Abstract
Helicobacter pylori is a Gram-negative pathogen that colonizes the gastric epithelium of 50–60% of the world’s population. Approximately one-fifth of the infected individuals manifest severe diseases such as peptic ulcers or gastric cancer. H. pylori infection has proven difficult to cure despite intensive antibiotic treatment. One possible reason for the relatively high resistance to antimicrobial therapy is the ability of H. pylori to reside inside host cells. Although considered by most as an extracellular pathogen, H. pylori can invade both gastric epithelial cells and immunocytes to some extent. The intracellular survival of H. pylori has been implicated in its ability to persist in the stomach, evade host immune responses and resist eradication by membrane-impermeable antibiotics. Interestingly, recent evidence suggests that macroautophagy, a cellular self-degradation process characterized by the formation of double-membraned autophagosomes, plays an important role in determining the intracellular fate of H. pylori. Detailed understanding of the interaction between H. pylori and host cell autophagic processes is anticipated to provide novel insights into the molecular mechanisms of macroautophagy and H. pylori pathogenesis, opening new avenues for the therapeutic intervention of autophagy-related and H. pylori-related disorders.
Introduction
H. pylori is one of the most common human bacterial pathogens, and infection causes a wide array of gastric disorders, including simple gastritis, peptic ulcer or gastric malignancies.Citation1 In view of the strong correlation of H. pylori infection with gastric cancer in humans, the bacterium was designated a class I carcinogen by the International Agency for Research on Cancer in 1994.Citation2 Individuals chronically infected with H. pylori typically show marked inflammation characterized by the substantial infiltration of neutrophils and macrophages into the gastric epithelial layer.Citation3H. pylori also induces significant T cell responses.Citation4 However, for reasons that remain to be understood, the pronounced host immune responses triggered fail to clear the infection. It has been proposed that this inefficiency in bacterial clearance may be due to the ability of H. pylori to take refuge within gastric epithelial cells, which allows the bacterium to escape eradication by immunocytes and extracellularly-acting antibiotics. H. pylori can also survive in phagocytes.Citation5,Citation6 Accumulating evidence suggests that the bacterium can perturb normal phagocytic pathways in order to prevent or delay its degradation during infection.Citation3,Citation5,Citation7
The precise mechanisms by which H. pylori exploits host cell machineries for intracellular survival are poorly understood. Over the last decade, several research groups have independently reported that infection by H. pylori can induce macroautophagy.Citation6,Citation8-Citation11 Macroautophagy is a conserved process used by eukaryotic cells to maintain cellular homeostasis and defend against invading microbial pathogens.Citation12 Although mostly targeting intracellular bacteria, macroautophagy can also act against those bacteria regarded as being extracellular when they manage to enter the eukaryotic cell.Citation13 It has been proposed that H. pylori, once internalized and sequestered in double-membraned autophagosomes, can use these compartments as a replicative niche.Citation6,Citation8,Citation10 H. pylori has been reported also to evade the autophagic machinery by downregulating the expression of autophagic proteins.Citation11 The subversion of, or subjection to, host autophagic machinery by H. pylori adds further complexity to the already multifaceted pathogenesis of this bacterium. This review presents an overview of the current findings on how autophagic processes have an impact upon the intracellular fate of H. pylori and discusses their implications for its pathogenesis.
Helicobacter pylori
H. pylori is a Gram-negative, spiral-shaped, flagellated, microaerophilic bacterium that causes a wide range of gastric diseases.Citation1 Though occurring worldwide, the prevalence of H. pylori infection is higher in developing countries compared with developed countries as determined by geographic, genetic and socioeconomic factors.Citation14 Infections are acquired mostly in childhood via the fecal-oral or oral-oral routes of transmission.Citation15
H. pylori is unique in that it resides within the gastric mucosal environment despite this being a highly acidic environment, which normally serves as a means of protection from bacterial pathogens. Once it has become established within this ecological niche, colonization leads to chronic inflammation of the gastric mucosa and the induction of histological gastritis. Many people infected with H. pylori remain asymptomatic until the bacterium evades the immune system to achieve persistent colonization.Citation16 Long-term H. pylori infection results in the more severe symptoms of gastric disorders including peptic ulcer, gastric adenocarcinoma and mucosal-associated lymphoid tissue (MALT) lymphoma.Citation1
Like most pathogens, H. pylori possesses multiple virulence factors, including urease, catalase, peptidoglycan, neutrophil-activating protein (NapA), cytotoxin-associated-gene A (CagA), the cag pathogenicity island (cag PAI), vacuolating toxin (VacA), and the outer membrane proteins SabA, BabA, AlpA and OipA (reviewed in refs. Citation1 and Citation17). Clinical isolates of H. pylori are broadly categorized into type I and type II, which differ in their virulence capacity and the expression of two cytotoxic proteins, CagA and VacA. Clinical presentation of infection with type I strains (which produce CagA and VacA) is generally more severe compared with infection with type II strains (which lack these two cytotoxins).Citation18 CagA and VacA modulate a range of host cell processes during infection, and both are of particular relevance to this review.
CagA, a 120- to 140-kDa protein, is encoded by the cagA gene located at one end of the cag pathogenecity island, a 40-kb stretch of the H. pylori chromosomal DNA.Citation1,Citation17 In addition to CagA, cag PAI encodes the structural components of a putative type IV secretion system,Citation19,Citation20 a transmembrane macromolecular apparatus thought to facilitate the translocation of CagA and other virulence factors into host cells.Citation21,Citation22 Once within the host cell, CagA is tyrosine phosphorylated at EPIYA (glutamate-proline-isoleucine-tyrosine-alanine) motifs by the members of Abl and Src kinase families.Citation23-Citation26 Subsequent to this phosphorylation, a number of signaling molecules are activated leading to changes in host cell morphology and cellular functions, including polymerization of cytoskeletal actin, disruption of cell-to-cell junctions, modulation of the inflammatory response, and induction of apoptosis.Citation27 The phosphorylated form of CagA has also been shown to induce gastric epithelial cell proliferation and carcinoma and is therefore considered a potential oncoprotein.Citation17 A proportion of translocated CagA molecules seems to remain unphosphorylated and their interaction with host cell molecules has been reported to stimulate mitogenic responses, pro-inflammatory reactions, disruption of cell-to-cell junctions, and alteration of cell polarity.Citation27
Another important virulence factor of H. pylori is VacA, which plays a role in the pathogenesis of both peptic ulcer and gastric cancer.Citation28,Citation29 VacA is a highly immunogenic toxin capable of forming vacuoles in host cells.Citation30 The 90-kDa toxin is produced from a 140-kDa protoxin encoded by the vacA gene.Citation31 This mature toxin is then further cleaved into two subunits: p33 (a 33-kDa N-terminal fragment) and p55 (a 55-kDa C-terminal fragment).Citation32,Citation33 Although initially p33 was reported as being required for vacuolization activityCitation34 and p55 as playing a role in cell-binding activity,Citation35 more recent reports have suggested that p33 is also involved in cell binding, and that p55 is critical for vacuolation.Citation36-Citation39 Although all strains of H. pylori possess a functional vacA gene, polymorphism in vacA has been observed in its encoded signal region (s), the middle region (m) and the more recently identified intermediate region (i).Citation40,Citation41 This sequence heterogeneity is linked to disease severity.Citation28 Apart from vacuole formation, VacA apparently exerts a number of other effects on host cells, including disruption of endosomal and lysosomal activity by forming channels on their membranes, disruption of mitochondrial function, retardation of T-cell proliferation, induction of apoptosis, and inflammatory modulation.Citation33
Our understanding of the roles of CagA and VacA in the pathogenesis of H. pylori infection has been continually increasing since their discovery and initial characterization. Recent findings have raised the possibility that they might also be involved in autophagy induction upon H. pylori infection of host cells.
Autophagy and its Role in Antimicrobial Defense
Autophagy is an evolutionarily conserved catabolic process in eukaryotes that maintains cellular homeostasis through the degradation of cargo such as long-lived proteins and damaged organelles. The resulting molecules can then be reused to help cells adapt to cellular stresses such as nutrient starvation, or growth factor depletion. There are three types of autophagy: (1) macroautophagy, (2) microautophagy and (3) chaperone-mediated autophagy, which differ in the process of cargo delivery to the lysosome and the nature of the cargo/substrate.Citation42,Citation43 Here we will concern ourselves with the process of macroautophagy (hereafter referred to as autophagy) which is characterized by the formation of double-membraned autophagosomes that sequester the cargo. The autophagosomes subsequently fuse with lysosomes to create a compartment where the degradation of cargo takes place.
