Abstract
Endoplasmic reticulum (ER) stress induces both autophagy and apoptosis yet the molecular mechanisms and pathways underlying the regulation of these two cellular processes in cells undergoing ER stress remain less clear. We report here that eukaryotic elongation factor-2 kinase (EEF2K) is a critical controller of the ER stress-induced autophagy and apoptosis in tumor cells. DDIT4, a stress-induced protein, was required for transducing the signal for activation of EEF2K under ER stress. We further showed that phosphorylation of EEF2K at Ser398 was essential for induction of autophagy, while phosphorylation of the kinase at Ser366 and Ser78 exerted an inhibitory effect on autophagy. Suppression of the ER stress-activated autophagy via silencing of EEF2K aggravated ER stress and promoted apoptotic cell death in tumor cells. Moreover, inhibiting EEF2K by either RNAi or NH125, a small molecule inhibitor of the enzyme, rendered tumor cells more sensitive to curcumin and velcade, two anticancer agents that possess ER stress-inducing action. Our study indicated that the DDIT4-EEF2K pathway was essential for inducing autophagy and for determining the fate of tumor cells under ER stress, and suggested that inhibiting the EEF2K-mediated autophagy can deteriorate ER stress and lead to a greater apoptotic response, thereby potentiating the efficacy of the ER stress-inducing agents against cancer.
Keywords: :
Introduction
The endoplasmic reticulum (ER) is an organelle that plays important roles in multiple cellular processes, including intracellular calcium homeostasis, protein secretion and lipid biosynthesis, all of which are required for cell survival and normal cellular functions.Citation1 An intact ER function is critical for correct folding of various proteins and for post-translational modifications such as glycosylation and disulfide bond formation. A variety of stimuli and pathological conditions can cause accumulation of unfolded and misfolded proteins in the ER, triggering an evolutionarily conserved response, termed the unfolded protein response (UPR), in order to re-establish homeostasis and to restore ER function.Citation2 The UPR is mediated by three ER membrane-associated proteins: endoplasmic reticulum to nucleus signaling 1 (ERN1), eukaryotic translation initiation factor 2-α kinase 3 (EIF2AK3), and activating transcription factor 6 (ATF6).Citation3 These proteins activate the signal-transduction pathways that alleviate the accumulation of misfolded proteins in the ER via enhancing the protein-folding capacity of the ER, inhibiting new protein synthesis, or accelerating the degradation of proteins. Nonetheless, if the function of the ER fails to be re-established, extensive or sustained, ER stress eventually induces cell death via apoptosis.
Autophagy, a cellular process mainly responsible for degrading long-lived proteins and subcellular organelles, is activated in response to stressful conditions such as starvation, hypoxia, irradiation and growth factor deprivation, and is involved in numerous physiological and pathological processes including cancer, neurodegeneration and aging.Citation4 It has been shown recently that ER stress is also a stimulus that causes activation of autophagy, which plays a critical role in alleviating ER stress,Citation5 and ER-induced autophagy may have important implications in therapeutic intervention of diseases such as cancer. Nevertheless, how ER stress activates autophagy and the functional association between autophagy and apoptosis under ER stress remain less clear. Eukaryotic elongation factor-2 kinase (EEF2K), a calcium/calmodulin-dependent enzyme, is a negative regulator of protein synthesis. EEF2K phosphorylates EEF2 at Ser56, thereby disabling its role in mediating ribosome elongation during mRNA translation.Citation6 EEF2K is overexpressed in several types of malignancies including brain tumors and breast cancers.Citation7-Citation9 Our recent studies have revealed that EEF2K controls the switch between autophagy and apoptosis in cancer cells subjected to therapeutic stress.Citation10 Since EEF2K is activated in response to ER stress,Citation11,Citation12 and ER stress-inducing agents are being tested and developed as anticancer agents, in this study we intended to delineate the exact roles and regulation of EEF2K in the cellular response to ER stress, and to explore whether manipulating ER stress-induced autophagy via EEF2K can be exploited as an approach to reinforcing the effectiveness of ER stress-inducing agents against cancer. Our study demonstrated that DDIT4-mediated activation of EEF2K plays an important regulatory role in controlling the switch between autophagy and apoptosis in cancer cells undergoing ER stress, and that phosphorylation of the kinase at Ser398 is necessary for induction of autophagy. We also showed that suppression of EEF2K-mediated autophagy aggravates ER stress and shuttles more tumor cells into the apoptotic pathway, reinforcing the cytotoxicity of curcumin and velcade against tumor cells.
Results
Autophagy is an earlier response than apoptosis in tumor cells undergoing ER stress
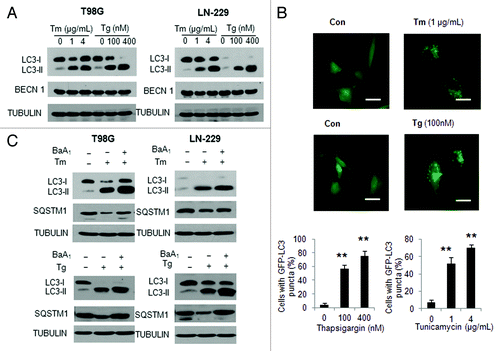
To better understand the relationship between activation of autophagy and apoptosis when tumor cells are undergoing ER stress, we first treated T98G and LN-229 human glioma cells with ER stress inducers thapsigargin (Tg) or tunicamycin (Tm), and then examined how ER stress caused by these two agents affects autophagic response. shows that treatment of glioma cells with Tg or Tm resulted in a dose-dependent elevation of the level of LC3-II, a marker of autophagy. The stimulatory effect of Tg and Tm on autophagy was confirmed by examining GFP-LC3 puncta, which showed an increase in the GFP-LC3 dots in the cells treated with Tg or Tm (), and by detecting the level of the selective autophagy substrate, SQSTM1, which showed a reduction by the treatments (). Since a increased level of LC3-II can be due to either stimulation of the LC3 conversion or the inhibition of the fusion of autophagosome with lysosome, in order to validate the effects of Tg and Tm on autophagy, we measured autophagic flux in the presence of bafilomycin A1, an inhibitor of autophagosome-lysosome fusion. illustrates that LC3-II and SQSTM1 significantly accumulated in the presence of bafilomycin A1 in the cells treated with Tg or Tm, indicating that autophagic flux was enhanced by ER stress ().
