Abstract
Gastrointestinal stromal tumor (GIST) is a prototype of mutant KIT oncogene-driven tumor. Prolonged tyrosine kinase inhibitor (TKI) treatment may result in a resistant phenotype through acquired secondary KIT mutation. Heat shock protein 90 (HSP90AA1) is a chaperone protein responsible for protein maturation and stability, and KIT is a known client protein of HSP90AA1. Inhibition of HSP90AA1 has been shown to destabilize KIT protein by enhancing its degradation via the proteasome-dependent pathway. In this study, we demonstrated that NVP-AUY922 (AUY922), a new class of HSP90AA1 inhibitor, is effective in inhibiting the growth of GIST cells expressing mutant KIT protein, the imatinib-sensitive GIST882 and imatinib-resistant GIST48 cells. The growth inhibition was accompanied with a sustained reduction of both total and phosphorylated KIT proteins and the induction of apoptosis in both cell lines. Surprisingly, AUY922-induced KIT reduction could be partially reversed by pharmacological inhibition of either autophagy or proteasome degradation pathway. The blockade of autophagy alone led to the accumulation of the KIT protein, highlighting the role of autophagy in endogenous KIT turnover. The involvement of autophagy in endogenous and AUY922-induced KIT protein turnover was further confirmed by the colocalization of KIT with MAP1LC3B-, acridine orange- or SQSTM1-labeled autophagosome, and by the accumulation of KIT in GIST cells by silencing either BECN1 or ATG5 to disrupt autophagosome activity. Therefore, the results not only highlight the potential application of AUY922 for the treatment of KIT-expressing GISTs, but also provide the first evidence for the involvement of autophagy in endogenous and HSP90AA1 inhibitor-induced KIT degradation.
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common type of mesenchymal neoplasms in the gastrointestinal tract.Citation1,Citation2 The annual incidence of GISTs is estimated to be approximately 10–20 per million individuals. Primary gain-of-function KIT mutations occur in 60– 80% of GISTs. The most common primary mutations occur in the juxta-membrane domain (exon 11), occasionally in the extracellular domain (exon 9), but rarely in the ATP-binding domain (exon 13/14) and the activation loop domain (exon 17).Citation3-Citation6 Clinically, the tyrosine kinase inhibitor (TKI), Imatinib Mesylate (IM; Gleevec®, Novartis Pharma), has been approved as a first-line therapy for metastatic or unresectable GIST. However, 50% of patients with initially IM-responsive GISTs suffer from disease progression within 2 years of beginning treatment.Citation4 The best-recognized mechanisms underlying the development of acquired IM resistance include the acquisition of secondary mutations in exon 13, 14 or 17 of KIT or the overexpression of wild-type KIT.Citation7,Citation8 Currently, sunitinib malate (SU; Sutent®, Pfizer, Inc.) is the FDA-approved second-line treatment for IM-resistant GISTs. Like IM, SU acts primarily by inhibiting KIT phosphorylation.Citation9,Citation10 Unfortunately, SU is not effective for IM-resistant GISTs with secondary KIT mutations in exon 17, as indicated by the difference in median progression-free survival for patients with secondary exon 13/14 and exon 17 mutations of 7.8 and 2.1 months, respectively.Citation10,Citation11 Apparently, alternative therapeutic strategies and novel practicable agents are urgently needed for these patients.
Although several TKIs have been investigated in clinical trials, only limited therapeutic activity is exhibited in IM/SU-resistant GISTs. In addition, the acquisition of TKI-resistant KIT genotypes can be evolved after repetitive exposure to new TKI therapy. Therefore, it is imperative to explore novel therapeutic strategies that can overcome the inevitable problem of TKI resistance in GIST patients, irrespective of the specific mutational activation mechanisms. Two potential strategies are to silence KIT gene transcription and to enhance the cellular degradation of constitutively active KIT.Citation12 Fumo et al. have discovered that KIT is stabilized and protected from protein degradation by heat shock protein 90 (HSP90AA1), and that HSP90AA1 inhibition reduces the levels of wild-type or D816V-mutated KIT protein on mast cells.Citation13 Bauer et al. also find that 17-AAG, an inhibitor of HSP90AA1, can substantially reduce the protein levels of both phosphorylated and total KIT in IM-sensitive and IM-resistant GIST cell lines.Citation14 Moreover, HSP90AA1 overexpression is an indicator of poor prognosis that correlates with several adverse parameters, suggesting this protein as a therapeutic target for patients with high-risk IM-resistant GISTs.Citation15
HSP90AA1 is a molecular chaperone involved in the conformational maturation, stability and function of its substrates, or “client proteins,” within a cell. Many HSP90AA1 client proteins, including receptor tyrosine kinases, signaling molecules and kinases, and transcription factors, participate in a variety of critical cellular processes, such as signal transduction, survival, proliferation, invasion, metastasis, angiogenesis, cell cycle regulation and apoptosis.Citation16 Nevertheless, HSP90AA1 has not been not pursued as a drug target until the discovery and characterization of naturally-derived inhibitors of HSP90AA1, such as geldanamycin (GA) and radicicol (RD).Citation17,Citation18 The prevalence of a high-affinity form of HSP90AA1 in tumor cells, the critical roles played by oncogenic client proteins in cancer cells and the greater HSP90AA1 dependency of cancer cells as compared with normal tissues have been proposed as rationales for the selectivity of HSP90AA1 inhibitors for cancer vs. normal cells.Citation16,Citation19 Furthermore, Kamal et al. and Vilenchik et al. have demonstrated that the inhibition of HSP90AA1 selectively kills cancer cells compared with the effect for normal cells.Citation20,Citation21 The inhibition of HSP90AA1 causes most client proteins to be degraded by the ubiquitin-proteasome pathway.Citation14,Citation17,Citation22 However, the role of the autophagy pathway in HSP90AA1 inhibition-induced protein degradation remains unclear.
The GA analog 17-AAG was the first HSP90AA1 inhibitor to undergo clinical trials.Citation23,Citation24 Further clinical trials have been initiated with 17-AAG as a single agent or in combination therapies in numerous cancer types, including multiple myeloma, lymphoma, breast cancer, prostate cancer and kidney cancer.Citation25-Citation28 HSP90AA1 inhibitors are now a major focus of cancer therapies and as novel compounds because their ability for interfering with multiple oncogenic pathways is thought to confer a broad antitumor activity and less susceptibility to acquired drug resistance. However, despite the high antitumor activity and progress in clinical treatment of 17-AAG, this compound has several potential limitations, including poor solubility, limited bioavailability, hepatotoxicity and extensive metabolism by polymorphic enzymes.Citation29,Citation30 In the present study, the next-generation HSP90AA1 inhibitor AUY922, which inhibited HSP90AA1 with higher affinity in vitro, was examined for its antitumor activity in mutant KIT-expressing GIST48 and GIST882 cell lines.Citation31 Moreover, the detailed mechanisms of AUY922-induced KIT downregulation were investigated at the posttranslational level. These results represent an important step toward identifying the optimal therapy for IM-resistant GISTs.