The molecular mechanism of autophagy
The machinery and molecular pathways of autophagy are complex and not fully elucidated. The overall process as currently understood can be divided into several distinct steps including induction and nucleation of the phagophore (the precursor to the autophagosome), cargo recognition, expansion of the phagophore and completion of the autophagosome, vesicle maturation/fusion with the lysosome, and finally cargo degradation (). Each of these steps involves a number of ATG (autophagy-related) proteins.Citation12
Figure 1. Autophagy in mammalian cells. After induction of autophagy by the inhibition of MTOR in response to various stimuli, the ULK complex is activated and translocated to the rough ER followed by subsequent recruitment of the class III PtdIns3K complex (which includes BECN1) and the generation of its substrate PtdIns3P. Effector proteins (e.g., ZFYVE1, WIPIs) are then recruited to the ER and eventually result in the formation of a phagophore/omegasome, which develops into the autophagosome. Next, cargo is recruited to the autophagic machinery possibly via a ligand-receptor-scaffold complex that ultimately interacts with LC3-II. Two ubiquitin-like conjugation systems (ATG12–ATG5 and LC3–PE) are required for the expansion of the phagophore and completion of the autophagosome. Finally, the maturation of autophagosomes takes place when they fuse with acidic lysosomes to form autolysosomes where the cargo is degraded by lysosomal enzymes.
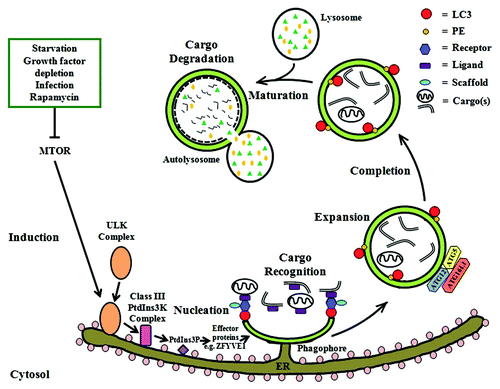
The initiation of autophagy requires the inhibition of mechanistic target of rapamycin (MTOR), a serine/threonine protein kinase, triggered by conditions including starvation, growth factor depletion, infection or by the inhibitory action of the bacteria-derived macrolide rapamycin.Citation43 Inhibition of MTOR then promotes the downstream activation of ULK1/2 (Unc-51-like kinase) that phosphorylates ATG13 and RB1CC1; the significance of which is yet to be understood.Citation44 Next, C12orf44/ATG101 binds to and stabilizes ATG13, and all of these proteins together form the MTOR substrate termed the ULK complex (ULK1/2-ATG13-RB1CC1-C12orf44). The ULK complex translocates (by a mechanism still not clear in mammalian cells) from the cytosol to the endoplasmic reticulum (ER) where the nucleation of autophagosome formation is thought to occur. The association of the ULK complex with the ER activates an ER-localized autophagy-specific class III phosphatidylinositol 3-kinase (PtdIns3K) complex.Citation43 This complex is composed of at least PIK3C3/VPS34, PIK3R4/VPS15, BECN1 and ATG14. BECN1 regulates the induction of autophagy, and is in turn regulated by BCL2 (B-cell CLL/lymphoma 2) in a nutrient-dependent manner. Under nutrient-rich conditions, BCL2 inhibits autophagy by binding to BECN1, whereas upon starvation BCL2 dissociates from BECN1, inducing autophagy.Citation12 The resulting PtdIns3K complex generates phosphatidylinositol 3-phosphate (PtdIns3P) on the outer membrane of the ER, which in turn recruits other effector proteins such as ZFYVE1Citation45 and WIPIs,Citation46 which are required for the formation of an ER-associated omegasome.Citation44 This initial structure eventually forms the omegasome from which a double-membraned autophagosome is developed.Citation43 The origin of the autophagosomal membrane is not known and more than one source might be involved. Several studies have suggested that the main source of this membrane is the ER, as electron tomography shows a connection between the omegasome and ER.Citation47,Citation48 However, later studies have also presented evidence that supports other possible membrane sources, such as the Golgi apparatus,Citation49 mitochondriaCitation50 and plasma membrane.Citation51
The mechanism of cargo selection and its subsequent delivery into the autophagosomal structure remain unclear. A very recent concept of “receptor protein complexes” has been proposed by Mijaljica et al.Citation52 to describe the mechanism of cargo recognition. According to this concept, a ligand(s) present on the surface of cargo is recognized by a receptor (e.g., SQSTM1/p62, NBR1) in the cytosol, either directly or first linked with ubiquitin before being recognized by a ubiquitin-binding receptor. The ligand-bound receptor then interacts with a scaffold, an autophagic protein (e.g., Atg11 in yeast), which brings cargo into close proximity with the autophagy machinery. In mammalian cells, the final step involves the recognition of microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3), a ubiquitin-like protein, by the ligand-bound receptor via a specific LC3-interacting region present on the receptor.Citation52
Expansion of the phagophore and complete enclosure of the autophagosome require two ubiquitin-like conjugation systems: the ATG12–ATG5 protein conjugate that interacts with ATG16L1 to form a large complex via the dimerization of ATG16L1,Citation44,Citation53,Citation54 and the covalently-linked LC3–phosphatidylethanolamine (PE) conjugate that is initially present on both the outer and inner membrane of the autophagosome.Citation55,Citation56 This conjugated form of LC3, also known as LC3-II, is the only known protein marker that associates with phagophores and autophagosomes throughout the autophagy process in mammalian cells.Citation57 Hence, the detection of LC3-II, either by immunofluorescence microscopy (as GFP-LC3 puncta) or by western blot (as the conversion from cytosolic LC3-I to LC3-II), has been widely used as a specific marker of autophagic activity.Citation57 However, such results have to be interpreted carefully, as LC3 can aggregate within cells or be incorporated in inclusion bodies in an autophagy-independent manner.Citation58 LC3 is also involved in the pathogenesis of Chlamydia trachomatis, independent of autophagic processes, as a microtubule-associated proteinCitation59 and in human skeletal homeostasis.Citation60 Most notably, LC3 can also be recruited to single-membrane phagosomes in a process termed LC3-associated phagocytosis (LAP), which shares many similarities with autophagy,Citation61,Citation62 a phenomenon to be discussed in further detail below.
The final event in autophagy is the maturation of autophagosomes, during which autophagosomes fuse with acidic lysosomes to form autolysosomes. The latter contain lysosomal proteases whose activity results in the degradation of sequestered cargos.Citation12 Together, all these steps constitute the autophagic flux, a continuous intracellular flow from autophagosome generation to the degradation of the cargo and the release of the breakdown products back into the cytosol.