Figure 1. Effect of tunicamycin and thapsigargin on autophagy in human glioma cells. (A) LN-229 or T98G cells were treated with Tm or Tg for 24 h. At the end of treatment, the levels of LC3 and BECN1 were examined by western blot. TUBULIN was used as a loading control. (B) LN-229 cells were transfected with a GFP-LC3 plasmid, followed by treatment with 1 μg/ml Tm or 100 nM Tg for 24 h. At the end of treatment, the cells were inspected at 60× magnification for numbers of GFP-LC3 puncta (scale bar: 100 μM). Bars represent the percentage of cells with ≥ 10 GFP-LC3 puncta. At least 100 cells were scored in each treatment. *p < 0.05; **p < 0.01, t-test, Tg or Tm vs. vehicle. (C) LN-229 or T98G cells were treated with 1 μg/ml Tm or 100 nM Tg for 24 h in the absence or presence of 10 nM of bafilomycin A1. At the end of treatment, the levels of LC3 and SQSTM1 were examined by western blot. TUBULIN was used as a loading control.
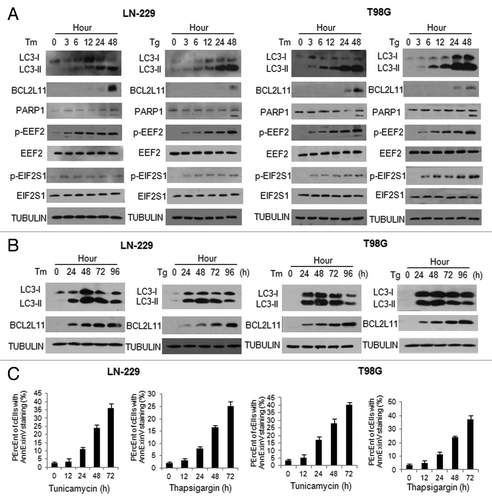
As ER stress also triggers apoptosis, we next compared the time course of apoptosis activation with that of autophagy activation in cells treated with Tg or Tm. shows that the autophagy marker, LC3-II, began to accumulate by 3 h following the drug treatment, and reached a highest level at 48 h. In contrast, BCL2L11 and cleaved PARP1 did not appear until 24 to 48 h following the treatment with Tg or Tm (). These results indicated that autophagy was activated early after the start of treatment, while apoptosis was induced at a later stage of ER stress. Notably, the phosphorylation of EEF2, a negative regulator of protein synthesis, and the phosphorylation of EIF2S1, an important downstream effector of ER stress, were elevated by treatment with Tg or Tm in a time-dependent manner (), suggesting that phosphorylation of these two proteins was likely associated with activation of autophagy and apoptosis under ER stress. To further determine the relationship between autophagy and apoptosis activated by ER stress, we extended our observation of these two cellular processes to 96 h. We found that autophagy induced by ER stress reached a peak at 48 h, and began to decline thereafter (). Of note, when autophagy receded, apoptosis was further activated, as evidenced by an increase in BCL2L11 protein level () and in annexin V staining (). These results indicated that persistent ER stress promoted a switch from autophagy to apoptosis.
Figure 2. Activation of autophagy in tumor cells was an earlier response to ER stress than apoptosis. LN-229 or T98G cells were treated with Tm (1 μg/ml) or Tg (100 nM) for different periods of time. At the end of treatment, (A and B) the levels of LC3, BCL2L11, PARP1, p-EEF2. EIF2S1 and p-EIF2S1 were examined by western blot. TUBULIN was used as a loading control; (C) Apoptosis was determined by flow cytometric analyses of annexin staining.
EEF2K is required for induction of autophagy in tumor cells treated with ER stressors
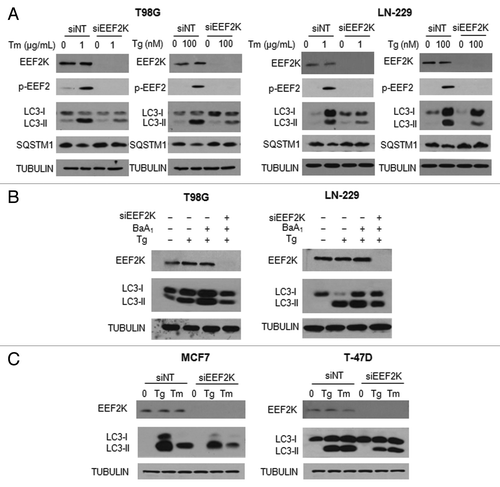
As treatment with Tm or Tg caused an increase in phosphorylation of EEF2 (), we next sought to determine whether there was an association between upregulation of phospho-EEF2 and activation of autophagy. EEF2K is the enzyme that phosphorylates EEF2, thus we examined the effects of knocking down this kinase on ER stress-induced autophagy in glioma cells. shows that silencing of EEF2K expression led to a loss of phospho-EEF2 and markedly blunted autophagic response in the tumor cells treated with Tg or Tm, as determined by a decrease in LC3-II and an increase in SQSTM1. To validate the effect of EEF2K on induction of autophagy, we measured autophagic flux in the cells with silencing of EEF2K expression, following treatment with Tg. shows that inhibition of EEF2K blocked the accumulation of LC3-II in the presence of bafilomycin A1 in the cells treated with Tg, further supporting that the phosphorylation of EEF2 is involved in the induction of autophagy. The effect of EEF2K expression on ER stress-induced autophagy was also observed in the human breast cancer cell lines MCF7 and T-47D (). These experiments demonstrated a regulatory role for EEF2K in autophagic response to ER stress.