Results
AUY922 downregulated phospho- and total KIT expression
First we investigated whether AUY922, which had a high affinity to HSP90AA1, could reduce phospho- and total KIT expression in COS-1 cells expressing its wild-type or mutant proteins. Experiments using transient transfection of COS-1 cells with KIT constructs containing either a wild-type, or mutant KIT gene (mimicking primary GISTs with or without secondary mutations in clinical GIST samples such as exon 11V560D or Δ555–576, exon 17N822K, exon 11V560D/17N822K or exon 11Δ555–576/17N822K, respectively) in the presence of AUY922 for the indicated time courses and doses were performed. In COS-1 cells transfected with the wild-type and KIT exon 11V560D, exon 11Δ555–576, exon 17N822K, exon 11V560D/17N822K or exon 11Δ555–576/17N822K mutants, AUY922 downregulated both phospho- and total KIT protein in dose- (0.01–1 μM) and time- (0.5–8 h) dependent manners ().
Figure 1. AUY922 reduced KIT expression in GIST48, GIST882, and COS-1 cells. COS-1 transfected with KIT constructs (A and B) or LacZ (C) were incubated with indicated doses of AUY922 for 8 h, lysed, and then analyzed KIT or LacZ by immunoblotting. GIST48 (D) and GIST882 (E) cells were treated with AUY922 as indicated by doses and times and analyzed by immunoblotting against phospho- and total KIT, MAPK1/3, AKT or HSPA1A proteins. (F) GIST48 cells were incubated with AUY922 or 17-AAG for 24 h, replaced with growth media for indicated times, harvested and then analyzed by immunoblotting.
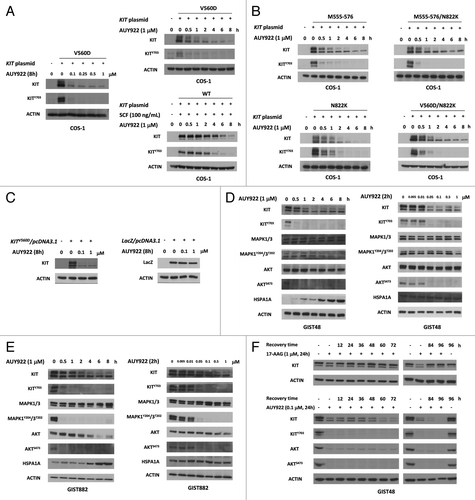
To exclude the possibility that AUY922 induced KIT downregulation in COS-1 due to the influence of the KIT plasmid on the CMV promoter, the LacZ gene was constructed into pcDNA3.1 and acted as a control plasmid. COS-1 cells were transfected with the LacZ/pcDNA3.1 or KITV560D/pcDNA3.1 plasmid and treated with 1 μM AUY922 for 6 h. The expression level of KIT was greatly reduced, while that of LacZ was not significantly affected by AUY922 treatment of either 0.1 or 1 μM (), indicating that downregulation of KIT by AUY922 in COS-1 cells was a protein-specific event.
The findings were further validated in GIST cells in the IM-sensitive GIST882 and IM-resistant GIST48 cells expressing mutant KIT proteins. Because HSPA1A, also known as HSP70, accumulation is known as a functional index of HSP90AA1 inhibition, we also measured the protein level of HSPA1A. These results showed that 1 μM AUY922 elevated the level of HSPA1A expression in a time-dependent manner, indicating that AUY922 effectively inhibited the HSP90AA1 activity. This inhibitory effect of AUY922 on HSP90AA1 activity also caused reduction of the phospho- and total KIT protein levels in both GIST48 and GIST882 cells treated with AUY922 for 0.5–8 h (). Consistently, its downstream signaling molecules, such as MAPK1/3 and AKT, were inactivated by the AUY922 treatment after 0.5–2 h. In the dose-dependent experiments, a 2 h exposure to 0.05–1 μM AUY922 downregulated the expression levels of phospho- and total KIT proteins in both GIST48 and GIST882 cell lines. The minimum concentrations of AUY922 for completely abolishing the phospho-KIT in GIST cells were at 0.05–0.1 μM. With the reduced levels of phospho-KIT, the phospho-MAPK1/3 and phospho-AKT levels were also decreased by AUY922 treatment in GIST882 cells, whereas the phospho-AKT level was found to be decreased only in GIST48 cells. In addition, the exposure of 0.1 μM AUY922 for 24 h was able to sustainably reduce the levels of total and phospho-KIT as compared with the effect of 1 μM 17-AAG in GIST48 cells, as shown in . These findings indicated that AUY922 is a potent and durable anticancer drug for reducing KIT expression in GIST cells expressing mutant forms of KIT.
The cytotoxic effect of AUY922 in GIST48 and GIST882 cells
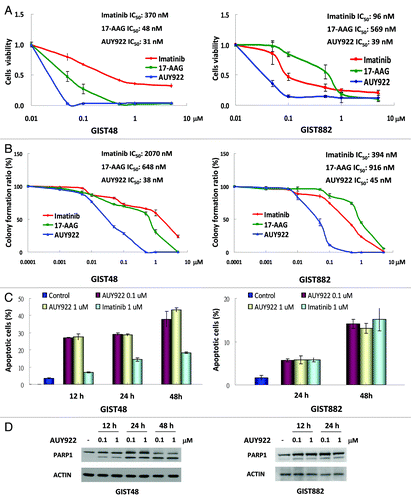
To examine the cytotoxicity of AUY922, GIST48 and GIST882 cells were incubated with this drug at the indicated doses and analyzed by cell viability assay () and clonogenic assay (). Both 17-AAG and IM acted as controls. The IC50 values of AUY922, 17-AAG, and IM were 31, 38, and 370 nM, respectively, in GIST48 cells and 39, 569, and 96 nM, respectively, in GIST882 cells (). showed that the IC50 results of clonogenic assay were 38, 648, and 2070 nM for AUY922, 17-AAG, and IM, respectively, in GIST48 cells, and 45, 916, and 394 nM for them, respectively, in GIST882 cells. We then incubated GIST48 or GIST882 cells with 0.1 or 1 μM AUY922 for 12–48 h to determine whether AUY922 could induce cell apoptosis. IM was included as a control. These data showed that the apoptotic cell populations, as identified by Annexin V-positive and PI-negative staining, were increased from 27–40% in GIST48 cells and from 5–12% in GIST882 cells (accompanied by extended exposure time from 12–48 h) (). Moreover, PARP1 cleavage, another apoptotic marker, was observed after AUY922 treatment for 12 h in GIST 48 cells and 24 h in GIST882 cells, and cleavage was markedly increased at 48 h (). In accordance with the finding of 0.1 μM of AUY922 was able to completely abolish the phospho-KIT in both cell lines, the cell apoptotic effects of 0.1 μM AUY922 were equivalent to those of 1 μM in both Annexin V staining and PARP1 cleavage studies.