As mentioned above, LC3 may be recruited to the phagosomal membrane in a nonclassical autophagic process named LAP, in addition to the classical LC3 recruitment to phagophore membrane as occurs in canonical autophagy. LAP was first described as a phenomenon in professional phagocytes where, upon ingestion of cargo containing toll-like receptor (TLR) ligands, LC3 is rapidly recruited to the surface of these phagosomes within 15 min, as opposed to the 2–24 h in conventional TLR-induced autophagy.Citation61,Citation62 This intracellular process has been implicated in the infection pathway for several bacterial pathogens, including Escherichia coli,Citation63 Mycobacterium marinumCitation64 and Burkholderia pseudomallei.Citation65 Detection of LC3-II is therefore not in itself sufficient to identify autophagy. The distinction between autophagy and LAP is critical, and research that looks into these differences is only beginning to emerge. So far, known differences between the two pathways are the membrane nature of their compartment (single-membrane phagosomes in LAP vs. double-membrane autophagosomes in autophagy),Citation62 and the requirement for the ULK1 protein in autophagy but not in LAP.Citation66 Currently, the most readily applicable technique able to distinguish between canonical autophagy and LAP is transmission electron microscopy (TEM), which enables determination of the ultrastructure of the bacteria-containing vesicles.Citation62
Roles of autophagy in infection and immunity
Over the past few years, much attention has been paid to a selective form of autophagy termed xenophagy that targets intracellular pathogens as cargo. This form of autophagy plays a vital role in the innate and adaptive immune system responses to infection by intracellular and extracellular pathogens.Citation42,Citation43 The overall process of xenophagy uses the machinery of autophagy, except no scaffold has been so far identified for xenophagic cargo (i.e., pathogen) recognition.Citation52 Generally, the induction of autophagy by pathogenic bacteria is triggered via virulence factors such as pathogen-associated molecular patterns, toxins and other effector proteins.Citation67 Upon induction of autophagy, three possible consequences have been observed ().Citation13 First, autophagy restricts the growth of bacteria as seen with Salmonella enterica serovar Typhimurium and M. tuberculosis. The mycobacterial lipoprotein LpqH activates autophagy that targets bacteria-containing phagosomes,Citation68 whereas Salmonella-containing vacuoles are targeted to autophagosomes.Citation69 In both cases, bacteria are degraded in the autophagosomes. Second, some bacteria can escape autophagic degradation, as found for Shigella flexneri and Listeria monocytogenes. Binding of the S. flexneri protein IcsB with IcsA blocks the latter protein from linking with ATG5 (a component of one of the ubiquitin-like conjugation systems), thereby preventing sequestration of the bacteria in autophagosomes,Citation70 whereas L. monocytogenes protein internalin K (InlK) inhibits autophagic recognition by binding to the host major vault protein complex.Citation71 Third, some bacteria can utilize autophagic vesicles as their intracellular niche. Such examples include Staphylococcus aureus and Anaplasma phagocytophilum, both of which can both replicate inside autophagosomes.Citation72,Citation73 Interestingly, all of these outcomes have been reported to occur during H. pylori infection. For example, after induction of autophagy by H. pylori,Citation6,Citation8-Citation11 the bacterium can evade autophagy by downregulating autophagic proteins;Citation11 alternatively the bacterium can exploit autophagosomes as their intracellular niche,Citation6,Citation8,Citation10 or be degraded in autolysosomes.Citation8,Citation10 Each of these scenarios will be discussed in detail below.
Table 1. Association of host cell autophagic processes with different pathogenic bacteria
Interaction Between Helicobacter pylori and Host Autophagic Processes
Recent studies have suggested that H. pylori can induce autophagy in gastric epithelial cellsCitation9-Citation11 and professional phagocytes.Citation6,Citation8 An increase in the level of GFP-LC3 puncta and conversion of LC3-I to LC3-II has been observed in the event of H. pylori infection, indicating that autophagic processes were induced in the presence of the bacterium. However, the precise mechanisms by which H. pylori activate host autophagic processes have not yet been elucidated, although different bacterial and host factors have been implicated (). The use of different host cell lines and bacterial strains has produced inconsistent results, indicating that H. pylori may affect autophagy in a host cell/bacterial strain-dependent manner. In the remainder of this review, we will discuss current findings and their implications, and propose future research directions for deciphering the molecular mechanisms by which H. pylori interacts with host autophagic processes.
Table 2. Host and bacterial factors involved in H. pylori- associated autophagic processes and the intracellular survival of H. pylori
Internalization of H. pylori by gastric epithelial cells and professional phagocytes
The facultative intracellular nature of H. pylori has become increasingly evident in the literature, with many studies reporting that the bacterium can invade, survive and multiply in both epithelial cells and professional phagocytes in vitro and in vivo.Citation77 Multiple lines of evidence for the in vivo invasion of H. pylori into gastric cells of infected patients have been reported.Citation78-Citation81 In vitro, H. pylori enters a number of human epithelial cell lines including the gastric adenocarcinoma cell line, AGS,Citation82,Citation83 the cervix carcinoma cell line, HEp2,Citation84 and the human embryonic kidney cell line, HEK293.Citation85 The entry of H. pylori into host gastric epithelial cells possibly occurs by an endocytic mechanism that is regulated by various host signal transduction events.Citation82 Although neither CagA nor VacA was found to be essential for entry of H. pylori into AGS cells,Citation83 recently it has been proposed that H. pylori invasin NudA influences bacterial entry into these cells.Citation86 The bacterium is capable of surviving in acidic late endosomal vacuoles of AGS cells.Citation82,Citation83 This intracellular niche could explain the recurrence of H. pylori infection after treatment with extracellularly-acting antibiotics, since the bacterium could repopulate the extracellular environment from its refuge inside host gastric epithelial cells after the antibiotics are removed.Citation83
Unlike epithelial cells, which do not naturally internalize H. pylori, professional phagocytes actively participate in the uptake of this bacterium. In mouse macrophages, type I H. pylori strains (VacA+CagA+) are phagocytosed less rapidly than type II strains (VacA-CagA-).Citation5 Moreover, type I but not type II strains remain viable for at least 24 h within large, homotypically fused, bacteria-containing phagosomes called megasomes.Citation5 These observations highlight the potential of H. pylori to resist phagocytic killing. It has also been suggested that the chronic nature of H. pylori infection is in part due to the failure of professional phagocytes to mount an effective immune response against the pathogen. Although the mechanism involved in the intracellular fate of H. pylori is not yet fully understood, recent findings that indicate the ability of H. pylori to modulate host autophagy pathways could partly explain the impaired efficiency of intracellular bacterial clearance.
H. pylori-induced autophagy in gastric epithelial cells
Terebiznik et al.Citation9 first reported that infection of AGS cells with the well-characterized H. pylori 60190 (ATCC 49503) strain triggers the induction of autophagy, evidence of which was sought by ultrastructural observation of the bacteria-containing compartment using TEM (), conversion of cytosolic LC3-I to autophagosome-associated LC3-II using western blots, and visualization of LC3 puncta using immunofluoresence microscopy. The phenomenon appears to be dependent on the host autophagic markers ATG5 and ATG12 as formation of LC3 puncta was visibly increased in H. pylori-infected Atg5+/+ mouse embryonic fibroblast (MEF) cells compared with infected Atg5-knockout (atg5−/−) MEFs. Also, fewer GFP-LC3 puncta and decreased levels of LC3-I to LC3-II conversion were observed in infected ATG12-knockdown AGS cells compared with control cells. Bacterial virulence factors including urease, CagA and the type IV secretion system machinery appear to be dispensable in inducing autophagosome formation during H. pylori infection in vitro. In contrast, VacA is required for H. pylori-induced autophagy, as both an isogenic vacA-deficient mutant and its corresponding bacteria-free culture supernatant fail to induce autophagosome formation. Incubation of AGS cells with purified VacA toxin shows increased abundance of GFP-LC3 puncta and conversion of LC3-I to LC3-II compared with untreated control cells, suggesting that VacA alone is sufficient to induce autophagy. Moreover, the pore-forming activity of VacA is found to be crucial for the induction of autophagy, as decreased formation of GFP-LC3 puncta and LC3-II is observed with the isogenic vacA mutants, P19A and G14A, which are defective in vacuolating and membrane channel-forming activities. Interestingly, the induction of autophagy is suggested to limit the stability of intracellular VacA in AGS cells.Citation9 Degradation of intracellular VacA over a 24 h period requires ATG12,Citation9 indicating that autophagy might play a role in protecting host cells from the toxic effects of VacA. Together, these results possibly represent a complex scenario where VacA induces autophagy, which in turn limits the extent of toxin-mediated cellular damage by the degradation of VacA, thereby conferring protection to host cells against H. pylori infection ().
Table 3. Demonstration for H. pylori-containing autophagosomes in different host cell types
Figure 2.H. pylori-associated autophagy in gastric epithelial cells. Upon induction of autophagy by H. pylori in gastric epithelial cells, internalized bacteria are able to multiply inside the autophagosome and are degraded after fusion of the autophagosome with a lysosome. H-pylori-induced autophagy seems to depend on ATG5 and ATG12. The miRNA MIR30B impairs autophagy via downregulation of ATG12 and BECN1. VacA can induce autophagy after internalization mediated by LRP1/other unknown receptors and is degraded within the autophagosome. Acute exposure to VacA induces formation of autophagosomes that results in the degradation of VacA and might contribute to bacterial clearance, whereas chronic exposure to VacA results in the accumulation of SQSTM1 and formation of defective autolysosomes, which lack CTSD. The latter has been proposed to impair cargo degradation and contribute to intracellular survival of H. pylori.