Figure 3. Effect of EEF2K expression on ER stress-induced autophagy in tumor cells. (A) LN-229 or T98G cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with Tg or Tm for 24 h. The levels of EEF2K, p-EEF2, LC3 and SQSTM1 were examined by western blot. TUBULIN was used as a loading control. (B) LN-229 or T98G cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with Tg for 24 h in the presence or absence of bafilomycin A1. The levels of EEF2K and LC3 were examined by western blot. TUBULIN was used as a loading control. (C) MCF7 or T-47D cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with 100 nM Tg or 1 μg/ml Tm for 24 h. EEF2K and LC3 were examined by western blot. TUBULIN was used as a loading control.
DDIT4 is essential for activation of EEF2K and induction of autophagy under ER stress
To explore how EEF2K is activated in response to ER stress, we tested the role of DDIT4, a stress-induced protein that acts as a negative regulator of MTOR,Citation13 as the activity of EEF2K is regulated by the MTOR-RPS6KB-mediated phosphorylation at Ser366 and Ser78. shows that treatment of tumor cells with Tg not only upregulated DDIT4 protein but also activated EEF2K, as determined by the level of phosphorylation of its substrate, EEF2. Strikingly, when Tg-induced upregulation of DDIT4 was repressed by siRNA against this stress-induced gene, EEF2K expression and the activity of this kinase (level of phospho-EEF2) were markedly downregulated (). Repression of DDIT4 by siRNA also suppressed the autophagic response induced by Tg treatment, as determined by the decreased amount of the autophagy marker LC3-II (). We further found that in the presence of Tg, downregulation of EEF2K, when expression of DDIT4 was silenced, was rescued by MG132, an inhibitor of proteasome (), suggesting that prevention of the ER stress-induced upregulation of DDIT4 promoted the degradation of EEF2K by the proteasome. To demonstrate the effect of altered DDIT4 level on MTOR activity, we next examined the phosphorylation of RPS6KB and EIF4EBP1, two down-stream effectors of MTOR, in the cells with knockdown of DDIT4 under ER stress. As shown in , suppression of DDIT4 expression relieved the inhibition of MTOR in cells subjected to ER stress, as evidenced by a restoration of phosphorylation RPS6KB and EIF4EBP1. To further demonstrate the role of MTOR in regulating the DDIT4-EEF2K pathway under ER stress, we compared the p-EEF2 level in the cells transfected with a control plasmid or with a plasmid of RHEB, a positive regulator of MTOR, under ER stress. Activation of EEF2K by ER stress was reduced when MTOR activity was rescued by enforced expression of RHEB (Fig. S1), suggesting that the effect of DDIT4 on EEF2K was mediated through MTOR under ER stress.
Figure 4. Silencing of DDIT4 expression blocked activation of EEF2K and suppressed autophagy in tumor cells treated with Tg. (A) LN-229 or T98G cells were transfected with a nontargeting RNA or a siRNA targeting DDIT4, followed by treatment with Tg for 24 h. The levels of DDIT4, EEF2K, EEF2, p-EEF2 and LC3 were examined by western blot. TUBULIN was used as a loading control. (B) LN-229 and T98 G cells were transfected with a nontargeting RNA or a DDIT4-targeted siRNA, followed by treatment with Tg for 24 h in the presence or absence of MG132. The levels of DDIT4 and EEF2K were examined by western blot. TUBULIN was used as a loading control. (C) LN-229 or T98G cells were transfected with a nontargeting RNA or a siRNA targeting DDIT4, followed by treatment with Tg for 24 h. The levels of DDIT4, RPS6KB, p-RPS6KB, EIF4EBP1 and p-EIF4EBP1 were examined by western blot. TUBULIN was used as a loading control. (D) LN-229 or T98G cells were transfected with a nontargeting RNA or a siRNA targeting ATF4, followed by treatment with Tg for 24 h. The levels of ATF4, DDIT4 and LC3 were examined by western blot. TUBULIN was used as a loading control.
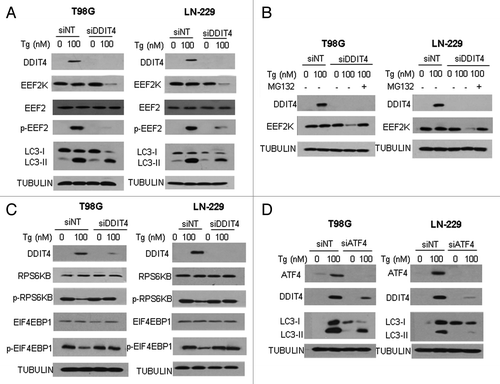
To determine how ER stress upregulates DDIT4, we examined the role of ATF4, as ATF4 has been reported to play a role in induction of DDIT4.Citation14-Citation16 We found that treatment of cells with Tg not only induced expression of DDIT4 and activated autophagy, but also caused an upregulation of ATF4 (); silencing of ATF4 expression blocked the induction of DDIT4 and autophagy by Tg (). These results suggest that ATF4, which is known to be upregulated when EIF2S1 is phosphorylated,Citation17,Citation18 plays a critical role in the induction of DDIT4 expression under ER stress.