Figure 2. AUY922 showed antitumor activity in GIST48 and GIST882 cells. GIST48 and GIST882 cells were incubated with AUY922, 17-AAG, and IM, respectively, with indicated doses. IC50 was determined by cell viability assay (A) or clonogenic assay (B), respectively. GIST48 and GIST882 cells were treated with 0.1 or 1 μM AUY922 or 1 μM IM for 12–48 h and then analyzed by Annexin V staining (C) or immunoblotting against PARP1 (D). All experiments were repeated at least three times. The data are expressed as the mean ± SE of two or more independent experiments.
The enhancement of KIT protein degradation by AUY922
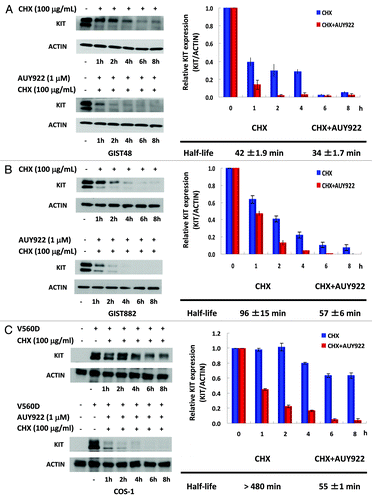
We have demonstrated that AUY922 could downregulate KIT expression in cells expressing its wild-type or mutant forms, regardless of their IM sensitivity. To examine whether AUY922 might enhance KIT protein degradation, GIST48, GIST882 or COS-1 cells harboring KIT mutants were treated with the protein synthesis inhibitor cycloheximide (CHX) alone or in combination with AUY922, and then the KIT protein levels were analyzed by western blotting analysis. The half-life of the KIT protein in each cell lines was estimated after normalization of ACTIN control. The data showed that the KIT half-lives in CHX-treated cells vs. AUY922/CHX combination-treated cells were 42 ± 1.9 min vs. 34 ± 1.7 min, 96 ± 15 min vs. 57 ± 6 min, and > 480 min vs. 55 ± 1 min for GIST48, GIST882, and COS-1V560D cells, respectively (). These consistent results demonstrated that AUY922 treatment could accelerate KIT protein degradation, as other HSP90AA1 inhibitors, for instance, 17-AAG and IPI-504, did.
Figure 3. The effect of AUY922 on KIT protein degradation in GIST48, GIST882, and COS-1 cells. GIST48 (A), GIST882 (B), and COS-1 cells expressing mutant KITV560D (C) were treated with 100 μg/mL CHX alone or in combination with 1 μM AUY922 for the indicated time. Cells were lysed and analyzed by immunoblotting against KIT. KIT expression was normalized to ACTIN and compared with the untreated control. The half-life of the KIT protein was estimated based on the relative KIT expression ratio. The data are expressed as the mean ± SE of two or more independent experiments.
AUY922-induced autophagy led to KIT reduction
To further distinguish which protein degradation pathway(s) were involved in AUY922-induced KIT degradation, cells were pretreated with the proteasome degradation inhibitor MG-132 or the autophagy inhibitor 3-MA for 4 h and followed by co-incubation with 1 μM of AUY922 for further 6 h. These results showed that downregulation of KIT protein by AUY922 could be partially rescued by MG-132 or 3-MA in both GIST48 and GIST882 cells (). The findings were further validated by alternative inhibitors of proteasome degradation (lactacystin) and autophagy (bafilomycin A1), as shown in , and also confirmed in COS-1 cells expressing mutant KIT (). Taken together, these results demonstrated that both autophagy- and proteasome-mediated degradation pathways played roles in the AUY922-induced KIT protein downregulation in GIST48, GIST882, and COS-1 cells.
Figure 4A–E. Autophagy was involved in AUY922-induced KIT protein degradation in GIST48, GIST882, and COS-1 cells. GIST48 and GIST882 were pretreated with 20 μM MG-132, 10 mM 3-MA (A), 1 μM lactacystin or 20 nM bafilomycin A1 (B) for 4 h and then treated with 1 μM AUY922 for an additional 8 h. The cells were lysed and analyzed by immunoblotting against phospho- and total KIT. (C) GIST48 and GIST882 cells were treated with 1 μM AUY922 as indicated time, lysed, and then analyzed by immunoblotting against MAP1LC3A/B. Cells were transfected with 150 nM siRNA targeting BECN1 (D) or ATG5 (E) for 72 h and then treated with 1 μM AUY922 for another 2 h.
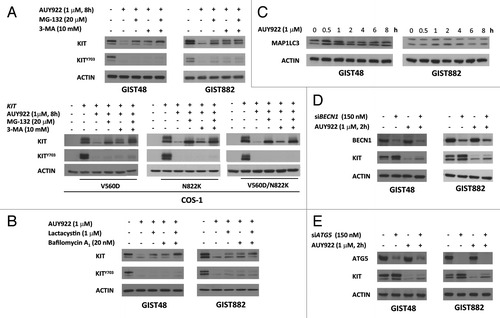
To further confirm the involvement of autophagy in the KIT downregulation induced by AUY922, we first examined MAP1LC3B accumulation, an index of autophagy activity, after AUY922 treatment. Following exposure to 1 μM of AUY922, a sustained increase in the MAP1LC3B level was noted from 0.5–8 h in GIST48 cells; in contrast, the MAP1LC3B level was increased from 1–4 h but decreased to the basal level afterward in GIST882 cells. Furthermore, attenuating the expression of BECN1 or ATG5, the essential molecules for autophagosome formation, by their gene-specific siRNA resulted in a partial rescue of the AUY922-induced KIT expression downregulation in both GIST48 and GIST882 cells (). By confocal microscopic examination of immunofluorescence staining of GIST cells, we found that there was no obvious nonspecific staining in normal rabbit, goat or mouse antibody-incubated cells. After treatment, AUY922 led unequivocally to more puncta of MAP1LC3B and more aggregates of SQSTM1 staining, as well as KIT condensation and aggregation, as shown in . In addition, aggregation of SQSTM1 and accumulation of MAP1LC3B- or acridine orange (AO)-labeled autophagosomes were colocalized with KIT in the autophagosomes visualized as yellow dots. These results indicate that AUY922 promoted autophagy and translocation of KIT to the autophagosomes.
Figure 4F–K. Cell lysates were extracted and analyzed by immunoblotting against BECN1, ATG5 and KIT. GIST48 and GIST882 cells were treated with 1 μM AUY922 for 1 h and then stained with KIT, MAP1LC3B (F and G), AO (H and I) or SQSTM1 (J and K). After immunostaining, cells were visualized by confocal microscopy and images were acquired through the Cy2, Rhodamine or DAPI channels (600×). The data were representative ones of 5 fields/pictures of each sample. The inserted figure in the corner showed magnified (1500×) and representative cells of each image.