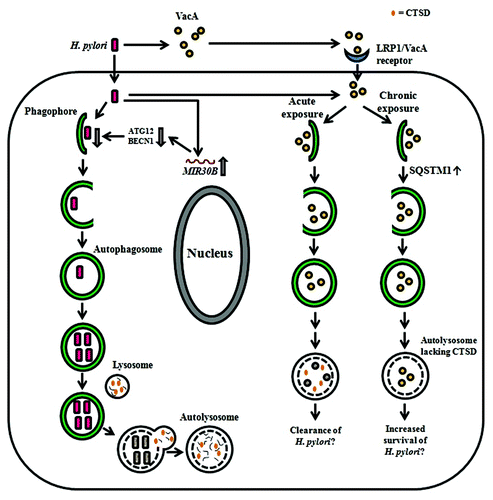
The study of Raju et al.Citation74 added further complexity to the scenario presented above. It showed that prolonged exposure (24 h) of AGS cells to bacterial culture supernatant of VacA+ H. pylori 60190 results in the accumulation of defective autophagosomes with impaired capacity for protein hydrolysis. Based on these observations, it was proposed that the host autophagic machinery reacts differently in response to acute and chronic exposure to the VacA toxin ().Citation74,Citation87 These authors found that AGS cells treated with VacA+ H. pylori culture supernatant contain reduced levels of the key lysosomal protease CTSD/cathepsin D, in autolysosomes compared with untreated cells. The maturation of proCTSD to the proteolytically active CTSD requires the acidic environment in endosomes and lysosomes.Citation88,Citation89 One study thus attributed the reduced amount of mature CTSD in VacA-treated HeLa cells to the action of VacA in partially neutralizing the acidic pH of the endosomal and lysosomal lumens thereby inhibiting the maturation of proCTSD.Citation76 Contrasting results from other studies have, however, indicated that lysosomal acidification is not affected in VacA-treated AGS cellsCitation74 and that VacA-induced bacteria-containing vacuoles are acidic,Citation90 although it remains to be ascertained whether the VacA-induced bacteria-containing vacuoles represent autophagosomes. The current view then is that VacA-induced autophagosomes acquire an acidic pH upon fusion with lysosomes, but are defective in their ability to degrade cargo as they lack CTSD.Citation74,Citation90 Whatever the mechanism of CTSD depletion in VacA-induced autophagosomes, the autophagic degradation of cargo cannot be completed, and these conditions thereby render these compartments permissible to H. pylori intracellular survival and replication. This notion is in complete agreement with the observed effects of VacA on the accumulation of SQSTM1.Citation74 SQSTM1 is a major cargo ubiquitin-binding receptor in mammalian cells that is degraded by autophagy;Citation91,Citation92 deficiencies in the autophagy pathway lead to marked accumulation of SQSTM1.Citation93,Citation94 The formation of SQSTM1 aggregates is significantly enhanced in VacA+ H. pylori culture supernatant-treated AGS cells compared with cells treated with VacA- H. pylori culture supernatant.Citation74 Also, increased SQSTM1 staining is observed in gastric biopsies of patients infected with H. pylori strains expressing the toxigenic s1m1 form of VacA compared with those associated with nontoxigenic strains.Citation74 These findings provide further evidence that chronic exposure to VacA impairs autophagic degradation.Citation87
Building on the proposition that VacA is necessary and sufficient for the induction of autophagy, Yahiro et al.Citation75 have identified a low density lipoprotein receptor-related protein 1 (LRP1) that mediates the binding and internalization of VacA with the subsequent induction of autophagy in AZ-521, a human gastric adenocarcinoma cell line. The LRP1 protein belongs to a family of low density lipoproteins that is highly involved in endocytosis of diverse biological ligands, leading to the signaling of important metabolic pathways downstream.Citation95 Furthermore, in agreement with Terebiznik et al.,Citation9 the channel-forming activity of VacA is required for VacA-induced autophagy in AZ-521 cells.Citation75 Nevertheless, the results do not show a statistically significant increase in LC3-II levels in cells treated with active wild-type VacA compared with heat-inactivated VacA. It is noteworthy that the pore-forming activity of VacA has been reported to alter the membrane permeability of mitochondria, leading to the subsequent release of cytochrome c.Citation39 These effects of VacA could potentially lead to induction of mitophagy, a selective form of autophagy which targets damaged or superfluous mitochondria for degradation, and also to apoptosis.Citation96 It would therefore be of interest to further explore the potential links between VacA-induced apoptosis and VacA-induced mitophagy.
VacA-induced autophagy in AZ-521 cells appears not to follow the canonical pathway of autophagy in which the PtdIns3K complex and BECN1 are involved in the nucleation of autophagosomes, since the process is not inhibited in the presence of 3-methyladenine (which inhibits the class III PtdIns3K), or in BECN1 knockdown cells.Citation75 Additional experiments using ATG5 and ATG12 knockdown cells could be performed to further test this hypothesis.
Apart from VacA-induced autophagy, H. pylori also employs other strategies to evade or subvert the autophagy process in gastric epithelial cells. It has been demonstrated that the expression of microRNA MIR30B increases during infection of various human gastric epithelial cell lines (including AGS) and gastric mucosal tissue with H. pylori. This event appears to be unique to H. pylori infection since infection with other pathogens, e.g., E. coli strains DH5α and O157:57, or treatment with autophagy modulators, e.g., rapamycin and 3-methyladenine do not have any effect on MIR30B expression.Citation11 The use of MIR30B mimics and inhibitors shows that the upregulation of MIR30B expression during H. pylori infection of AGS cells downregulates host cell autophagy as indicated by reduced conversion of LC3-I to LC3-II.Citation11 This effect of MIR30B has been proposed to enhance the intracellular survival and replication of H. pylori in host cells. Using a bioinformatics tool to search for potential MIR30B targets, the mRNAs encoding two important autophagy proteins, ATG12 and BECN1, were identified to be the direct targets of MIR30B ().Citation11 These observations correlate with those made in H. pylori-positive human samples that the expression level of MIR30B is inversely proportional to that of ATG12 or BECN1.
The role of BECN1 in H. pylori-induced autophagy as reported by Tang et al.Citation11 seemingly contradicts the suggestion by Yahiro et al.Citation75 that BECN1 does not play a role in VacA-induced autophagy. The discrepancy of results between these two studies could be due to the fact that Yahiro et al.Citation75 used the VacA toxin purified from the H. pylori strain 60190 whereas Tang et al.Citation11 used viable bacteria of the H. pylori strain 26695 as the “trigger” of autophagy. Moreover these two studies used different cell lines; AZ-521Citation75 and AGS,Citation11 respectively. The emerging notion that H. pylori induction of autophagy may be bacterial-strain dependent undoubtedly requires further investigation. Another interesting finding of the study by Tang et al.Citation11 was that overexpression of MIR30B increases the formation of VacA-dependent vacuoles in H. pylori-infected cells. However, the molecular basis for the link between compromised autophagy and increased vacuolization in H. pylori infection remains unclear. It would also be of great interest to investigate the effects of different H. pylori virulence factors on MIR30B expression in different host cell types.
H. pylori-induced autophagy in professional phagocytes
The evidence for the association of H. pylori infection with autophagy in professional phagocytes is less straightforward compared with that reported for epithelial cells, as the interpretation of the reports is complicated by the diversity of cell lines and H. pylori strains used.
