the activity of PRKAA and AKT. Inhibition of the AKT-TSC-MTORC1 pathway by ER stress overrode the downregulation of autophagy by inhibited PRKAA, with a net result of enhanced autophagy. We have provided a pathway by which HCV induced autophagy via ER stress. Further studies will be required to determine if HCV protein(s) or RNA directly activates the UPR, how UPR regulates AKT and other possible pathways from ER stress to autophagy in HCV infection. We also found metformin could inhibit HCV replication not only by activating PRKAA but also by activating AKT. Additional studies will be needed to better define this mechanism.
Materials and Methods
Cells, antibodies and reagents
The human hepatoma cell line Huh7 was a gift from Dr. Yue Wang (Institute for Viral Disease Control and Prevention, China CDC) and was cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, 11995) supplemented with 10% fetal bovine serum (Life Technologies), 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mM non-essential amino acids (Life Technologies, 11140) (‘complete DMEM’). Huh7 harboring the full-genomic replicon of HCV-N strain (genotype 1b) was a gift from Dr. Stanley Lemon (University of Texas Medical Branch) and maintained in complete DMEM plus 500 μg/ml G418 (Sigma-Aldrich, G8168). Huh7.5.1 cell line, a highly permissive cell clone for HCV replication (described in ref. Citation84), was provided by Dr. Francis V. Chisari (The Scripps Research Institute) and grown in complete DMEM with additional 2 mM L-glutamate (Life Technologies, 25030). All experiments were performed on Huh7 cells. Huh7.5.1 was only used in HCV propagation. The 293FT cell line was purchased from Invitrogen and maintained in complete DMEM with additional 2 mM L-glutamine.
The following antibodies were obtained from Cell Signaling Technology: anti-ACACA (3676), anti-Phospho-ACACA (S79) (3661), anti-AKT (9272), anti-Phospho-AKT (T308) (9275), anti-Phospho-AKT (S473) (4060), anti-Phospho-AKT Substrate (RXXS*/T*) (9614), anti-PRKAA (2603), anti-Phospho-PRKAA (T172) (2535), anti-Phospho-PRKAA (S485) (4185), anti-TSC2 (4308), anti-MTOR (2983), anti-Phospho-MTOR (S2448) (2971), anti-ULK1 (4776), anti-Phospho-ULK1 (S757) (6888), anti-p70 RPS6K (2708, also recognizes p85 RPS6K), anti-Phospho-p70 RPS6K (T389) [9234, also recognizes Phospho-p85 RPS6K (T412)], anti-EIF2AK3 (3192), anti-Phospho-EIF2AK3 (T980) (3179), anti-EIF2S1 (2103), anti-Phospho-EIF2S1 (S51) (3398), anti-HSPA5/GRP78 (3183), anti-DDIT3/CHOP (2895) and anti-ERN1 (3294). Anti-ACTB (A3853), anti-LC3B (L7543) and anti-SQSTM1 (P0067) antibodies were obtained from Sigma-Aldrich. Anti-ATF6 (sc-22799), anti-HCV core protein (sc-57800) and anti-XBP1 (sc-7160) antibodies were obtained from Santa Cruz. IRdye 680RD labeled anti-rabbit IgG (926-68071) and 800CW labeled anti-mouse IgG (926-32210) secondary antibodies were obtained from LI-COR Biotechnology.
Chloroquine (CQ, C6628), 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside (AICAR, A9978), metformin (D150959) and 4-phenylbutyric acid (4-PBA, P21005) were purchased from Sigma-Aldrich and solubilized in sterilized H2O. AKT Inhibitor VIII (A6730), Compound C (P5499) and thapsigargin (Tg, T9033) were also obtained from Sigma-Aldrich and solubilized in dimethyl sulfoxide (DMSO, Sigma-Aldrich, D8148). All regents were stored at −20°C in single use aliquots. Recombinant human platelet-derived growth factor-BB (PDGF-BB, 100-14B) was obtained from PeproTech, reconstituted in complete DMEM, and stored in aliquots at −80°C.