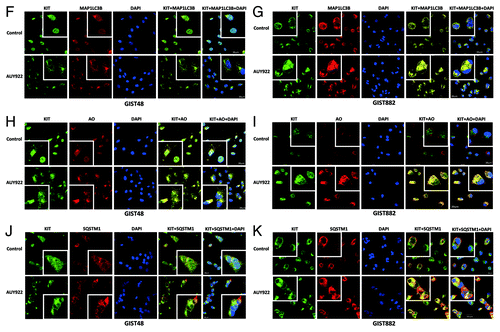
Autophagy was also involved in endogenous KIT turnover in GIST cells
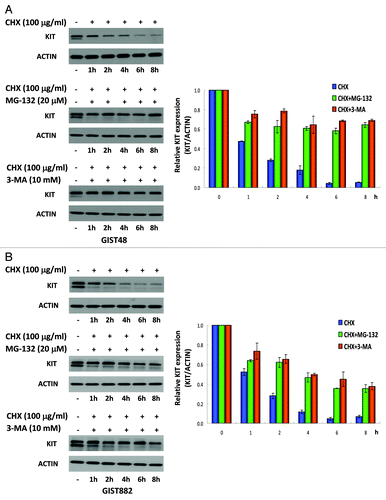
Interestingly, we found that knockdown of BECN1 or ATG5 upregulated KIT expression () and colocalization of KIT proteins with SQSTM1-, MAP1LC3B- or AO-labeled autophagosomes in untreated GIST48 or GIST882 cells (). These phenomena implied that autophagy may participate in endogenous KIT protein turnover. Hence, we used CHX alone or in combination with either MG-132 or 3-MA to examine the protein turnover of KIT. showed that endogenous KIT protein level was rapidly reduced to 48–50% of its initial level at 1 h and continually degraded to 5–7% at 8 h in GIST48 and GIST882 cells treated with CHX alone. However, the cotreatment with inhibitor of either autophagy or proteasome degradation pathways sustained KIT protein levels at 70% and 40% at 8 h after translational blockade in GIST48 and GIST882 cells, respectively. These data support our previous observation that autophagy-mediated degradation was also involved in endogenous KIT protein turnover.
Figure 5. KIT protein turnover in GIST48 and GIST882 cells. GIST48 (A) and GIST882 (B) cells were incubated with 100 μg/mL CHX alone or in combination with 20 μM MG-132 or 10 mM 3-MA for the indicated times. Cells were lysed and analyzed by immunoblotting against KIT. KIT expression was normalized to ACTIN and compared with untreated controls. The data are expressed as the mean ± SE of two or more independent experiments.
Discussion
In the present study, we demonstrated that a novel resorcinylic pyrazole/isoxazole amide class of HSP90AA1 inhibitor, AUY922, had a much higher ability to downregulate the KIT protein () and to inhibit the growth of GIST cells () as compared with 17-AAG. In addition, autophagy was one of the molecular mechanisms partially involved in the endogenous and AUY922-induced KIT degradation. In 2006, Bauer et al. report the potential use of HSP90AA1 inhibitor as a treatment option for TKI-resistant GIST and show that HSP90AA1 inhibition would result in KIT ubiquitination followed by proteasome-mediated degradation, and GIST cells apoptosis.Citation14 Several HSP90AA1 inhibitors, including analogs of GA 17-DMAG, IPI-504 and its pro-drug IPI-493, purine derivative BIIB021 and synthetic STA-9090, have been evaluated and demonstrated their antitumor activities on TKI-resistant GIST cells in preclinical models.Citation32-Citation35 Unfortunately, further clinical studies have failed to demonstrate their potential clinical usefulness in patients with TKI-refractory GIST. For instance, a previous randomized, phase III trial of IPI-504 vs. placebo control has been terminated early because of high incidence of unexpected treatment-related severe adverse events, including renal failure, liver failure, metabolic acidosis and cardiopulmonary arrest in experimental arm.Citation36 Additionally, paired biopsy sample analysis from first stage of STA-9090 in a phase II trial has shown that weekly administration of STA-9090 is unable to sustainably inhibit the activation of KIT and its downstream pathways in patients with TKI-refractory advanced GIST.Citation37 In the present study, we demonstrated that AUY922 could effectively downregulate the expression of both phospho- and total KIT in time- and dose-dependent manners, as most of other HSP90AA1 inhibitors did. However, AUY922 appeared to be more potent than 17-AAG to inhibit the growth of both IM-sensitive GIST882 and IM-resistant GIST48 cells, as shown in . The minimum concentration to abolish expression of the phospho-KIT and the IC50 to inhibit the growth of both GIST882 and GIST48 cell lines was approximately 50 nM. In addition, a 24 h exposure was able to induce sustainable downregulation of the phospho- and total KIT protein for more than 96 h in GIST48 cells (). These findings along with its prolonged half-life in human pharmacokinetic study would support the use of weekly administration as an optimal schedule for AUY922 to treat patients with TKI-resistant GIST (NCT01389583 and NCT01404650).Citation38
Based on the study of Bauer et al., the underlying mechanisms mediated by the HSP90AA1 inhibition-induced KIT downregulation are thought to involve enhanced degradation of KIT via an ubiquitin-dependent proteasome pathway. In the present study, we found that 3-MA could partially reverse the effects of AUY922 on KIT protein degradation in both GIST cells with endogenous mutant KIT expression and COS-1 cells transfected with KIT-containing plasmid, as MG-132 did. The involvement of autophagy in the AUY922-induced KIT degradation was further validated by experiments with an alternative autophagy inhibitor, knockdown of the expression of either BECN1 or ATG5 using their specific siRNAs, and colocalization of KIT protein with autophagosomes in confocal microscopy after fluorescent immunostaining. Our findings were also supported by recent studies that ubiquitinated proteins can lead to either proteasome- or autophagy-mediated degradation, and there is crosstalk between these two protein-degradation pathways.Citation39,Citation40
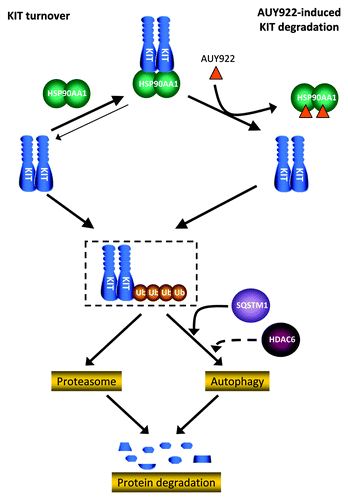
Although proteasome and autophagy pathways are assumed to be two independent degradation systems, increasing evidence suggests that there are numerous intersections between these two degradation machineries. As an example, SNCA/α-synuclein, is degraded by both the proteasome and autophagy after the ubiquitination of the co-chaperone carboxyl terminus of HSPA1A-interacting protein (STUB1).Citation41 Ubiquitination, a major characteristic of proteasome-mediated degradation, demonstrates that proteins ubiquitinated on K63 chains are recognized and degraded through the autophagic machinery.Citation42 Histone deacetylases 6 (HDAC6) and SQSTM1 are two of molecular links between the two systems. HDAC6 could interact with polyubiquitinated proteins and rescue defects of the proteasomal degradation by increasing autophagy.Citation43 On the other hand, SQSTM1 could act as a cargo receptor delivering ubiquitinated substrates to autophagosome, named as a selective autophagy.Citation40,Citation44-Citation48 In our study, and , and showed that the amount of KIT was still reduced through either autophagy or other degradation pathways such as lysosome, when the proteasome pathway was blocked, and vice versa. indicated that SQSTM1 might shuttle KIT to autophagosome. Based on the reports in the literature and our findings, summarizes the mechanisms involved in KIT degradation. It shows that most KIT proteins were protected by HSP90AA1 from degradation and few were degraded via the proteasome and autophagy pathways, as part of endogenous protein turnover. While HSP90AA1 function was inhibited by AUY922, KIT was released, supposed to be ubiquinated, and degraded via proteasomal and autophagy machineries. The SQSTM1 and HDAC6 might be involved in delivering ubiquinated KIT to autophagosome to induce selective autophagy. In addition, other protein degradation pathway, as lysosome, might also play a role in KIT degradation. This highlighted the importance of clarifying the involvement of autophagy in KIT turnover and HSP90AA1 inhibitor-induced degradation.