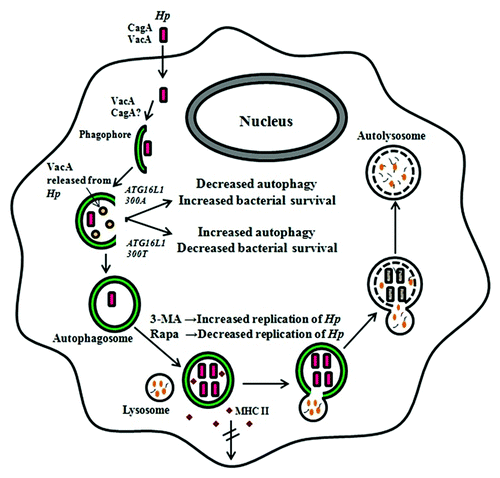
The first report of H. pylori-induced autophagy in professional phagocytes was published by Wang et al.,Citation6 who observed using TEM that H. pylori could reside in double-membrane autophagosomal structures within host cells. Using gentamicin protection assays to investigate the replication of intracellular H. pylori, these authors showed that although several clinical isolates of H. pylori could invade human monocytic THP-1 or U937 cells and murine macrophage RAW264.7 cells, the bacteria multiplied only in the human cells, suggesting that the mouse macrophages were not permissive to bacterial replication. At first sight these results seem logical, as H. pylori is an exclusively human pathogen.Citation97 However, the origin of the host cell was not found to be critical when these authors later showed that the same isolates of H. pylori could multiply in autophagosomes within mouse bone marrow-derived dendritic cells (BMDCs).Citation8 Alternatively, the use of different murine cell types in these studies could explain the variation. The infection by H. pylori of mouse BMDCs, which are antigen-presenting cells, impairs the transport of major histocompatibility complex class II molecules from the cytoplasm to the plasma membrane and the proliferation of antigen-specific T cells in wild-type, but not in TLR2- and TLR4-deficient cells.Citation8
A consistent observation in both phagocytesCitation8 and gastric epithelial cellsCitation10 is that the degradation of H. pylori is found to occur in the resulting autolysosome after 24 h of infection. This finding leaves unanswered the question of whether the autophagic processes induced during H. pylori infection up to this point in time benefit bacterial survival or restrict bacterial growth.
In contrast to the direct role of VacA in autophagy induction in gastric epithelial cells, both CagA and VacA are found to be important in the survival and multiplication of intracellular H. pylori in macrophagesCitation6 and BMDCs.Citation8 Both CagA- and VacA-deficient mutants are more quickly eliminated than wild-type H. pylori (clinical isolate HP238) 12–24 h after invasion of these cells. This purported involvement of CagA in H. pylori-induced autophagy in macrophages contradicts the observations made in gastric epithelial cells where CagA is found to be unnecessary,Citation9 but again this inconsistency might arise from the use of different cell types and bacterial strains. Given that the genomic sequences of many clinical isolates, including HP238, have not been determined and hence their complete genetic makeup remains unknown, it would be worthwhile to use genetically well-characterized standard laboratory strains in parallel studies to validate the results.
In addition to bacterial factors, host autophagic proteins have also been demonstrated to affect H. pylori-induced autophagy in phagocytic cells. A single nucleotide polymorphism in the ATG16L1 gene has been identified as a causal risk factor for Crohn disease,Citation98 an inflammatory bowel disorder which shares an intriguing association with H. pylori infections.Citation99 While functional ATG16L1 is involved in one of the two ubiquitin-like conjugation systems necessary for autophagosome formation (see above), the ATG16L1T300A risk variant has been postulated to hinder bacterial capture and clearance during autophagy, hence increasing the risk of Crohn disease.Citation100 Using peripheral blood monocytes (PBMCs), Raju et al.Citation74 found that the single nucleotide polymorphism corresponding to T300A in ATG16L1 is associated with reduced autophagy in response to VacA toxin during H. pylori infection, as depicted in . When PBMCs from individuals possessing the 300A risk allele are treated with VacA+ H. pylori culture supernatant, the autophagic response is significantly impaired compared with infected PBMCs carrying the 300T allele. In a population study of Caucasian cohorts, subjects who are homozygous for the risk 300A allele, compared with those homozygous for the protective 300T allele, show increased susceptibility to infection by toxigenic H. pylori strains that express the s1m1 form of VacA.Citation74 Collectively, these findings suggest that individuals harboring the ATG16L1300A allele have a compromised autophagic response and subsequently increased susceptibility to H. pylori infection.Citation87 This suggestion implicates the ATG16L1300A allele as a novel host genetic risk factor for H. pylori infection.
Figure 3.H. pylori-associated autophagy in professional phagocytes. In professional phagocytes such as monocytes, macrophages and BMDCs, internalized bacteria are able to multiply inside the autophagosome. VacA and probably CagA are involved in this process. Bacteria are eventually degraded in the autophagic vesicles. Bacteria inhibit the transport of major histocompatibility complex class II molecules to the cell membrane in wild-type, but not in TLR2- and TLR4-deficient BMDCs. In PMBCs, the ATG16L1300A risk variant for Crohn disease is associated with decreased autophagy, leading to increased bacterial intracellular survival and the individual’s increased susceptibility to H. pylori infections. The events depicted however do not discount the possibility that H. pylori induces LC3-associated phagocytosis (LAP) rather than canonical autophagic pathways. 3-MA, 3-methyladenine; Hp, H, pylori; MHC, major histocompability complex; Rapa, rapamycin; the double-crossed arrow indicates blocking.
NOD2, a eukaryotic intracellular pattern recognition receptor that recognizes bacterial muramyl dipeptide, has also been suggested as contributing to bacterial autophagy.Citation101 It interacts with ATG16L1 at the point of Shigella flexneri host cell entry, where it presumably acts as a sensor and nucleating factor to recruit the autophagy machinery for sequestration of intracellular bacteria within autophagosomes.Citation101 This process is impaired in bone marrow-derived macrophages isolated from mice homozygous for the Crohn disease-associated NOD2fs allele,Citation101 indicating that polymorphism in NOD2 influences bacterial autophagy. In the context of H. pylori infection, NOD2 mediates H. pylori-stimulated NFKB activation in infected host cells, whereas the Crohn disease-associated NOD2 variant R702W fails to mediate such a response.Citation102 The R702W mutation in NOD2 is also strongly associated with gastric MALT lymphoma, a disorder that is frequently associated with H. pylori infection.Citation102 Whether NOD2 is involved in recruiting ATG16L1 to the site of H. pylori host cell entry and hence triggers autophagy of internalized H. pylori is an intriguing proposition that remains to be investigated. TLR4, another pattern recognition receptor, stimulates autophagy in macrophages activated by bacterial lipopolysaccharide (LPS) via a mechanism that involves ubiquitination of BECN1 by TNF receptor-associated factor 6, E3 ubiquitin protein ligase (TRAF6)Citation103 and dissociation of BECN1 from BCL2.Citation104 H. pylori LPS activates TLR4 in gastric pit cells,Citation105 and polymorphism in TLR4 is associated with increased risk of gastric cancer.Citation106 It would thus be of interest to examine whether activation of TLR4 by H. pylori LPS might trigger autophagy via BECN1-dependent pathways and whether such pathways play a role in gastric carcinogenesis. Apart from NOD2 and TLR4, polymorphism in yet another autophagy-associated gene, IRGM (immunity-related GTPase family, M) has also been reported to influence susceptibility to gastric cancer.Citation107 While IRGM is known to be involved in the activation of autophagy in Crohn disease and mycobacterial infection,Citation108 future studies are required to ascertain whether polymorphism in IRGM correlates with the extent of autophagy induction in H. pylori infection and H. pylori-induced gastric diseases. These various findings indicate that further examination of the contribution of polymorphisms in other autophagy-associated genes to H. pylori pathogenesis is warranted.
H. pylori-induced autophagic pathways: canonical or noncanonical?
As discussed earlier, it is crucial to determine whether intracellular H. pylori is subject to canonical autophagy, or other unconventional autophagy-associated processes. Although not mentioned in the literature, some findings point toward the possibility that H. pylori might also induce LAP. In most studies, the induction of autophagy was determined only by the increased level of GFP-LC3 puncta and/or increased conversion of LC3-II from LC3-I, and no ultrastructural evidence of autophagosomes as the bacteria-containing compartment was presented. Moreover, the electron micrographs provided by some studies did not show very clear evidence of double-membrane autophagosomes (). Therefore, in future studies a more definitive distinction between H. pylori-induced canonical autophagy vs. H. pylori-induced LAP should be a priority for investigators.