Production, titration and infection of HCV JFH1 strain (genotype 2a)
The plasmid pcDNA6/TR-Tight/JFH1-FL/AR harboring JFH1 strain cDNA (described previously in ref. Citation85) was transfected into Huh7.5.1 cells. At 24 h post-transfection, 5 μg/ml blasticidin (Life Technologies, R21001) was added to the cells and the selected pool was cultured for 10 d before collecting the supernatant containing HCV. To generate HCV stock, the supernatant collected above was used to infect Huh7.5.1 at the multiplicity of infection (MOI) 0.01. The infected cells reached confluence on day 4 postinfection and were split at a ratio of 1:6. The supernatant was collected and supplemented with 20 mM HEPES buffer (pH 7.5) on day 8 or 9 postinfection and centrifuged at 2000 × g, 4°C, to remove cellular debris. The supernatant was concentrated using Amicon Ultra 100 kDa centrifugal filter devices (Millipore, UFC910096) and aliquoted for storage at −80°C.
The titration of infectious HCV using immunofluorescence staining for HCV core protein was performed as described previously.Citation49 The titer was expressed as focus-forming units per milliliter (FFU/ml) by calculating the average number of HCV core-positive foci at the highest dilutions.
For experiments in this study, 2 × 105 Huh7 cells were infected (MOI = 10) for 12 h in a 100-mm culture dish with HCV stock supplemented with 4 μg/ml polybrene (Sigma-Aldrich, H9268). The infected cells were collected for analysis on day 3 or 5 postinfection.
Lentivirus packaging, titration and transduction
For expressing myristoylated AKT1 (Myr-AKT) and kinase dead AKT1 (AKT mut, T308A, S473A) in Huh7 cells, AKT1 genes were amplified from a commercial AKT1 cDNA clone (Origene Technologies, SC116883) by PCR and inserted into the lentiviral transfer vector pLVX-IRES-ZsGreen1 (Clontech, 632187). For gene knockdown experiments, the short hairpin RNA (shRNA) transfer plasmids for TSC2, EIF2AK3, ATF6, ERN1 and a nontarget negative control from the MISSION TRC library (Sigma-Aldrich) were used. For packaging lentiviruses, 6 μg lentiviral packaging mix (Sigma-Aldrich, SHP001) and 3 μg transfer vector mentioned above were cotransfected into 4 × 106 293FT cells with 27 μl Fugene HD according to the manual (Roche, 12134200). The supernatants containing lentiviruses were collected, filtered and stored at −80°C as aliquots. The titer of lentiviruses was measured with the Lenti-X p24 Rapid Titer Kit following the product’s manual (Clontech, 632200). For transduction, Huh7 cells were incubated with viral stocks supplemented with 4 μg/ml polybrene for 3 h, and then supplied with fresh medium. Cell samples were collected for analysis 72 h after transduction. Specific shRNA clones and infection MOI were selected according to the target downregulation efficiency and the absence of cytotoxic effects. The following shRNA clones were used in this study: TSC2, TRCN0000040182, EIF2AK3, TRCN0000001401, ATF6, TRCN0000017855 and ERN1, TRCN0000000530.
SDS-PAGE and western blot
Cells were lysed in lysis buffer (50 mM TRIS-HCl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate and 0.1% sodium dodecyl sulfate) containing a protease inhibitor cocktail (Roche, 04693132001) and a phosphatase inhibitor cocktail (Roche, 04906845001). The lysates were centrifuged at 16000 × g, 4°C, and the protein concentration of the supernatant was measured with a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, 23225). Equal amounts of total protein were electrophoresed in a sodium dodecyl sulfate-polyacrylamide gel and then transferred onto BioTrace nitrocellulose membrane (Pall, 66485). After blocking with 5% BSA in phosphate buffered saline (PBS) at a (pH 7.4, the membranes were incubated with primary antibodies [1 μg/ml diluted in PBS with 5% BSA (Sigma-Aldrich, A7906) and 0.1% Tween-20 (Sigma-Aldrich, P1379)], followed by incubation with Irdye-labeled secondary antibodies mentioned above. Finally, the membrane was scanned using an Odyssey Infrared Imaging System (LI-COR Biosciences). Only brightness and contrast were adjusted on the images. At least three independent experiments were performed and a representative result was shown. For the grayscale analysis, blot bands were analyzed and calculated using Quantity One software (Bio-Rad). For statistic analysis, standard error and p value (Student’s t-test) were calculated from at least three independent experiments using Prism 5 software (GraphPad Software Inc.). p < 0.05 was considered significant.