Figure 6. Scheme of KIT protein degradation pathway. Ub, ubiquitin
Autophagy is a self-degradation process that contributes to the turnover of long-lived proteins as well as to the physiological elimination of old or damaged organelles to providing cells nutrition support under various stresses. It has also been linked to the various pathogenesis, including tumorigenesis.Citation49 Recently, autophagy has been shown to be involved in the degradation of oncoproteins, for instance, all trans-retinoid acid-mediated degradation of promyelocytic leukemia-retinoic acid receptor-α (PRAM1) fusion oncoprotein in acute promyelocytic leukemias (APL) and oxazoline analog of apratoxin A (oz-apraA)-induced degradation of EGFR. All-trans retinoic acid (ATRA) is the current first-line treatment for APL. It could induce PRAM1 degradation, differentiation of leukemic cells, and clinical remission. Isakson et al. have found that autophagy is the major pathway accounting for both the basal turnover as well as the ATRA-induced degradation of PRAM1.Citation50 Wang et al. have further demonstrated that blockage of autophagy by RNA interference against to autophagy essential proteins and by pharmacological inhibitor 3-MA can prevent PRAM1 degradation and subsequently, differentiation of human myeloid leukemic cells.Citation51 Recently, by recognizing a conserved KFERQ-like motif in EGFR, Shen et al. propose and have demonstrated that oz-apraA-induced HSP90AA1 client EGFR degradation occurs through chaperone-mediated autophagy (CMA).Citation52 Our study demonstrated that AUY922 treatment could destabilize KIT through both proteasome- and autophagy-dependent pathways. The reduction of total and phospho-KIT expression level was associated with inactivation of downstream AKT and MAPK1/3 signaling and consequently caused cell apoptosis as previously reported.Citation12,Citation53 The mechanisms of AUY922-induced KIT degradation include both AKT inactivation-induced activation of autophagy and HSP90AA1 inhibition-induced increase of unchaperoned, polyubiquitinated KIT accumulation in autophagosome. These findings suggest the potential combination of AUY922 and autophagy inducer in the treatment of TKI-resistant, KIT expressing GIST.
In treating patients with advanced GIST, IM resistance is categorized into two patterns. In the first, 10–20% of patients experience disease progression within 3–6 mo after the initiation of IM treatment. This is classified as primary or early resistance and is usually associated with either a KIT mutation in exon 9, a D842V mutation in platelet-derived growth factor receptor α (PDGFRA), or overexpression of wild-type KIT and PDGFRA. Meanwhile, late resistance is defined as progression occurring more than 6 mo after beginning the IM treatment. Late resistance can be associated with several mechanisms, including secondary KIT mutations, genomic KIT amplification, the activation of alternative survival pathways, altered pharmacokinetics or the elevated expression of drug efflux proteins.Citation54,Citation55 Therefore, the overexpression of wild-type KIT presents another challenge for therapy. Our studies showed that AUY22 could effectively downregulate not only mutant KIT but also wild-type KIT, implying that it could overcome IM resistance in GIST patients whose tumors express KIT with double mutations or even wild-type.
In conclusion, we found that AUY922 treatment induced downregulation of KIT protein and apoptosis in both IM-sensitive and -resistant GIST cells. In addition to proteasome-dependent pathway, autophagy-mediated protein degradation may also play an important role in both endogenous and HSP90AA1 inhibitor induced KIT degradation in GIST. Our data supported the potential use of AUY922 in treating TKI-refractory, KIT-expressing GIST and highlighted a possible role for autophagy in HSP90AA1 mediated KIT protein degradation.
Materials and Methods
Chemicals and antibodies
AUY922 and IM were kindly supplied by Novartis. CHX (C1988), MG-132 (C2211), lactacystin (L6785), 3-MA (M9281), bafilomycin A1 (B1793), 17-AAG (A8476), AO (158550), protease inhibitor cocktail (P8340), PMSF (P7626), sodium fluoride (S7920), sodium orthovanadate (S6508), bovine serum albumin (A7906), CelLytic cell lysis reagent (C2978), and rabbit anti-MAP1LC3A/B (L7543) were purchased from Sigma Aldrich. Primary antibodies against KIT (A4502) or phospho-KIT (Tyr703) (44-492) were obtained from DAKO and Invitrogen, respectively. Antibodies against MAPK1/3 (9102), phospho-MAPK1/3 (Tyr204/Thr202) (9101), AKT (4691), phospho-AKT (Ser473) (9271), BECN1 (3738), and ATG5 (2630) were purchased from Cell Signaling Technology. Normal rabbit IgG (SC-2027), normal goat IgG (SC-2028), normal mouse IgG (SC-2025), goat anti-MAP1LC3B (SC-16755), rabbit anti-PARP1 (SC-7150), and mouse anti-SQSTM1 (SC-28359) were obtained from Santa Cruz Biotechnology. Antibodies against HSPA1A (SPA-810) and actin (MAB1501) were obtained from Stressgen and Millipore, respectively. Horseradish peroxidase-labeled secondary antibodies and CyTM2 goat anti-rabbit (111-225-144), rhodamine donkey anti-goat (705-025-147), and rhodamine goat anti-mouse (115-025-146) antibodies were purchased from Jackson ImmunoResearch.
Construction of the KIT mutants
Messenger RNA (mRNA) was isolated from the peripheral blood mononuclear cells of a healthy volunteer. A 2.9 kB full-length complimentary DNA (cDNA) of wild-type KIT was obtained by reverse transcription PCR (RT-PCR) using primers matching sequences in exon 1 and 21 of KIT and was cloned into pcDNATM3.1/Zeo (Invitrogen, V86020). The sequence fidelity of KIT was then confirmed by bidirectional sequencing. Common single and/or double KIT mutants were constructed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, 200518), while a 63-bp deletion of exon 11Δ555–576 was generated by slicing overlap extension PCR, as described previously.Citation56 The primers used for site-directed mutagenesis and SOE are listed in .