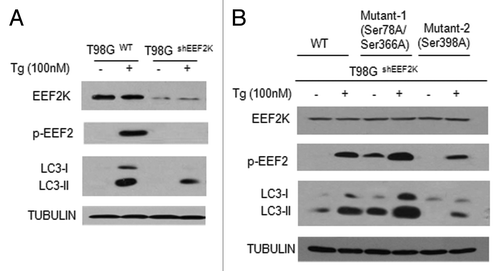
To further demonstrate the roles of the DDIT4/MTOR-mediated phosphorylation of EEF2K in regulating autophagy induced by ER stress, we constructed the phosphorylation-defective mutants of EEF2K using site-directed mutagenesis. In these mutants, the codons for Ser366, Ser78 and Ser398 are changed to an alanine codon. We transfected the T98GshEEF2K cells with the expression plasmids of the phosphorylation mutants, and then measured the effects of Tg on autophagy in the cells expressing these phosphorylation-defective mutants. As shown in , treatment with Tg elicited a stronger autophagic response in T98Gwt than in T98GshEEF2K cells, as demonstrated by a higher level of LC3-II in T98Gwt than in T98GshEEF2K cells. Notably, LC3-II level was higher in T98GshEEF2K cells expressing the Ser366A/Ser78A mutant than in T98GshEEF2K cells expressing a wild-type EEF2K both in the absence or presence of Tg (), suggesting that disabling phosphorylation of EEF2K at Ser78 and Ser366 enhanced autophagic activity likely through maintaining EEF2K activity. By contrast, a defect in phosphorylation at Ser398, which is known to be phosphorylated by PRKAA2, weakened the autophagic response to Tg treatment (), suggesting that phosphorylation at Ser366/Ser78 and Ser398 has opposite effects on autophagy induced by ER stress. Indeed, PRKAA2 was activated by ER stress, as examined by phosphorylation of PRKAA2, and the PRKAA2 inhibitor, Compound C, blocked the activation of EEF2K by ER stress (Fig. S2A). MAPK14, another downstream protein kinase activated by ER stress, did not show any effect on EEF2K activity (Fig. S2B).
Figure 5. Phosphorylation of EEF2K at Ser78/Ser366 and Ser398 differentially regulated autophagy. (A) T98GWT cells or T98GshEEF2K cells were treated with 100 nM Tg for 24 h. The levels of EEF2K, p-EEF2 and LC3 were detected by western blot. TUBULIN was used as a loading control. (B) T98GshEEF2K cells transfected with a wild-type EEF2K or the phosphorylation-defective mutants of EEF2K were treated with 100 nM Tg for 24 h. Mutant 1: the codons for Ser366 and Ser78 were changed to an alanine codon by site-directed mutagenesis; Mutant 2: the codon for Ser398 was changed to an alanine codon. The levels of EEF2K, p-EEF2 and LC3 were detected by western blot. TUBULIN was used as a loading control.
Inhibition of EEF2K aggravates ER stress and augments ER stress-induced apoptosis in tumor cells
To further understand the role of EEF2K in cellular response to ER stress, we determined the effects of inhibiting EEF2K on DDIT3 and phospho-EIF2S1, two downstream protein markers of ER stress. shows that DDIT3 and p-EIF2S1 were induced in tumor cells treated with Tg or Tm; when EEF2K expression was silenced by siRNA, DDIT3 and p-EIF2S1 were further upregulated, indicating that inhibition of EEF2K aggravated ER stress. As DDIT3 plays an important role in inducing apoptosis in cells undergoing ER stress, we next assessed how inhibition of EEF2K affects ER-stress-induced apoptosis. As shown in , depletion of EEF2K not only augmented apoptosis in glioma cells treated with Tg or Tm, as determined by an increased annexinannexin V staining, but also advanced the activation of apoptosis from 48 h to 12h following ER stress. Enhancement of ER stress-induced apoptosis by inhibition of EEF2K was also evidenced by an increase in BCL2L11 level and a decrease in BIRC5 level, with silencing of EEF2K expression, as compared with cells transfected with a nontargeting siRNA (). Similar effects of EEF2K on ER stress markers DDIT3 and p-EIF2S1, and on apoptosis (BCL2L11 level and annexin V staining) were observed in human breast cancer cell lines MCF7 and T-47D (). These results imply that EEF2K plays a critical role in mitigating ER stress, and in modulating the crosstalk between autophagy and apoptosis.
Figure 6A–C. Silencing of EEF2K expression augmented ER stress and resulted in increased apoptosis in tumor cells. (A) LN-229 and T98G cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with Tg or Tm for 24 h. EEF2K, DDIT3 and p-EIF2S1 were examined by western blot. TUBULIN was used as a loading control. (B) LN-229 and T98G cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with Tg or Tm for 48 h or 12 h. Apoptosis was determined by flow cytometric analyses of annexin staining. Each bar represents the mean ± SE of three experiments. (C) LN-229 or T98G cells were transfected with a nontargeting RNA or an EEF2K-targeted siRNA, followed by treatment with Tg or Tm. EEF2K, BCL2L11 and BIRC5 were examined by western blot. TUBULIN was used as a loading control.
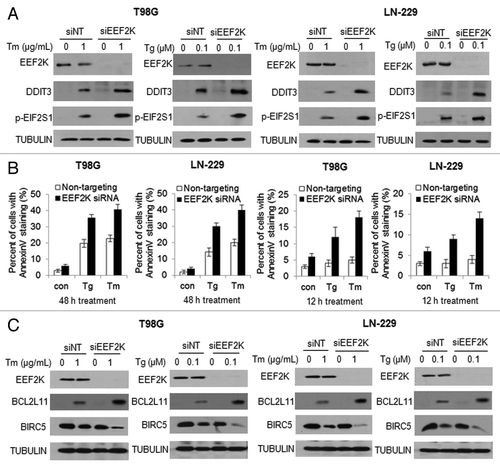
Figure 6D and E. Silencing of EEF2K expression augmented ER stress and resulted in increased apoptosis in tumor cells. MCF7 or T-47D cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with Tg or Tm for 48 h. (D) EEF2K, LC3, DDIT3, p-EIF2S1, BIRC5 and BCL2L11 were examined by western blot. TUBULIN was used as a loading control. (E) Apoptosis was determined by flow cytometric analyses of annexin staining. Each bar represents the mean ± SE of three experiments.