Immunofluorescence and confocal microscopy
For analysis of endogenous LC3B, cells were fixed using ice-cold methanol for 20 min and then permeabilized using 0.05% saponin (Sigma-Aldrich, 47036) in 2% FBS PBS for 30 min. After blocking, cells were incubated with LC3B antibody for 2 h and then incubated with HCV core antibody for another 2 h. Cells were then incubated with Dylight 488-conjugated anti-rabbit secondary antibody (Jackson ImmunoResearch, 711-485-152) and Rhodamine-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch, 715-295-150) for 1 h, followed by incubation with DAPI (Life Technologies, D1306) for nuclear staining.
For analysis of autophagy flux, we used the mTagRFP-mWasabi-LC3B construct, which was kindly provided by Dr. Jian Lin (College of Chemistry and Molecular Engineering, Peking University). Cells were fixed using 4% paraformaldehyde (Sigma-Aldrich, 16005) in PBS for 15 min on ice and permeabilized using 0.3% Triton X-100 (Sigma-Aldrich, T8787) and 2% FBS in PBS for 30 min. After blocking, cells were incubated with HCV core protein and then with Dylight 405-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch, 715-475-150) for 1 h. The images were analyzed using a TCS SP5 Laser Scanning Confocal Microscope and its software (Leica Microsystems CMS GmbH).
Flow cytometry
For analysis of apoptosis in HCV-infected Huh7 cells, the cells were detached by trypsinization and stained with an Annexin-V-FITC PI kit (NeoBioscience, FAK012) following the manual. Briefly, cells were incubated with Annexin-V-FITC for 10 min and then with PI for 5 min. The cells were analyzed immediately after staining using a FACS Canto II Flow Cytometry machine (Becton Dikinson) and Flowjo software (Tree Star, Inc.).
Real-time PCR
RNA from cell samples was extracted and purified using a SV Total RNA Isolation kit (Promega, Z3100). One microgram of total RNA was reverse transcribed in a 20 μl volume using a Reverse Transcription kit with random primers (Promega, A3500). One microliter of the resulting cDNA was used in real-time PCR with Power SYBR® Green PCR master mix (Life Technologies, 4367659). Each experiment was performed in triplicate. The following primers were used:
JFH1 sense: 5′-TCTGCGGAACCGGTGAGTA-3′,
JFH1 anti-sense: 5′-TCAGGCAGTACCACAAGGC-3′,
GAPDH sense: 5′-TGGGCTACACTGAGCACCAG-3′,
GAPDH anti-sense: 5′-AAGTGGTCGTTGAGGGCAAT-3′.
The reaction was run on a 7900HT Fast Real-Time PCR machine (Life Technologies) and the result was analyzed using RQ manager software (Life Technologies).
| Abbreviations: | ||
| 4-PBA | = | 4-phenylbutyric acid |
| ACACA | = | acetyl-CoA carboxylase alpha |
| ACTB | = | actin, beta |
| AICAR | = | 5-aminoimidazole-4-carboxamide 1-β-D-ribofuranoside |
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| ATF6 | = | activating transcription factor-6 |
| CQ | = | chloroquine |
| DDIT3 | = | DNA-damage-inducible transcript 3 |
| EIF2AK3 | = | eukaryotic translation initiation factor 2-alpha kinase 3 |
| EIF2S1 | = | eukaryotic translation initiation factor 2, subunit 1 alpha |
| ERN1 | = | endoplasmic reticulum to nucleus signal-1 |
| HCV | = | hepatitis C virus |
| HSPA5 | = | heat shock 70kDa protein 5 |
| LC3B | = | MAP1LC3B, microtubule-associated protein 1 light chain 3 beta |
| LC3B-I | = | unlipidated form of LC3B |
| LC3B-II | = | phosphatidylethanolamine-conjugated form of LC3B |
| MTOR | = | mechanistic target of rapamycin |
| MTORC1 | = | MTOR complex 1 |
| Myr-AKT | = | myristoylated AKT |
| PDGF-BB | = | platelet-derived growth factor-BB |
| PI | = | propidium iodide |
| PIK3CA | = | phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha |
| PRKAA | = | protein kinase, AMP-activated, alpha |
| RPS6K | = | ribosomal protein S6 kinase |
| SQSTM1 | = | sequestosome 1 |