Table 1. Primers used for mutagenesis of KIT
Cell lines and transient transfection
GIST882 and GIST48 cells encoding the exon 13K642E and exon 11V560D/17D820A mutant KIT oncoprotein, respectively, were gifts from Dr. Jonathan Fletcher (Harvard Medical School). GIST882 cells were maintained in RPMI 1640 containing 20% fetal bovine serum (FBS). GIST48 cells were incubated in F10 with 15% FBS, 0.5% Mito+ serum extender (BD Biosciences, 355006) and 1% pituitary extract bovine (BD Biosciences, 354123). COS-1 cell line was obtained from Dr. Shih (Neng-Yao laboratory, National Health Research Institute) and maintained in DMEM containing 10% FBS. COS-1 cells were transfected using Lipofectamine 2000® (Invitrogen, 11668-019) according to the manufacturer’s protocol. In brief, COS-1 cells were grown to approximately 90% confluence in 6-well plates and then admixed with 2 μg of plasmid KIT/pcDNA3.1 and 2 μl of Lipofectamine 2000® for 6 h. Transfected cells were allowed to recover in DMEM plus 10% FBS for 18 h and were then treated with AUY922 or other inhibitors.
RNA interference
Small interfering RNA (siRNA) specific to human BECN1 (1299003) and negative control siRNA were purchased from Invitrogen. Specific siRNA targeted to Human ATG5 was synthesized by Invitrogen, using published sequences 5-GGACGAAUUCCAACUUGUU-3.Citation57 GIST882 and GIST48 cells were transfected with BECN1, ATG5 or negative control siRNA using Amaxa electroporation (Amaxa Biosystems, VCA-1003) or Lipofectamine 2000®, respectively, according to the manufacturer’s protocol. Briefly, 2 × 106 trypsinized GIST882 cells were transfected with annealed RNA duplexes, transferred to 6-well culture plates and grown in 2 ml antibiotic-free RPMI 1640 for 72 h. GIST48 cells among 90% confluence in the 6-well plates were transfected with indicated dose of annealed RNA duplexes mixed with 2 μl of Lipofectamine 2000® for 6 h, and then replaced with serum-containing complete medium for 72 h. The influence of siRNA on BECN1 or ATG5 protein expression was confirmed by immunoblotting with specific antibodies.
Cell viability assay
For this assay, 4 × 104 GIST882 or 2 × 104 GIST48 cells were seeded per well in 24-well plates. GIST882 and GIST48 Cells were exposed to various concentrations of drugs for 96 h and 72 h, respectively. The methylene blue dye assay was used to evaluate the effects of each drug on the relative number of viable cells.Citation58 Data were measured with a SpectraMax M5 microplate reader (Molecular Device) at 595 nm and normalized to the DMSO-only control group. The drug concentration that inhibited cell growth by 50% (IC50) was determined after plotting growth relative to the untreated controls. All experimental points were measured in duplicate wells for each plate and replicated in at least three plates.
Clonogenic assay
One × 103 of GIST882 or GIST48 cells in logarithmic growth phase were seeded per well in 6-well plates. GIST48 and GIST882 cells were incubated with indicated doses of drugs for 1 d, replaced with growth media for further 14 and 21 d, respectively, and stained with 50% ethanol containing 0.5% methylene blue for 5 h. The plates were washed five times with water and allowed to air-dry. Colonies were countered manually. The IC50 value resulting from 50% inhibition of cell growth was determined after plotting growth relative to the untreated controls. Each value represented the average of at least three independent experiments run in triplicates.
Immunoblotting studies
Cells were lysed in CelLyticTM M Cell Lysis Reagent containing protease inhibitor cocktail, PMSF, sodium fluoride and sodium orthovanadate. The protein concentration was determined using the Bradford method (Bio-Rad, 500-0006). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed after loading equal amounts of protein into each lane. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes for immunoblotting. After blocking with bovine serum albumin, the membranes were blotted by adding primary antibodies against KIT, KITY703, MAPK1/3, MAPK1Y204/3T202, AKT, AKTS473, HSPA1A, MAP1LC3A/B, PARP1 or ACTIN, followed by the appropriate secondary antibodies. Protein bands were detected by enhanced chemiluminescence (ECL) (PerkinElmer, NEL104001EA), developed by autoradiography and quantified using 1Dscan EX gel analysis software (Scanalytics). The half-life of the KIT protein in each group was estimated after plotting KIT protein expression relative to the untreated control. The data are expressed as the mean ± SE.
Fluorescent immunostaining and acridine orange staining
For fluorescent immunostaining, GIST822 and GIST48 cells were fixed with paraformaldehyde, permeabilized with Triton X-100, and then blocked with 5% BSA in PBS at 4°C overnight. Cells were incubated with primary antibodies against KIT, MAP1LC3B, and SQSTM1, respectively. Cells for nonspecific staining control were incubated with normal rabbit, goat, and mouse antibodies, respectively. The cells were washed three times with PBS, incubated with AO or a fluorescently labeled secondary antibody, washed three times with PBS and then examined under a confocal microscope (Olympus, FV1000, Confocal Laser Scanning Biological Microscope).
Flow cytometry
Apoptosis was determined by staining cells with Annexin V-FITC (BD Biosciences, 556419) and PI (BD Biosciences, 556463) according to the manufacturer’s protocol, followed by flow cytometry analysis. In brief, GIST882 and GIST48 cells were incubated with AUY922 or IM for the indicated time and dose and then trypsinized. Samples containing 1 × 105 cells were washed with cold PBS and resuspended in 100 μl binding buffer. Then, 2 μl Annexin V-FITC and 5 μl PI were added to the cells and incubated for 15 min at RT in the dark. An additional 400 μl binding buffer was added to the reaction prior to analysis.
| Abbreviations: | ||
| 3-MA | = | 3-methyladenine |
| 17-AAG | = | 17-N-allylamino-17-demethoxygeldanamycin |
| AO | = | acridine orange |
| AKT | = | v-akt murine thymoma viral oncogene homolog |
| APL | = | acute promyelocytic leucemias |
| ATG5 | = | autophagy-related 5 |
| ATRA | = | all-trans retinoic acid |
| AUY922 | = | NVP-AUY922 |
| BECN1 | = | Beclin 1 |
| CHX | = | cycloheximide |
| CMA | = | chaperone-mediated autophagy |
| EGFR | = | epidermal growth factor receptor |
| GA | = | geldanamycin |
| GIST | = | gastrointestinal stromal tumor |
| HDAC6 | = | histone deacetylase 6 |
| HSP90AA1 | = | heat shock protein 90 |
| HSPA1A | = | heat shock 70kDa protein 1A |
| IC50 | = | the drug concentration that inhibited cell growth by 50% |
| IM | = | Imatinib Mesylate |
| KIT | = | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog |
| MAP1LC3A/B | = | microtubule-associated protein 1 light chain 3 alpha and beta |
| MAPK1/3 | = | mitogen-activated protein kinase 1 and 3 |
| oz-apraA | = | oxazoline analogue of apratoxin A |
| PARP1 | = | poly (ADP-ribose) polymerase 1 |
| PDGFRA | = | platelet-derived growth factor receptor alpha |
| PRAM1 | = | PML-RARA regulated adaptor molecule 1 |
| SCF | = | stem cell factor |
| STUB1 | = | STIP1 homology and U-box containing protein 1 |
| SQSTM1 | = | sequestosome 1 |
| SU | = | sunitinib malate |
| TKI | = | tyrosine kinase inhibitor |
| Ub | = | ubiquitin |
| WT | = | wild type |
Acknowledgments
The authors are grateful to the administration and laboratory support from National Institute of Cancer Research, National Health Research Institute, and funding support from National Research Program for Biopharmaceuticals (100TMP009-1 and 101TMP-1033-1), and also administration and personnel support from the Excellence Cancer Research Center Program (DOH101-TD-C-111-004), Department of Health, Executive Yuan, Taiwan.