Concluding Remarks and Future Directions
Research related to H. pylori-induced autophagic processes is still in its infancy and many questions remain to be answered (). Although multiple host and bacterial factors have been found to be associated with this process, detailed understanding of the molecular mechanisms of autophagy induction by H. pylori is lacking. Given the multifaceted pathogenesis of the bacterium and the pleiotropic actions of its virulence factors (e.g., CagA and VacA), it is highly likely that the interplay between H. pylori and the host autophagic machinery involves complex mechanisms. The design of definitive experiments that could dissect the specific contribution of different host factors and bacterial virulence factors to the presumably dynamic process of H. pylori-induced autophagy will be a key challenge in the field. Furthermore, as there is evidence that H. pylori-associated autophagy is dependent on host cell types and bacterial strains, caution should be taken not to generalize the ability of H. pylori to elicit autophagic responses. In order to better understand the causal relationship between H. pylori-induced LC3 puncta formation and the potential autophagic processing of H. pylori, the physical presence of H. pylori within LC3 II-containing autophagosomes could be investigated in greater detail with the use of correlative light and electron microscopy.Citation109 Last, given the critical distinction between autophagy and LAP, future research efforts should be directed toward better defining these two phenomena in the context of H. pylori infection.
Table 4. Outstanding questions
The mounting resistance of H. pylori to antibiotics is anticipated to pose a significant challenge for eradicating H. pylori infections in the coming decades. It is increasingly believed that H. pylori-induced autophagy provides the bacterium a protective mechanism whereby autophagic vesicles serve as ecological niches for H. pylori to replicate inside the host cell. Thus, the use of such an intracellular refuge for H. pylori, albeit short-lived, may represent an important strategy for H. pylori to avoid killing by extracellularly acting antibiotics. A better understanding of the molecular mechanisms by which H. pylori modulates autophagy to its advantage could open new avenues for the development of more effective therapeutics against H. pylori infection. Furthermore, it is as yet uncertain whether autophagy serves as an effective front line of host defense against intracellular H. pylori. If indeed the autophagy mechanism can lead to effective clearance of internalized bacteria, it is tempting to speculate that a combinatorial therapy of intracellularly-acting antibiotics and autophagy inducers might be able to boost the innate immunity of host cells to fight against H. pylori infection and minimize the onset of H. pylori-induced diseases.
| Abbreviations: | ||
| ATG | = | autophagy-related |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BECN1 | = | Beclin 1, autophagy-related |
| BMDCs | = | bone marrow-derived dendritic cells |
| cag PAI | = | cag pathogenicity island |
| CagA | = | cytotoxin-associated-gene A |
| CTSD | = | cathepsin D |
| ER | = | endoplasmic reticulum |
| GFP | = | green fluorescent protein |
| LAP | = | LC3-associated phagocytosis |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| LRP1 | = | low density lipoprotein receptor-related protein 1 |
| LPS | = | lipopolysaccharide |
| MALT | = | mucosal-associated lymphoid tissue |
| MEFs | = | mouse embryonic fibroblasts |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NOD2 | = | nucleotide-binding oligomerization domain containing 2 |
| PBMCs | = | peripheral blood monocytes |
| PE | = | phosphatidylethanolamine |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| siRNA | = | small interfering RNA |
| SQSTM1 | = | sequestosome 1 |
| TEM | = | transmission electron microscopy |
| TLR | = | toll-like receptor |
| ULK | = | unc-51-like kinase |
| VacA | = | vacuolating toxin |
| WIPI | = | WD repeat domain, phosphoinositide-interacting |
| ZFYVE1 | = | zinc finger FYVE domain-containing protein 1 |
Acknowledgments
This work is supported by the National Health and Medical Research Council. NSD is the recipient of an Endeavor Postgraduate Award (PhD) from the Department of Education, Employment and Workplace Relations, Australian Government. We apologize to authors whose work has not been cited due to space limitations.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev 2006; 19:449 - 90; http://dx.doi.org/10.1128/CMR.00054-05; PMID: 16847081
- Vogiatzi P, Cassone M, Luzzi I, Lucchetti C, Otvos L Jr., Giordano A. Helicobacter pylori as a class I carcinogen: physiopathology and management strategies. J Cell Biochem 2007; 102:264 - 73; http://dx.doi.org/10.1002/jcb.21375; PMID: 17486575
- Allen LA. Modulating phagocyte activation: the pros and cons of Helicobacter pylori virulence factors. J Exp Med 2000; 191:1451 - 4; http://dx.doi.org/10.1084/jem.191.9.1451; PMID: 10790419
- Bergman MP, D’Elios MM. Cytotoxic T cells in H. pylori-related gastric autoimmunity and gastric lymphoma. J Biomed Biotechnol 2010; 2010:104918; http://dx.doi.org/10.1155/2010/104918; PMID: 20617132
- Allen LA, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med 2000; 191:115 - 28; http://dx.doi.org/10.1084/jem.191.1.115; PMID: 10620610
- Wang YH, Wu JJ, Lei HY. The autophagic induction in Helicobacter pylori-infected macrophage. Exp Biol Med (Maywood) 2009; 234:171 - 80; http://dx.doi.org/10.3181/0808-RM-252; PMID: 19064937
- Allen LA. Phagocytosis and persistence of Helicobacter pylori.. Cell Microbiol 2007; 9:817 - 28; http://dx.doi.org/10.1111/j.1462-5822.2007.00906.x; PMID: 17346311
- Wang YH, Gorvel JP, Chu YT, Wu JJ, Lei HY. Helicobacter pylori impairs murine dendritic cell responses to infection. PLoS One 2010; 5:e10844; http://dx.doi.org/10.1371/journal.pone.0010844; PMID: 20523725
- Terebiznik MR, Raju D, Vázquez CL, Torbricki K, Kulkarni R, Blanke SR, et al. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 2009; 5:370 - 9; http://dx.doi.org/10.4161/auto.5.3.7663; PMID: 19164948
- Chu YT, Wang YH, Wu JJ, Lei HY. Invasion and multiplication of Helicobacter pylori in gastric epithelial cells and implications for antibiotic resistance. Infect Immun 2010; 78:4157 - 65; http://dx.doi.org/10.1128/IAI.00524-10; PMID: 20696835
- Tang B, Li N, Gu J, Zhuang Y, Li Q, Wang HG, et al. Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori.. Autophagy 2012; 8:1045 - 57; http://dx.doi.org/10.4161/auto.20159; PMID: 22082964
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67 - 93; http://dx.doi.org/10.1146/annurev-genet-102808-114910; PMID: 19653858
- Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol 2012; 22:R540 - 5; http://dx.doi.org/10.1016/j.cub.2012.06.001; PMID: 22790007
- Bruce MG, Maaroos HI. Epidemiology of Helicobacter pylori infection. Helicobacter 2008; 13:Suppl 1:1 - 6; http://dx.doi.org/10.1111/j.1523-5378.2008.00631.x; PMID: 18783514
- Goh KL, Chan WK, Shiota S, Yamaoka Y. Epidemiology of Helicobacter pylori infection and public health implications. Helicobacter 2011; 16:Suppl 1:1 - 9; http://dx.doi.org/10.1111/j.1523-5378.2011.00874.x; PMID: 21896079
- Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med 2002; 347:1175 - 86; http://dx.doi.org/10.1056/NEJMra020542; PMID: 12374879
- Wroblewski LE, Peek RM Jr., Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 2010; 23:713 - 39; http://dx.doi.org/10.1128/CMR.00011-10; PMID: 20930071
- Xiang Z, Censini S, Bayeli PF, Telford JL, Figura N, Rappuoli R, et al. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect Immun 1995; 63:94 - 8; PMID: 7806390
- Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, et al. Analyses of the cag pathogenicity island of Helicobacter pylori.. Mol Microbiol 1998; 28:37 - 53; http://dx.doi.org/10.1046/j.1365-2958.1998.00770.x; PMID: 9593295