| Tg | = | thapsigargin |
| TSC1/2 | = | tuberous sclerosis 1/2 |
| ULK1 | = | UNC-51-like kinase 1 |
| XBP1 | = | X-box binding protein 1 |
Additional material
Download Zip (6 MB)Acknowledgments
The work is sponsored by grants (2009ZX10004-303) Eleven-fifth Mega-Scientific project on prevention and treatment of AIDS, viral hepatitis and other infectious diseases from P.R. China. Thanks Dr. Jian Lin from Peking University for providing the mTagRFP-mWasabi-LC3B construct. Special thanks to Dr. Tim Burkers for critically reading and revising the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/22791
References
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005; 5:558 - 67; http://dx.doi.org/10.1016/S1473-3099(05)70216-4; PMID: 16122679
- Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001; 345:41 - 52; http://dx.doi.org/10.1056/NEJM200107053450107; PMID: 11439948
- Raimondi S, Bruno S, Mondelli MU, Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J Hepatol 2009; 50:1142 - 54; http://dx.doi.org/10.1016/j.jhep.2009.01.019; PMID: 19395111
- Dal Maso L, Franceschi S. Hepatitis C virus and risk of lymphoma and other lymphoid neoplasms: a meta-analysis of epidemiologic studies. Cancer Epidemiol Biomarkers Prev 2006; 15:2078 - 85; http://dx.doi.org/10.1158/1055-9965.EPI-06-0308; PMID: 17119031
- White DL, Ratziu V, El-Serag HB. Hepatitis C infection and risk of diabetes: a systematic review and meta-analysis. J Hepatol 2008; 49:831 - 44; http://dx.doi.org/10.1016/j.jhep.2008.08.006; PMID: 18814931
- Pereira AA, Jacobson IM. New and experimental therapies for HCV. Nat Rev Gastroenterol Hepatol 2009; 6:403 - 11; http://dx.doi.org/10.1038/nrgastro.2009.92; PMID: 19575025
- Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol 2007; 5:453 - 63; http://dx.doi.org/10.1038/nrmicro1645; PMID: 17487147
- Egger D, Wölk B, Gosert R, Bianchi L, Blum HE, Moradpour D, et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J Virol 2002; 76:5974 - 84; http://dx.doi.org/10.1128/JVI.76.12.5974-5984.2002; PMID: 12021330
- Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 2003; 77:5487 - 92; http://dx.doi.org/10.1128/JVI.77.9.5487-5492.2003; PMID: 12692249
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8:519 - 29; http://dx.doi.org/10.1038/nrm2199; PMID: 17565364
- Kim R, Emi M, Tanabe K, Murakami S. Role of the unfolded protein response in cell death. Apoptosis 2006; 11:5 - 13; http://dx.doi.org/10.1007/s10495-005-3088-0; PMID: 16374548
- Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem 2006; 281:30299 - 304; http://dx.doi.org/10.1074/jbc.M607007200; PMID: 16901900
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741 - 52; http://dx.doi.org/10.1038/nrm2239; PMID: 17717517
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861 - 73; http://dx.doi.org/10.1101/gad.1599207; PMID: 18006683
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol 2008; 82:2241 - 9; http://dx.doi.org/10.1128/JVI.02093-07; PMID: 18077704
- Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008; 48:1054 - 61; http://dx.doi.org/10.1002/hep.22464; PMID: 18688877
- Sir D, Liang C, Chen WL, Jung JU, Ou JH. Perturbation of autophagic pathway by hepatitis C virus. Autophagy 2008; 4:830 - 1; PMID: 18635950
- Estrabaud E, De Muynck S, Asselah T. Activation of unfolded protein response and autophagy during HCV infection modulates innate immune response. J Hepatol 2011; 55:1150 - 3; http://dx.doi.org/10.1016/j.jhep.2011.04.025; PMID: 21723841
- Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest 2011; 121:37 - 56; http://dx.doi.org/10.1172/JCI41474; PMID: 21135505
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181:497 - 510; http://dx.doi.org/10.1083/jcb.200712064; PMID: 18443221
- Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy 2009; 5:649 - 62; http://dx.doi.org/10.4161/auto.5.5.8249; PMID: 19287211
- Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297 - 305; http://dx.doi.org/10.1074/jbc.M900573200; PMID: 19258318
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981 - 91; http://dx.doi.org/10.1091/mbc.E08-12-1248; PMID: 19211835
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992 - 2003; http://dx.doi.org/10.1091/mbc.E08-12-1249; PMID: 19225151
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett 2010; 584:1287 - 95; http://dx.doi.org/10.1016/j.febslet.2010.01.017; PMID: 20083114
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132 - 41; http://dx.doi.org/10.1038/ncb2152; PMID: 21258367
- Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One 2010; 5:e15394; http://dx.doi.org/10.1371/journal.pone.0015394; PMID: 21072212
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456 - 61; http://dx.doi.org/10.1126/science.1196371; PMID: 21205641
- Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008; 32:Suppl 4 S7 - 12; http://dx.doi.org/10.1038/ijo.2008.116; PMID: 18719601
- Hardie DG. AMPK and Raptor: matching cell growth to energy supply. Mol Cell 2008; 30:263 - 5; http://dx.doi.org/10.1016/j.molcel.2008.04.012; PMID: 18471972
- Aicher LD, Campbell JS, Yeung RS. Tuberin phosphorylation regulates its interaction with hamartin. Two proteins involved in tuberous sclerosis. J Biol Chem 2001; 276:21017 - 21; http://dx.doi.org/10.1074/jbc.C100136200; PMID: 11290735
- Manning J, Beutler K, Knepper MA, Vehaskari VM. Upregulation of renal BSC1 and TSC in prenatally programmed hypertension. Am J Physiol Renal Physiol 2002; 283:F202 - 6; PMID: 12060603
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003; 115:577 - 90; http://dx.doi.org/10.1016/S0092-8674(03)00929-2; PMID: 14651849
- Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 2009; 37:217 - 22; http://dx.doi.org/10.1042/BST0370217; PMID: 19143635
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 2011; 13:1016 - 23; http://dx.doi.org/10.1038/ncb2329; PMID: 21892142
- Alavian SM, Ande SR, Coombs KM, Yeganeh B, Davoodpour P, Hashemi M, et al. Virus-triggered autophagy in viral hepatitis - possible novel strategies for drug development. J Viral Hepat 2011; 18:821 - 30; http://dx.doi.org/10.1111/j.1365-2893.2011.01530.x; PMID: 22093031
- Eng KE, Panas MD, Karlsson Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy 2010; 6:634 - 41; http://dx.doi.org/10.4161/auto.6.5.12112; PMID: 20458170
- Hanslick JL, Lau K, Noguchi KK, Olney JW, Zorumski CF, Mennerick S, et al. Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol Dis 2009; 34:1 - 10; http://dx.doi.org/10.1016/j.nbd.2008.11.006; PMID: 19100327
- Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, Wei Y, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012; 8:1215 - 26; http://dx.doi.org/10.4161/auto.20284; PMID: 22647982
- Mizui T, Yamashina S, Tanida I, Takei Y, Ueno T, Sakamoto N, et al. Inhibition of hepatitis C virus replication by chloroquine targeting virus-associated autophagy. J Gastroenterol 2010; 45:195 - 203; http://dx.doi.org/10.1007/s00535-009-0132-9; PMID: 19760134
- Hoch RV, Soriano P. Roles of PDGF in animal development. Development 2003; 130:4769 - 84; http://dx.doi.org/10.1242/dev.00721; PMID: 12952899
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 2008; 22:1276 - 312; http://dx.doi.org/10.1101/gad.1653708; PMID: 18483217
- Guévin C, Manna D, Bélanger C, Konan KV, Mak P, Labonté P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010; 405:1 - 7; http://dx.doi.org/10.1016/j.virol.2010.05.032; PMID: 20580051
- Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 2009; 106:14046 - 51; http://dx.doi.org/10.1073/pnas.0907344106; PMID: 19666601
- Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009; 5:937 - 45; http://dx.doi.org/10.4161/auto.5.7.9243; PMID: 19625776
- Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, et al. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem 2012; 287:18036 - 43; http://dx.doi.org/10.1074/jbc.M111.320085; PMID: 22496373
- Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011; 53:406 - 14; http://dx.doi.org/10.1002/hep.24073; PMID: 21274862
- Ikeda M, Yi M, Li K, Lemon SM. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J Virol 2002; 76:2997 - 3006; http://dx.doi.org/10.1128/JVI.76.6.2997-3006.2002; PMID: 11861865
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261 - 74; http://dx.doi.org/10.1016/j.cell.2007.06.009; PMID: 17604717
- Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem 1997; 66:807 - 22; http://dx.doi.org/10.1146/annurev.biochem.66.1.807; PMID: 9242925
- Pearson RB, Kemp BE. Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol 1991; 200:62 - 81; http://dx.doi.org/10.1016/0076-6879(91)00127-I; PMID: 1956339
- Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002; 14:381 - 95; http://dx.doi.org/10.1016/S0898-6568(01)00271-6; PMID: 11882383
- Kumar CC, Madison V. AKT crystal structure and AKT-specific inhibitors. Oncogene 2005; 24:7493 - 501; http://dx.doi.org/10.1038/sj.onc.1209087; PMID: 16288296
- Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009; 30:588 - 98; http://dx.doi.org/10.1016/j.immuni.2009.02.009; PMID: 19362021
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004; 11:381 - 9; http://dx.doi.org/10.1038/sj.cdd.4401373; PMID: 14685163
- Qin L, Wang Z, Tao L, Wang Y. ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6:239 - 47; http://dx.doi.org/10.4161/auto.6.2.11062; PMID: 20104019
- de Almeida SF, Picarote G, Fleming JV, Carmo-Fonseca M, Azevedo JE, de Sousa M. Chemical chaperones reduce endoplasmic reticulum stress and prevent mutant HFE aggregate formation. J Biol Chem 2007; 282:27905 - 12; http://dx.doi.org/10.1074/jbc.M702672200; PMID: 17626021
- Giri S, Nath N, Smith B, Viollet B, Singh AK, Singh I. 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 2004; 24:479 - 87; http://dx.doi.org/10.1523/JNEUROSCI.4288-03.2004; PMID: 14724246
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 2001; 108:1167 - 74; PMID: 11602624
- Mankouri J, Tedbury PR, Gretton S, Hughes ME, Griffin SD, Dallas ML, et al. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc Natl Acad Sci U S A 2010; 107:11549 - 54; http://dx.doi.org/10.1073/pnas.0912426107; PMID: 20534540
- Nakashima K, Takeuchi K, Chihara K, Hotta H, Sada K. Inhibition of hepatitis C virus replication through adenosine monophosphate-activated protein kinase-dependent and -independent pathways. Microbiol Immunol 2011; 55:774 - 82; http://dx.doi.org/10.1111/j.1348-0421.2011.00382.x; PMID: 21895746
- George A, Panda S, Kudmulwar D, Chhatbar SP, Nayak SC, Krishnan HH. Hepatitis C virus NS5A binds to the mRNA cap-binding eukaryotic translation initiation 4F (eIF4F) complex and up-regulates host translation initiation machinery through eIF4E-binding protein 1 inactivation. J Biol Chem 2012; 287:5042 - 58; http://dx.doi.org/10.1074/jbc.M111.308916; PMID: 22184107
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 2002; 76:13001 - 14; http://dx.doi.org/10.1128/JVI.76.24.13001-13014.2002; PMID: 12438626
- Sainz B Jr., Barretto N, Uprichard SL. Hepatitis C virus infection in phenotypically distinct Huh7 cell lines. PLoS One 2009; 4:e6561; http://dx.doi.org/10.1371/journal.pone.0006561; PMID: 19668344
- Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol 2012; 86:8705 - 12; http://dx.doi.org/10.1128/JVI.00616-12; PMID: 22674982
- Banerjee S, Saito K, Ait-Goughoulte M, Meyer K, Ray RB, Ray R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J Virol 2008; 82:2606 - 12; http://dx.doi.org/10.1128/JVI.01672-07; PMID: 18160431