Disclosure of Potential Conflicts of Interest
Research funds were from National Health Research Institutes (L.-T.C.) and National Research Program for Biopharmaceuticals (NRPB), Department of Health, Executive Yuan, Taiwan (L.-T.C., C.-C.Y., C.-F.L.). Research compounds (AUY922 and imatinib) were kindly supplied by Novartis (L.-T.C.; honorarium and research grant from Novartis, Merck Serono, and TTY (L.-T.C.).
References
- Nishida T, Hirota S. Biological and clinical review of stromal tumors in the gastrointestinal tract. Histol Histopathol 2000; 15:1293 - 301; PMID: 11005253
- Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet 2007; 369:1731 - 41; http://dx.doi.org/10.1016/S0140-6736(07)60780-6; PMID: 17512858
- Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001; 61:8118 - 21; PMID: 11719439
- van der Zwan SM, DeMatteo RP. Gastrointestinal stromal tumor: 5 years later. Cancer 2005; 104:1781 - 8; http://dx.doi.org/10.1002/cncr.21419; PMID: 16136600
- Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 2008; 3:557 - 86; http://dx.doi.org/10.1146/annurev.pathmechdis.3.121806.151538; PMID: 18039140
- Lasota J, Miettinen M. Clinical significance of oncogenic KIT and PDGFRA mutations in gastrointestinal stromal tumours. Histopathology 2008; 53:245 - 66; http://dx.doi.org/10.1111/j.1365-2559.2008.02977.x; PMID: 18312355
- Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006; 24:4764 - 74; http://dx.doi.org/10.1200/JCO.2006.06.2265; PMID: 16954519
- Wang WL, Conley A, Reynoso D, Nolden L, Lazar AJ, George S, et al. Mechanisms of resistance to imatinib and sunitinib in gastrointestinal stromal tumor. Cancer Chemother Pharmacol 2011; 67:Suppl 1 S15 - 24; http://dx.doi.org/10.1007/s00280-010-1513-8; PMID: 21181476
- Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al, EORTC Soft Tissue and Bone Sarcoma Group, Italian Sarcoma Group, Australasian GastroIntestinal Trials Group. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006; 42:1093 - 103; http://dx.doi.org/10.1016/j.ejca.2006.01.030; PMID: 16624552
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006; 368:1329 - 38; http://dx.doi.org/10.1016/S0140-6736(06)69446-4; PMID: 17046465
- Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008; 26:5352 - 9; http://dx.doi.org/10.1200/JCO.2007.15.7461; PMID: 18955458
- Sambol EB, Ambrosini G, Geha RC, Kennealey PT, Decarolis P, O’connor R, et al. Flavopiridol targets c-KIT transcription and induces apoptosis in gastrointestinal stromal tumor cells. Cancer Res 2006; 66:5858 - 66; http://dx.doi.org/10.1158/0008-5472.CAN-05-2933; PMID: 16740725
- Fumo G, Akin C, Metcalfe DD, Neckers L. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells. Blood 2004; 103:1078 - 84; http://dx.doi.org/10.1182/blood-2003-07-2477; PMID: 14551138
- Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 2006; 66:9153 - 61; http://dx.doi.org/10.1158/0008-5472.CAN-06-0165; PMID: 16982758
- Li CF, Huang WW, Wu JM, Yu SC, Hu TH, Uen YH, et al. Heat shock protein 90 overexpression independently predicts inferior disease-free survival with differential expression of the alpha and beta isoforms in gastrointestinal stromal tumors. Clin Cancer Res 2008; 14:7822 - 31; http://dx.doi.org/10.1158/1078-0432.CCR-08-1369; PMID: 19047110
- Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res 2009; 15:9 - 14; http://dx.doi.org/10.1158/1078-0432.CCR-08-0132; PMID: 19118027
- Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol 1996; 10:705 - 12; http://dx.doi.org/10.1210/me.10.6.705; PMID: 8776730
- Sharma SV, Agatsuma T, Nakano H. Targeting of the protein chaperone, HSP90, by the transformation suppressing agent, radicicol. Oncogene 1998; 16:2639 - 45; http://dx.doi.org/10.1038/sj.onc.1201790; PMID: 9632140
- Weinstein IB, Joe A. Oncogene addiction. Cancer Res 2008; 68:3077 - 80, discussion 3080; http://dx.doi.org/10.1158/0008-5472.CAN-07-3293; PMID: 18451130
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003; 425:407 - 10; http://dx.doi.org/10.1038/nature01913; PMID: 14508491
- Vilenchik M, Solit D, Basso A, Huezo H, Lucas B, He H, et al. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90. Chem Biol 2004; 11:787 - 97; http://dx.doi.org/10.1016/j.chembiol.2004.04.008; PMID: 15217612
- Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS, et al. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther 2004; 3:551 - 66; PMID: 15141013
- Banerji U, O’Donnell A, Scurr M, Pacey S, Stapleton S, Asad Y, et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 2005; 23:4152 - 61; http://dx.doi.org/10.1200/JCO.2005.00.612; PMID: 15961763
- Goetz MP, Toft D, Reid J, Ames M, Stensgard B, Safgren S, et al. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol 2005; 23:1078 - 87; http://dx.doi.org/10.1200/JCO.2005.09.119; PMID: 15718306
- Vaishampayan UN, Burger AM, Sausville EA, Heilbrun LK, Li J, Horiba MN, et al. Safety, efficacy, pharmacokinetics, and pharmacodynamics of the combination of sorafenib and tanespimycin. Clin Cancer Res 2010; 16:3795 - 804; http://dx.doi.org/10.1158/1078-0432.CCR-10-0503; PMID: 20525756
- Heath EI, Gaskins M, Pitot HC, Pili R, Tan W, Marschke R, et al. A phase II trial of 17-allylamino-17- demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Prostate Cancer 2005; 4:138 - 41; http://dx.doi.org/10.3816/CGC.2005.n.024; PMID: 16197617
- Ronnen EA, Kondagunta GV, Ishill N, Sweeney SM, Deluca JK, Schwartz L, et al. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs 2006; 24:543 - 6; http://dx.doi.org/10.1007/s10637-006-9208-z; PMID: 16832603
- Solit DB, Osman I, Polsky D, Panageas KS, Daud A, Goydos JS, et al. Phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with metastatic melanoma. Clin Cancer Res 2008; 14:8302 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-08-1002; PMID: 19088048