Suppression of autophagy enhances the cytotoxic effect of ER stressors in tumor cells
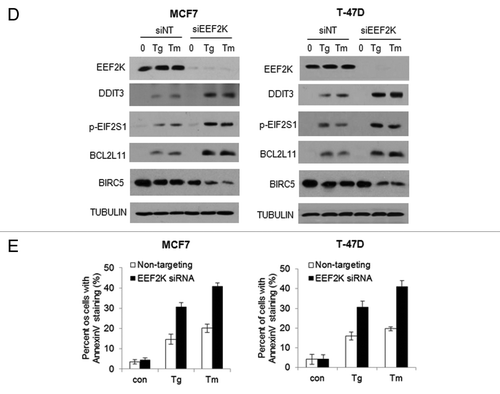
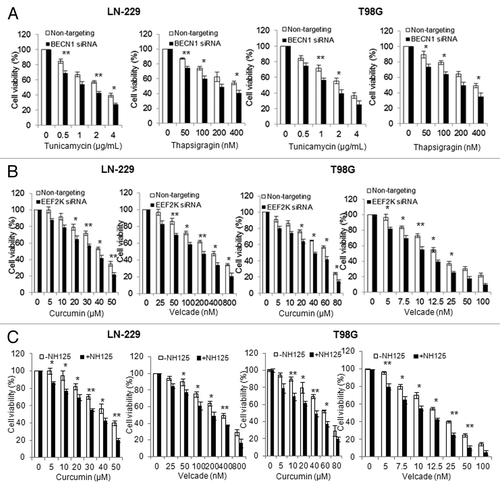
It is known that activation of autophagy is either cytoprotective or cytopathic, depending on context. To determine whether ER stress-induced autophagy plays a prosurvival or prodeath role, we silenced the expression of BECN1, a key regulator of autophagy, and then compared the viability of these cells with the cells without silencing of BECN1 expression following treatment with Tg or Tm. shows that inhibition of autophagy via silencing of BECN1 expression enhanced the cytotoxicity of Tg or Tm against glioma cells, suggesting that autophagy acts as a survival mechanism under ER stress. Since ER stress-inducing agents have been tested and utilized as therapeutics for treatment of cancer,Citation19-Citation21 we next explored the therapeutic implication of the EEF2K-mediated autophagy. As inhibiting EEF2K blunted the autophagy induced by ER stress (), we evaluated the effects of silencing this kinase on sensitivity of tumor cells to curcumin and velcade, two compounds that possess ER stress-inducing effect and antitumor activity.Citation20,Citation22 shows that the cytotoxicity of curcumin or velcade against T98G and LN-229 cells was significantly greater when EEF2K was silenced, as compared with that in the cells transfected with a nontargeting RNA. Sensitization of tumor cells to curcumin or velcade could also be achieved by cotreatment with 0.25 µM of NH125, a small molecule inhibitor of EEF2K (). These results suggest that targeting EEF2K can reinforce the efficacy of ER stress-inducing drugs against human tumor cells.
Figure 7. Inhibition of EEF2K increased the cytocidal activity of curcumin and velcade against tumor cells. (A) LN-229 or T98G cells were transfected with a nontargeting siRNA or a BECN1-targeted siRNA, followed by treatment with a series of concentrations of Tg or Tm for 48 h. At the end of treatment, cell viability was measured by MTT assay. (B) LN-229 or T98G cells were transfected with a nontargeting RNA or a siRNA targeting EEF2K, followed by treatment with a series of concentrations of curcumin or velcade for 48 h. Cell viability was measured by MTT assay. (C) LN-229 or T98G cells were treated with a series of concentrations of curcumin or velcade for 48 h in the presence or absence of 0.25 μM of NH125. At the end of treatment, cell viability was measured by MTT assay. Results shown were mean ± SE of three experiments.
Discussion
In this study, we demonstrated that ER stress-induced activation of EEF2K and autophagy are mediated, at least in part, through the ATF4-DDIT4 pathway, and that the regulation of EEF2K by DDIT4 is mediated by its effect on MTOR signaling. We also showed that inhibition of the EEF2K-regulated autophagy deteriorated ER stress, promotes apoptosis, and sensitized tumor cells to the cytotoxicity of agents that have ER stress-inducing action (curcumin and velcade). Our results not only added new insights into pathways involved in the regulation of ER stress-activated autophagy and apoptosis, but also provided new clues on how to reinforce the efficacy of the anticancer drugs whose mechanism of action involves induction of ER stress.