- Mannová P, Beretta L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: control of cell survival and viral replication. J Virol 2005; 79:8742 - 9; http://dx.doi.org/10.1128/JVI.79.14.8742-8749.2005; PMID: 15994768
- Park CY, Jun HJ, Wakita T, Cheong JH, Hwang SB. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J Biol Chem 2009; 284:9237 - 46; http://dx.doi.org/10.1074/jbc.M808773200; PMID: 19204002
- Peng L, Liang D, Tong W, Li J, Yuan Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J Biol Chem 2010; 285:20870 - 81; http://dx.doi.org/10.1074/jbc.M110.112045; PMID: 20439463
- Jahan S, Khaliq S, Ijaz B, Ahmad W, Hassan S. Role of HCV Core gene of genotype 1a and 3a and host gene Cox-2 in HCV-induced pathogenesis. Virol J 2011; 8:155; http://dx.doi.org/10.1186/1743-422X-8-155; PMID: 21457561
- Cheng Y, Yang JM. Survival and death of endoplasmic-reticulum-stressed cells: Role of autophagy. World J Biol Chem 2011; 2:226 - 31; http://dx.doi.org/10.4331/wjbc.v2.i10.226; PMID: 22031845
- Merquiol E, Uzi D, Mueller T, Goldenberg D, Nahmias Y, Xavier RJ, et al. HCV causes chronic endoplasmic reticulum stress leading to adaptation and interference with the unfolded protein response. PLoS One 2011; 6:e24660; http://dx.doi.org/10.1371/journal.pone.0024660; PMID: 21949742
- Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW, et al. ER stress inhibits mTORC2 and Akt signaling through GSK-3β-mediated phosphorylation of rictor. Sci Signal 2011; 4:ra10; http://dx.doi.org/10.1126/scisignal.2001731; PMID: 21343617
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230 - 9; http://dx.doi.org/10.1038/sj.cdd.4401984; PMID: 16794605
- Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters KA, Proll SC, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog 2010; 6:e1000719; http://dx.doi.org/10.1371/journal.ppat.1000719; PMID: 20062526
- Dreux M, Chisari FV. Autophagy proteins promote hepatitis C virus replication. Autophagy 2009; 5:1224 - 5; http://dx.doi.org/10.4161/auto.5.8.10219; PMID: 19844160
- Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci U S A 2011; 108:4788 - 93; http://dx.doi.org/10.1073/pnas.1100844108; PMID: 21383122
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214 - 26; http://dx.doi.org/10.1016/j.molcel.2008.03.003; PMID: 18439900
- Ferraris P, Blanchard E, Roingeard P. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J Gen Virol 2010; 91:2230 - 7; http://dx.doi.org/10.1099/vir.0.022186-0; PMID: 20484561
- Ishida H, Tatsumi T, Hosui A, Nawa T, Kodama T, Shimizu S, et al. Alterations in microRNA expression profile in HCV-infected hepatoma cells: involvement of miR-491 in regulation of HCV replication via the PI3 kinase/Akt pathway. Biochem Biophys Res Commun 2011; 412:92 - 7; http://dx.doi.org/10.1016/j.bbrc.2011.07.049; PMID: 21802413
- Coito C, Diamond DL, Neddermann P, Korth MJ, Katze MG. High-throughput screening of the yeast kinome: identification of human serine/threonine protein kinases that phosphorylate the hepatitis C virus NS5A protein. J Virol 2004; 78:3502 - 13; http://dx.doi.org/10.1128/JVI.78.7.3502-3513.2004; PMID: 15016873
- Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. Am J Physiol Cell Physiol 2010; 298:C580 - 91; http://dx.doi.org/10.1152/ajpcell.00375.2009; PMID: 20018949
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 2005; 102:9294 - 9; http://dx.doi.org/10.1073/pnas.0503596102; PMID: 15939869
- Cai Z, Zhang C, Chang KS, Jiang J, Ahn BC, Wakita T, et al. Robust production of infectious hepatitis C virus (HCV) from stably HCV cDNA-transfected human hepatoma cells. J Virol 2005; 79:13963 - 73; http://dx.doi.org/10.1128/JVI.79.22.13963-13973.2005; PMID: 16254332