- Terradot L, Waksman G. Architecture of the Helicobacter pylori Cag-type IV secretion system. FEBS J 2011; 278:1213 - 22; http://dx.doi.org/10.1111/j.1742-4658.2011.08037.x; PMID: 21352491
- Backert S, Ziska E, Brinkmann V, Zimny-Arndt U, Fauconnier A, Jungblut PR, et al. Translocation of the Helicobacter pylori CagA protein in gastric epithelial cells by a type IV secretion apparatus. Cell Microbiol 2000; 2:155 - 64; http://dx.doi.org/10.1046/j.1462-5822.2000.00043.x; PMID: 11207572
- Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000; 287:1497 - 500; http://dx.doi.org/10.1126/science.287.5457.1497; PMID: 10688800
- Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ, Covacci A. c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol 2002; 43:971 - 80; http://dx.doi.org/10.1046/j.1365-2958.2002.02781.x; PMID: 11929545
- Selbach M, Moese S, Hauck CR, Meyer TF, Backert S. Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem 2002; 277:6775 - 8; http://dx.doi.org/10.1074/jbc.C100754200; PMID: 11788577
- Poppe M, Feller SM, Römer G, Wessler S. Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 2007; 26:3462 - 72; http://dx.doi.org/10.1038/sj.onc.1210139; PMID: 17160020
- Tammer I, Brandt S, Hartig R, König W, Backert S. Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 2007; 132:1309 - 19; http://dx.doi.org/10.1053/j.gastro.2007.01.050; PMID: 17408661
- Backert S, Tegtmeyer N, Selbach M. The versatility of Helicobacter pylori CagA effector protein functions: The master key hypothesis. Helicobacter 2010; 15:163 - 76; http://dx.doi.org/10.1111/j.1523-5378.2010.00759.x; PMID: 20557357
- Atherton JC, Cao P, Peek RM Jr., Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem 1995; 270:17771 - 7; http://dx.doi.org/10.1074/jbc.270.30.17771; PMID: 7629077
- Wada A, Yamasaki E, Hirayama T. Helicobacter pylori vacuolating cytotoxin, VacA, is responsible for gastric ulceration. J Biochem 2004; 136:741 - 6; http://dx.doi.org/10.1093/jb/mvh181; PMID: 15671482
- Cover TL, Blaser MJ. Purification and characterization of the vacuolating toxin from Helicobacter pylori.. J Biol Chem 1992; 267:10570 - 5; PMID: 1587837
- Telford JL, Ghiara P, Dell’Orco M, Comanducci M, Burroni D, Bugnoli M, et al. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J Exp Med 1994; 179:1653 - 8; http://dx.doi.org/10.1084/jem.179.5.1653; PMID: 8163943
- Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat Rev Microbiol 2005; 3:320 - 32; http://dx.doi.org/10.1038/nrmicro1095; PMID: 15759043
- Isomoto H, Moss J, Hirayama T. Pleiotropic actions of Helicobacter pylori vacuolating cytotoxin, VacA. Tohoku J Exp Med 2010; 220:3 - 14; http://dx.doi.org/10.1620/tjem.220.3; PMID: 20046046
- McClain MS, Iwamoto H, Cao P, Vinion-Dubiel AD, Li Y, Szabo G, et al. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J Biol Chem 2003; 278:12101 - 8; http://dx.doi.org/10.1074/jbc.M212595200; PMID: 12562777
- Reyrat JM, Lanzavecchia S, Lupetti P, de Bernard M, Pagliaccia C, Pelicic V, et al. 3D imaging of the 58 kDa cell binding subunit of the Helicobacter pylori cytotoxin. J Mol Biol 1999; 290:459 - 70; http://dx.doi.org/10.1006/jmbi.1999.2877; PMID: 10390344
- Torres VJ, Ivie SE, McClain MS, Cover TL. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J Biol Chem 2005; 280:21107 - 14; http://dx.doi.org/10.1074/jbc.M501042200; PMID: 15817461
- Ivie SE, McClain MS, Torres VJ, Algood HM, Lacy DB, Yang R, et al. Helicobacter pylori VacA subdomain required for intracellular toxin activity and assembly of functional oligomeric complexes. Infect Immun 2008; 76:2843 - 51; http://dx.doi.org/10.1128/IAI.01664-07; PMID: 18443094
- González-Rivera C, Gangwer KA, McClain MS, Eli IM, Chambers MG, Ohi MD, et al. Reconstitution of Helicobacter pylori VacA toxin from purified components. Biochemistry 2010; 49:5743 - 52; http://dx.doi.org/10.1021/bi100618g; PMID: 20527875
- Palframan SL, Kwok T, Gabriel K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front Cell Infect Microbiol 2012; 2:92; http://dx.doi.org/10.3389/fcimb.2012.00092; PMID: 22919683
- Van Doorn LJ, Figueiredo C, Mégraud F, Pena S, Midolo P, Queiroz DM, et al. Geographic distribution of vacA allelic types of Helicobacter pylori.. Gastroenterology 1999; 116:823 - 30; http://dx.doi.org/10.1016/S0016-5085(99)70065-X; PMID: 10092304
- Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh Hosseini M, et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007; 133:926 - 36; http://dx.doi.org/10.1053/j.gastro.2007.06.056; PMID: 17854597
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42; http://dx.doi.org/10.1016/j.cell.2007.12.018; PMID: 18191218
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323 - 35; http://dx.doi.org/10.1038/nature09782; PMID: 21248839
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107 - 32; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005; PMID: 21801009
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; http://dx.doi.org/10.1083/jcb.200803137; PMID: 18725538
- Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 2004; 23:9314 - 25; http://dx.doi.org/10.1038/sj.onc.1208331; PMID: 15602573
- Ylä-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009; 5:1180 - 5; http://dx.doi.org/10.4161/auto.5.8.10274; PMID: 19855179
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 2009; 11:1433 - 7; http://dx.doi.org/10.1038/ncb1991; PMID: 19898463
- Yen W-L, Shintani T, Nair U, Cao Y, Richardson BC, Li Z, et al. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J Cell Biol 2010; 188:101 - 14; http://dx.doi.org/10.1083/jcb.200904075; PMID: 20065092
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656 - 67; http://dx.doi.org/10.1016/j.cell.2010.04.009; PMID: 20478256
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010; 12:747 - 57; http://dx.doi.org/10.1038/ncb2078; PMID: 20639872
- Mijaljica D, Nazarko TY, Brumell JH, Huang WP, Komatsu M, Prescott M, et al. Receptor protein complexes are in control of autophagy. Autophagy 2012; 8:1701 - 5; http://dx.doi.org/10.4161/auto.21332; PMID: 22082964
- Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 1999; 18:3888 - 96; http://dx.doi.org/10.1093/emboj/18.14.3888; PMID: 10406794
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol 1999; 147:435 - 46; http://dx.doi.org/10.1083/jcb.147.2.435; PMID: 10525546
- Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, et al. A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488 - 92; http://dx.doi.org/10.1038/35044114; PMID: 14076752
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151 - 75; PMID: 18188003
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323 - 8; PMID: 17387262
- Al-Younes HM, Al-Zeer MA, Khalil H, Gussmann J, Karlas A, Machuy N, et al. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis.. Autophagy 2011; 7:814 - 28; http://dx.doi.org/10.4161/auto.7.8.15597; PMID: 21464618
- DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, et al. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 2011; 21:966 - 74; http://dx.doi.org/10.1016/j.devcel.2011.08.016; PMID: 22055344
- Sanjuan MA, Milasta S, Green DR. Toll-like receptor signaling in the lysosomal pathways. Immunol Rev 2009; 227:203 - 20; http://dx.doi.org/10.1111/j.1600-065X.2008.00732.x; PMID: 19120486
- Lai S, Devenish RJ. LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy. Cells 2012; 1:396 - 408; http://dx.doi.org/10.3390/cells1030396
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253 - 7; http://dx.doi.org/10.1038/nature06421; PMID: 18097414
- Lerena MC, Colombo MI. Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell Microbiol 2011; 13:814 - 35; http://dx.doi.org/10.1111/j.1462-5822.2011.01581.x; PMID: 21447143
- Gong L, Cullinane M, Treerat P, Ramm G, Prescott M, Adler B, et al. The Burkholderia pseudomallei type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS One 2011; 6:e17852; http://dx.doi.org/10.1371/journal.pone.0017852; PMID: 21412437