Several lines of evidence suggest that under various stresses, EEF2K activity may serve as an integrator in activating autophagy. As an inhibitor of peptide elongation, EEF2K may also participate in alleviating ER stress via shutting down protein synthesis. Nonetheless, how ER stress activates EEF2K remains a question. To explore the pathway linking ER stress and EEF2K activation, we tested the role of DDIT4, which was reported to be upregulated in response to ER stress.Citation15,Citation16 We found that silencing of DDIT4 expression causes a downregulation of EEF2K, prevents the phosphorylation of EEF2 (), and blunts autophagy in tumor cells undergoing ER stress, indicating that DDIT4 is involved in transducing the signal between ER stress and EEF2K activation. DDIT4 appears to be required for activation of EEF2K and autophagy under other stressful conditions, because similar effects of this stress-induced protein also were observed in the cells subjected to nutrient starvation (Fig. S3). Further, we found that induction of DDIT4 plays a role in prohibiting EEF2K protein from proteasomal degradation, as the proteasome inhibitor, MG132, abolishes the downregulation of EEF2K in the cells subjected to both silencing of DDIT4 and Tg treatment (). DDIT4 is known to negatively regulate MTOR activity;Citation23,Citation24 indeed, the ER stress-induced phosphorylations of RPS6KB and EIF4EBP1, two downstream effectors of MTOR, were significantly decreased when DDIT4 expression was repressed. Thus, the protective effect of DDIT4 on EEF2K protein degradation may be associated with the negative regulation of MTOR activity by DDIT4. Consistently, we have found that the half-life of the EEF2K protein is longer in cells harboring the Ser78/Ser366 mutant of the kinase than in cells with the wild-type enzyme, suggesting that the phosphorylation of EEF2K at Ser78/Ser366 by MTOR may promote the degradation of the kinase protein (unpublished data). We further found that the increase of p-EEF2 under ER stress was reduced by the enforced expression of RHEB, a positive regulator of MTOR, suggesting that DDIT4-MTOR-EEF2K pathway was activated in response to ER stress. EEF2K can either be inhibited or be activated via phosphorylation at different sites by various kinases. PRKAA2 phosphorylates EEF2K at Ser398 and activates the kinase,Citation25 whereas phosphorylation at Ser78 and Ser366 by the MTOR-RPS6KB pathway decreased the binding of CaM to EEF2K and reduces its activity.Citation26,Citation27 Phosphorylation at Ser369 of EEF2K by MAPK14 also causes its inactivation.Citation28 In this study, we demonstrated that phosphorylation of EEF2K by PRKAA2 at Ser398 was essential for induction of autophagy, but phosphorylation at Ser366 and Ser78 negatively regulated the cellular autophagy process (; Fig. S2). MAPK14 did not seem to play a role in ER stress-induced activation of EEF2K (Fig. S2). The precise mechanisms by which these phosphorylation sites of EEF2K influence autophagy remain to be delineated.
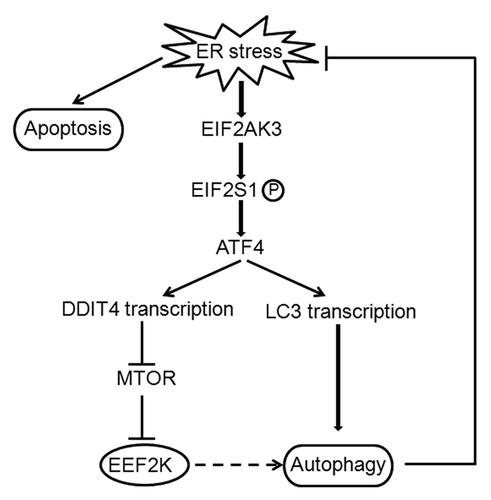
ATF4, an activating transcription factor, has been reported to play a role in the induction of DDIT4.Citation14-Citation16 Consistent with those observations, we found that both the levels of ATF4 and DDIT4 were elevated following Tg treatment, and silencing of ATF4 expression blocked the induction of DDIT4 (). ATF4 is known to be upregulated when EIF2S1 is phosphorylated,Citation18,Citation19 therefore, the increased phosphorylation of EIF2S1 observed in the cells treated with Tg or Tm () may account for the induction of ATF4 in the experiments shown in . Hence, these observations may pinpoint the EIF2S1-ATF4-DDIT4 pathway as an essential regulatory mechanism of ER stress-induced activation of EEF2K and autophagy. The EIF2AK-EIF2S1 pathway has been reported to mediate the ER stress-induced autophagy,Citation29,Citation30 and this role is regulated by ATF4, a downstream transcription factor of EIF2S1 and can increase the transcript of LC3.Citation31 Therefore, there may exist two independent pathways for the EIF2AK3-EIF2S1-ATF4-induced autophagy: the directed regulation of autophagy by ATF4-mediated LC3 transcription, and the indirect regulation of autophagy by the ATF4-regulated DDIT4-EEF2K pathway ().
Figure 8. Regulatory role of the EIF2AK3-EIF2S1-ATF4 pathway in autophagy in tumor cells undergoing ER stress. ER stress induces EIF2AK3-EIF2S1 phosphorylation and ATF4 activation, which induces LC3 transcription and stimulates autophagy. Activation of ATF4 also promotes autophagy by activating DDIT4, which increases EEF2K activity by inhibiting the negative regulation of MTOR on EEF2K. When autophagy is not sufficient to reduce ER stress, cells may undergo ER stress-mediated cell death by apoptosis. Full line: direct regulation; dotted line: indirect regulation.
Autophagy can exert either prosurvival or prodeath effect, depending on context. During ER stress, activation of autophagy is believed to play a role in alleviating ER stress and in favoring cell survival.Citation32,Citation33 Consistently, our study demonstrates that autophagy mediated by EEF2K was cytoprotective when tumor cells underwent ER stress, as inhibiting EEF2K blunted the autophagic response (), promoted apoptotic cell death (Fig. 6), and enhanced cytotoxicity of the drugs that have ER stress–inducing action, such as curcumin and velcade (). It is known that persistent ER stress leads to apoptosis. Recent studies also indicate that prosurvival pathways dominate the early ER stress response, whereas activation of apoptotic pathways occurs during the later stages of the stress.Citation34 We found that inhibition of EEF2K shifted the time course of apoptosis induction to an earlier stage (Fig. 6), suggesting that EEF2K played a regulatory role in the crosstalk between autophagy and apoptosis under ER stress, and targeting this kinase may be exploited as a means to enhance the effects of ER stressors. Indeed, we observed that inhibiting EEF2K sensitized tumor cells to curcumin or velcade, two antitumor agents that that possess ER stress-inducing action (). In both glioma cells and breast cancer cells, we observed that silencing of EEF2K expression enhanced apoptotic response under ER stress (Fig. 6), which was opposite to what was observed in EEF2K−/− MEFs.Citation11 Because tumor cells including glioma cells and breast cancer cells overexpress EEF2K, we believe that the antiapoptotic effect of EEF2K is specific to tumor cells.