- Egorin MJ, Rosen DM, Wolff JH, Callery PS, Musser SM, Eiseman JL. Metabolism of 17-(allylamino)-17-demethoxygeldanamycin (NSC 330507) by murine and human hepatic preparations. Cancer Res 1998; 58:2385 - 96; PMID: 9622079
- Kelland LR, Sharp SY, Rogers PM, Myers TG, Workman P. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90. J Natl Cancer Inst 1999; 91:1940 - 9; http://dx.doi.org/10.1093/jnci/91.22.1940; PMID: 10564678
- Brough PA, Aherne W, Barril X, Borgognoni J, Boxall K, Cansfield JE, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J Med Chem 2008; 51:196 - 218; http://dx.doi.org/10.1021/jm701018h; PMID: 18020435
- Floris G, Debiec-Rychter M, Wozniak A, Stefan C, Normant E, Faa G, et al. The heat shock protein 90 inhibitor IPI-504 induces KIT degradation, tumor shrinkage, and cell proliferation arrest in xenograft models of gastrointestinal stromal tumors. Mol Cancer Ther 2011; 10:1897 - 908; http://dx.doi.org/10.1158/1535-7163.MCT-11-0148; PMID: 21825009
- Floris G, Sciot R, Wozniak A, Van Looy T, Wellens J, Faa G, et al. The Novel HSP90 inhibitor, IPI-493, is highly effective in human gastrostrointestinal stromal tumor xenografts carrying heterogeneous KIT mutations. Clin Cancer Res 2011; 17:5604 - 14; http://dx.doi.org/10.1158/1078-0432.CCR-11-0562; PMID: 21737509
- Lin TY, Bear M, Du Z, Foley KP, Ying W, Barsoum J, et al. The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Exp Hematol 2008; 36:1266 - 77; http://dx.doi.org/10.1016/j.exphem.2008.05.001; PMID: 18657349
- Marcu MG, Schulte TW, Neckers L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J Natl Cancer Inst 2000; 92:242 - 8; http://dx.doi.org/10.1093/jnci/92.3.242; PMID: 10655441
- Demetri GD, Le Cesne A, von Mehren M, Chmielowski B, Bauer S, Chow WA, et al. Final results from a phase III study of IPI-504 (retaspimycin hydrochloride) versus placebo in patients (pts) with gastrointestinal stromal tumors (GIST) following failure of kinase inhibitor therapies. [abstract]. In: Proceedings of the ASCO Gastrointestinal Cancers Symposium; 2010 Jan 22–24; Orlando, FL. Alexandria (VA): ASCO; 2010. Abstract nr 64.
- Demetri GD, Heinrich MC, Chmielowski B, Morgan JA, George S, Bradley R, et al. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) in patients (pts) with metastatic and/or unresectable GIST. J Clin Oncol 2011; 29:Suppl abstr 10011
- Ide S, Motwani M, Jensen MR, Wang J, Huseinovic N, Stiegler P, et al. Pharmacodynamics and pharmacokinetics of AUY922 in a phase I study of solid tumor patients. J Clin Oncol 2009; 29:Suppl abstr 3533
- Willis MS, Townley-Tilson WH, Kang EY, Homeister JW, Patterson C. Sent to destroy: the ubiquitin proteasome system regulates cell signaling and protein quality control in cardiovascular development and disease. Circ Res 2010; 106:463 - 78; http://dx.doi.org/10.1161/CIRCRESAHA.109.208801; PMID: 20167943
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836 - 41; http://dx.doi.org/10.1038/ncb0910-836; PMID: 20811356
- Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 2005; 280:23727 - 34; http://dx.doi.org/10.1074/jbc.M503326200; PMID: 15845543
- Olzmann JA, Chin LS. Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy 2008; 4:85 - 7; PMID: 17957134
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007; 447:859 - 63; http://dx.doi.org/10.1038/nature05853; PMID: 17568747
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol 2010; 22:192 - 8; http://dx.doi.org/10.1016/j.ceb.2009.11.002; PMID: 19962293
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem 2011; 80:125 - 56; http://dx.doi.org/10.1146/annurev-biochem-052709-094552; PMID: 21548784
- Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ 2012; •••; http://dx.doi.org/10.1038/cdd.2012.72; PMID: 22722335
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726 - 34; http://dx.doi.org/10.1038/nrc1692; PMID: 16148885
- Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116:2324 - 31; http://dx.doi.org/10.1182/blood-2010-01-261040; PMID: 20574048
- Wang Z, Cao L, Kang R, Yang M, Liu L, Zhao Y, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011; 7:401 - 11; http://dx.doi.org/10.4161/auto.7.4.14397; PMID: 21187718
- Shen S, Zhang P, Lovchik MA, Li Y, Tang L, Chen Z, et al. Cyclodepsipeptide toxin promotes the degradation of Hsp90 client proteins through chaperone-mediated autophagy. J Cell Biol 2009; 185:629 - 39; http://dx.doi.org/10.1083/jcb.200810183; PMID: 19433452
- Ou WB, Zhu MJ, Demetri GD, Fletcher CD, Fletcher JA. Protein kinase C-theta regulates KIT expression and proliferation in gastrointestinal stromal tumors. Oncogene 2008; 27:5624 - 34; http://dx.doi.org/10.1038/onc.2008.177; PMID: 18521081
- Sleijfer S, Wiemer E, Seynaeve C, Verweij J. Improved insight into resistance mechanisms to imatinib in gastrointestinal stromal tumors: a basis for novel approaches and individualization of treatment. Oncologist 2007; 12:719 - 26; http://dx.doi.org/10.1634/theoncologist.12-6-719; PMID: 17602061
- Gounder MM, Maki RG. Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemother Pharmacol 2011; 67:Suppl 1 S25 - 43; http://dx.doi.org/10.1007/s00280-010-1526-3; PMID: 21116624
- Warrens AN, Jones MD, Lechler RI. Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest. Gene 1997; 186:29 - 35; http://dx.doi.org/10.1016/S0378-1119(96)00674-9; PMID: 9047341
- Chen KL, Chang WS, Cheung CH, Lin CC, Huang CC, Yang YN, et al. Targeting cathepsin S induces tumor cell autophagy via the EGFR-ERK signaling pathway. Cancer Lett 2012; 317:89 - 98; http://dx.doi.org/10.1016/j.canlet.2011.11.015; PMID: 22101325
- Finlay GJ, Baguley BC, Wilson WR. A semiautomated microculture method for investigating growth inhibitory effects of cytotoxic compounds on exponentially growing carcinoma cells. Anal Biochem 1984; 139:272 - 7; http://dx.doi.org/10.1016/0003-2697(84)90002-2; PMID: 6476363