Taken together, we report here that EEF2K was a critical regulator of autophagy and apoptosis in tumor cells subjected to ER stress, and that inhibiting this kinase can enhance sensitivity of tumor cells to ER stressors through promoting apoptotic cell death. Our study not only added new information on the regulation of autophagy and apoptosis under ER stress condition, but also provided a rationale of targeting EEF2K as a new strategy to reinforce the anticancer efficacy of ER stress-inducing agents.
Materials and Methods
Cell lines and culture
Human glioblastoma cell lines T98G (CRL-1690) and LN-229 (CRL-2611), and human breast cancer cell lines MCF7 (HTB-22) and T-47D (HTB-133), were purchased from American Type Culture Collection. T98GWT and T98GshEEF2K lines were generated by our group as described previously.Citation35 LN-229, MCF7 and T-47D cells were cultured in DMEM medium (Mediatech, Inc., 10-013-CV), and T98G, T98GWT and T98GshEEF2K cells were cultured in Ham’s F-10 (Mediatech, Inc., 10-070-CV): DMEM (10:1) medium. The media were supplemented with 10% fetal bovine serum (Invitrogen, 16000044) and 100 × penicillin-streptomycin solution (Mediatech, Inc., 30-002-CI) Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Reagents and antibodies
Thapsigargin (T9033), tunicamycin (T7765), bafilomycin A1 (B1793), curcumin (C1386) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, M2128) were purchased from Sigma. Velcade (B-1408) was purchased from LC Laboratories. NH125 (3439) was purchased from Tocris Bioscience. Antibodies to p-EEF2 (2331), EEF2 (2332), p-EIF2S1 (3398), RPS6KB (9202), p-RPS6KB (9205), p-EIF4EBP1 (9456), EIF4EBP1 (9452), BCL2L11 (2933), PARP1 (9542), EEF2K (3692), DDIT3 (2895), p-PRKAA2 (2531), PRKAA2 (2532), p-MAPK14 (9216), MAPK14 (9212), BECN1 (3738) and BIRC5 (2808) were purchased from Cell Signaling Technologies. LC3 (0231) antibody was purchased from nanoTools. α-TUBULIN (sc-23948) and ATF4 (sc-7583) antibodies were purchased from Santa Cruz. DDIT4 (10638-1-AP) antibody was purchased from PreteinTech Group, Inc. SQSTM1 (BML-PW 9860) antibody was purchased from Engo Life Science. Polyclonal goat anti-mouse immunoglobulins/HRP (P0447) and polyclonal goat anti-rabbit immunoglobulins/HRP (P0448) were purchased from Dako. Western lightning plus-ECL (NEL105001EA) was purchased from PerkinElmer.
Western blot analysis
Cells were lysed in mammalian protein extraction reagent (GenLantis, L300500) supplemented with a protease inhibitor cocktail (Roche, 04693132001) at room temperature for 5 min, followed by centrifugation at 14,000 × g for 10 min. Protein concentration of cell lysates were measured using the Pierce 660 nm protein assay reagent (Thermo scientific, 22660). Proteins (20–40 µg) were resolved on SDS-PAGE and transferred to PVDF membrane (Bio-Rad, 1620177). The PVDF membranes were then incubated with respective antibodies in 3% BSA (Santa Cruz, sc-2323)/ TBST at 4°C for overnight, followed by incubation with secondary antibody at room temperature for 1 h. The protein signals were detected by ECL method.
siRNA transfection
siRNA duplexes targeting BECN1, EEF2K, DDIT4, ATF4 or EIF2S1 were prepared by Dharmacon Research, Inc.. Scrambled siRNA was used as a control. Transfection of siRNA was performed according to the manufacturer’s protocol. Briefly, cells in exponential phase of growth were plated in six-well tissue culture plates at 1 × 105 cells per well, grown for 24 h, and then transfected with siRNA using Oligofectamine (Invitrogen, 12252-011) and OPTI-MEM I–reduced serum medium (Gibco, 31985-062). The concentrations of siRNAs were chosen based on dose-response studies.
Cellular viability assay
Cell viability was measured by MTT assay. Briefly, cells were plated at 5 × 103 cells per well in 96-well tissue culture plates, subjected to different treatment, and incubated at 37°C for 72 h in a humidified atmosphere containing 5% CO2. The formazan product, formed after 4 h incubation with MTT, was dissolved in DMSO (Acros Organics, AC61042-0010) and read at 570 nm on a Victor3 Multi Label plate reader (PerkinElmer).
Apoptosis assays
Apoptosis was determined by flow cytometric analysis of annexin V and 7-AAD staining. Briefly, 100 µl Guava Nexin reagent (Millipore, 4700-1140) was added to 1 × 105 cells (in 100 µl) and the cells were incubated with the reagent for 20 min at room temperature in the dark. At the end of incubation, the cells were analyzed by a Guava EasyCyte™ Plus FlowCytometry System (Millipore).
Statistical analysis
The difference between the samples with silencing of EEF2K and the samples without silencing of the enzyme, or the samples with inhibition of autophagy and the samples without inhibition of autophagy, was analyzed using a two-sample t-test.
Additional material
Download Zip (764.8 KB)Acknowledgments
This work was supported by grants from the US. Public Health Service R01CA135038 (J.-M.Y.) and R01DK13499 (L.S.J.), and from the Department of Defense BC103654 (Y.C.), Elsa Pardee Foundation (J.-M.Y.), and National Natural Sciences Foundation of China (NO. 81072146).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/ 22801
References
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 2008; 7:1013 - 30; http://dx.doi.org/10.1038/nrd2755; PMID: 19043451
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 1999; 13:1211 - 33; http://dx.doi.org/10.1101/gad.13.10.1211; PMID: 10346810
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010; 140:900 - 17; http://dx.doi.org/10.1016/j.cell.2010.02.034; PMID: 20303879
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 2005; 6:439 - 48; http://dx.doi.org/10.1038/nrm1660; PMID: 15928708
- Høyer-Hansen M, Jäättelä M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ 2007; 14:1576 - 82; http://dx.doi.org/10.1038/sj.cdd.4402200; PMID: 17612585
- Proud CG. Peptide-chain elongation in eukaryotes. Mol Biol Rep 1994; 19:161 - 70; http://dx.doi.org/10.1007/BF00986958; PMID: 7969104
- Parmer TG, Ward MD, Yurkow EJ, Vyas VH, Kearney TJ, Hait WN. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br J Cancer 1999; 79:59 - 64; http://dx.doi.org/10.1038/sj.bjc.6690012; PMID: 10408694
- Bagaglio DM, Cheng EH, Gorelick FS, Mitsui K, Nairn AC, Hait WN. Phosphorylation of elongation factor 2 in normal and malignant rat glial cells. Cancer Res 1993; 53:Suppl 2260 - 4; PMID: 8485712
- Cheng EH, Gorelick FS, Czernik AJ, Bagaglio DM, Hait WN. Calmodulin-dependent protein kinases in rat glioblastoma. Cell Growth Differ 1995; 6:615 - 21; PMID: 7647041
- Cheng Y, Ren X, Zhang Y, Patel R, Sharma A, Wu H, et al. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res 2011; 71:2654 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-10-2889; PMID: 21307130
- Boyce M, Py BF, Ryazanov AG, Minden JS, Long K, Ma D, et al. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ 2008; 15:589 - 99; http://dx.doi.org/10.1038/sj.cdd.4402296; PMID: 18188169
- Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy 2009; 5:393 - 6; http://dx.doi.org/10.4161/auto.5.3.7762; PMID: 19221463
- Ellisen LW. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle 2005; 4:1500 - 2; http://dx.doi.org/10.4161/cc.4.11.2139; PMID: 16258273
- Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, et al. SP600125 negatively regulates the mammalian target of rapamycin via ATF4-induced Redd1 expression. FEBS Lett 2009; 583:123 - 7; http://dx.doi.org/10.1016/j.febslet.2008.11.035; PMID: 19059405
- Whitney ML, Jefferson LS, Kimball SR. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem Biophys Res Commun 2009; 379:451 - 5; http://dx.doi.org/10.1016/j.bbrc.2008.12.079; PMID: 19114033
- Jin HO, Seo SK, Woo SH, Kim ES, Lee HC, Yoo DH, et al. Activating transcription factor 4 and CCAAT/enhancer-binding protein-beta negatively regulate the mammalian target of rapamycin via Redd1 expression in response to oxidative and endoplasmic reticulum stress. Free Radic Biol Med 2009; 46:1158 - 67; http://dx.doi.org/10.1016/j.freeradbiomed.2009.01.015; PMID: 19439225
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6:1099 - 108; http://dx.doi.org/10.1016/S1097-2765(00)00108-8; PMID: 11106749
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 2004; 101:11269 - 74; http://dx.doi.org/10.1073/pnas.0400541101; PMID: 15277680
- Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol 2009; 625:234 - 46; http://dx.doi.org/10.1016/j.ejphar.2009.06.064; PMID: 19835867
- Hill DS, Martin S, Armstrong JL, Flockhart R, Tonison JJ, Simpson DG, et al. Combining the endoplasmic reticulum stress-inducing agents bortezomib and fenretinide as a novel therapeutic strategy for metastatic melanoma. Clin Cancer Res 2009; 15:1192 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-08-2150; PMID: 19228725
- Armstrong JL, Corazzari M, Martin S, Pagliarini V, Falasca L, Hill DS, et al. Oncogenic B-RAF signaling in melanoma impairs the therapeutic advantage of autophagy inhibition. Clin Cancer Res 2011; 17:2216 - 26; http://dx.doi.org/10.1158/1078-0432.CCR-10-3003; PMID: 21270111
- Bakhshi J, Weinstein L, Poksay KS, Nishinaga B, Bredesen DE, Rao RV. Coupling endoplasmic reticulum stress to the cell death program in mouse melanoma cells: effect of curcumin. Apoptosis 2008; 13:904 - 14; http://dx.doi.org/10.1007/s10495-008-0221-x; PMID: 18493855
- Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res 2011; 71:4366 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-10-1769; PMID: 21540236
- Jin HO, Seo SK, Kim YS, Woo SH, Lee KH, Yi JY, et al. TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene 2011; 30:3792 - 801; http://dx.doi.org/10.1038/onc.2011.102; PMID: 21460850
- Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem 2004; 279:12220 - 31; http://dx.doi.org/10.1074/jbc.M309773200; PMID: 14709557
- Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol 2004; 24:2986 - 97; http://dx.doi.org/10.1128/MCB.24.7.2986-2997.2004; PMID: 15024086
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J 2001; 20:4370 - 9; http://dx.doi.org/10.1093/emboj/20.16.4370; PMID: 11500364
- Knebel A, Morrice N, Cohen P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J 2001; 20:4360 - 9; http://dx.doi.org/10.1093/emboj/20.16.4360; PMID: 11500363
- Fujita E, Kouroku Y, Isoai A, Kumagai H, Misutani A, Matsuda C, et al. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum Mol Genet 2007; 16:618 - 29; http://dx.doi.org/10.1093/hmg/ddm002; PMID: 17331981
- Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, et al. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 2007; 14:230 - 9; http://dx.doi.org/10.1038/sj.cdd.4401984; PMID: 16794605
- Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120:127 - 41; http://dx.doi.org/10.1172/JCI40027; PMID: 20038797
- Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 2007; 282:4702 - 10; http://dx.doi.org/10.1074/jbc.M609267200; PMID: 17135238
- Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 2006; 26:9220 - 31; http://dx.doi.org/10.1128/MCB.01453-06; PMID: 17030611
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007; 318:944 - 9; http://dx.doi.org/10.1126/science.1146361; PMID: 17991856
- Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res 2006; 66:3015 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-05-1554; PMID: 16540650