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 2011; 108:17396 - 401; http://dx.doi.org/10.1073/pnas.1113421108; PMID: 21969579
- Gong L, Devenish RJ, Prescott M. Autophagy as a macrophage response to bacterial infection. IUBMB Life 2012; 64:740 - 7; http://dx.doi.org/10.1002/iub.1070; PMID: 22815102
- Shin DM, Yuk JM, Lee HM, Lee SH, Son JW, Harding CV, et al. Mycobacterial lipoprotein activates autophagy via TLR2/1/CD14 and a functional vitamin D receptor signalling. Cell Microbiol 2010; 12:1648 - 65; http://dx.doi.org/10.1111/j.1462-5822.2010.01497.x; PMID: 20560977
- Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem 2006; 281:11374 - 83; http://dx.doi.org/10.1074/jbc.M509157200; PMID: 16495224
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science 2005; 307:727 - 31; http://dx.doi.org/10.1126/science.1106036; PMID: 15576571
- Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, et al. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog 2011; 7:e1002168; http://dx.doi.org/10.1371/journal.ppat.1002168; PMID: 21829365
- Schnaith A, Kashkar H, Leggio SA, Addicks K, Krönke M, Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem 2007; 282:2695 - 706; http://dx.doi.org/10.1074/jbc.M609784200; PMID: 17135247
- Niu H, Yamaguchi M, Rikihisa Y. Subversion of cellular autophagy by Anaplasma phagocytophilum.. Cell Microbiol 2008; 10:593 - 605; http://dx.doi.org/10.1111/j.1462-5822.2007.01068.x; PMID: 17979984
- Raju D, Hussey S, Ang M, Terebiznik MR, Sibony M, Galindo-Mata E, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 2012; 142:1160 - 71; http://dx.doi.org/10.1053/j.gastro.2012.01.043; PMID: 22333951
- Yahiro K, Satoh M, Nakano M, Hisatsune J, Isomoto H, Sap J, et al. Low-density lipoprotein receptor-related protein-1 (LRP1) mediates autophagy and apoptosis caused by Helicobacter pylori VacA. J Biol Chem 2012; 287:31104 - 15; http://dx.doi.org/10.1074/jbc.M112.387498; PMID: 22822085
- Satin B, Norais N, Telford J, Rappuoli R, Murgia M, Montecucco C, et al. Effect of helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J Biol Chem 1997; 272:25022 - 8; http://dx.doi.org/10.1074/jbc.272.40.25022; PMID: 9312109
- Dubois A, Borén T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell Microbiol 2007; 9:1108 - 16; http://dx.doi.org/10.1111/j.1462-5822.2007.00921.x; PMID: 17388791
- Ozbek A, Ozbek E, Dursun H, Kalkan Y, Demirci T. Can Helicobacter pylori invade human gastric mucosa?: an in vivo study using electron microscopy, immunohistochemical methods, and real-time polymerase chain reaction. J Clin Gastroenterol 2010; 44:416 - 22; PMID: 19904218
- Semino-Mora C, Doi SQ, Marty A, Simko V, Carlstedt I, Dubois A. Intracellular and interstitial expression of Helicobacter pylori virulence genes in gastric precancerous intestinal metaplasia and adenocarcinoma. J Infect Dis 2003; 187:1165 - 77; http://dx.doi.org/10.1086/368133; PMID: 12695995
- Oh JD, Karam SM, Gordon JI. Intracellular Helicobacter pylori in gastric epithelial progenitors. Proc Natl Acad Sci U S A 2005; 102:5186 - 91; http://dx.doi.org/10.1073/pnas.0407657102; PMID: 15795379
- Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori.. Gastroenterology 2007; 132:1009 - 23; http://dx.doi.org/10.1053/j.gastro.2007.01.049; PMID: 17383424
- Kwok T, Backert S, Schwarz H, Berger J, Meyer TF. Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect Immun 2002; 70:2108 - 20; http://dx.doi.org/10.1128/IAI.70.4.2108-2120.2002; PMID: 11895977
- Amieva MR, Salama NR, Tompkins LS, Falkow S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol 2002; 4:677 - 90; http://dx.doi.org/10.1046/j.1462-5822.2002.00222.x; PMID: 12366404
- Evans DG, Evans DJ Jr., Graham DY. Adherence and internalization of Helicobacter pylori by HEp-2 cells. Gastroenterology 1992; 102:1557 - 67; PMID: 1568565
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5:1166 - 74; http://dx.doi.org/10.1038/ni1131; PMID: 15489856
- Liu H, Semino-Mora C, Dubois A. Mechanism of H. pylori Intracellular Entry: An in vitro Study. Front Cell Infect Microbiol 2012; 2:13; PMID: 22919593
- Raju D, Hussey S, Jones NL. Crohn disease ATG16L1 polymorphism increases susceptibility to infection with Helicobacter pylori in humans. Autophagy 2012; 8:1387 - 8; http://dx.doi.org/10.4161/auto.21007; PMID: 22885761
- Rijnboutt S, Stoorvogel W, Geuze HJ, Strous GJ. Identification of subcellular compartments involved in biosynthetic processing of cathepsin D. J Biol Chem 1992; 267:15665 - 72; PMID: 1322403
- Delbrück R, Desel C, von Figura K, Hille-Rehfeld A. Proteolytic processing of cathepsin D in prelysosomal organelles. Eur J Cell Biol 1994; 64:7 - 14; PMID: 7957314
- Terebiznik MR, Vazquez CL, Torbicki K, Banks D, Wang T, Hong W, et al. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect Immun 2006; 74:6599 - 614; http://dx.doi.org/10.1128/IAI.01085-06; PMID: 17000720
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259 - 69; http://dx.doi.org/10.1016/j.molcel.2009.04.026; PMID: 19450525
- Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181 - 97; http://dx.doi.org/10.1016/S0076-6879(08)03612-4; PMID: 19200883
- Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, et al. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci 2006; 26:8057 - 68; http://dx.doi.org/10.1523/JNEUROSCI.2261-06.2006; PMID: 16885219
- Mizushima N, Hara T. Intracellular quality control by autophagy: how does autophagy prevent neurodegeneration?. Autophagy 2006; 2:302 - 4; PMID: 16874082
- Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest 2001; 108:779 - 84; PMID: 11560943
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 2007; 462:245 - 53; http://dx.doi.org/10.1016/j.abb.2007.03.034; PMID: 17475204
- Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113:321 - 33; PMID: 14755326
- Prescott NJ, Fisher SA, Franke A, Hampe J, Onnie CM, Soars D, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007; 132:1665 - 71; http://dx.doi.org/10.1053/j.gastro.2007.03.034; PMID: 17484864
- Luther J, Dave M, Higgins PD, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis 2010; 16:1077 - 84; http://dx.doi.org/10.1002/ibd.21116; PMID: 19760778
- Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One 2008; 3:e3391; http://dx.doi.org/10.1371/journal.pone.0003391; PMID: 18852889
- Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11:55 - 62; http://dx.doi.org/10.1038/ni.1823; PMID: 19898471
- Rosenstiel P, Hellmig S, Hampe J, Ott S, Till A, Fischbach W, et al. Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol 2006; 8:1188 - 98; http://dx.doi.org/10.1111/j.1462-5822.2006.00701.x; PMID: 16819970
- Shi CS, Kehrl JH. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 2010; 3:ra42; http://dx.doi.org/10.1126/scisignal.2000751; PMID: 20501938
- Shi CS, Kehrl JH. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J Biol Chem 2008; 283:33175 - 82; http://dx.doi.org/10.1074/jbc.M804478200; PMID: 18772134
- Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, Rokutan K. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun 2001; 69:4382 - 9; http://dx.doi.org/10.1128/IAI.69.7.4382-4389.2001; PMID: 11401977
- Fukata M, Abreu MT. Role of Toll-like receptors in gastrointestinal malignancies. Oncogene 2008; 27:234 - 43; http://dx.doi.org/10.1038/sj.onc.1210908; PMID: 18176605
- Burada F, Plantinga TS, Ioana M, Rosentul D, Angelescu C, Joosten LA, et al. IRGM gene polymorphisms and risk of gastric cancer. J Dig Dis 2012; 13:360 - 5; http://dx.doi.org/10.1111/j.1751-2980.2012.00602.x; PMID: 22713085
- Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006; 313:1438 - 41; http://dx.doi.org/10.1126/science.1129577; PMID: 16888103
- Razi M, Tooze SA. Correlative light and electron microscopy. Methods Enzymol 2009; 452:261 - 75; http://dx.doi.org/10.1016/S0076-6879(08)03617-3; PMID: 19200888
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119:753 - 66; http://dx.doi.org/10.1016/j.cell.2004.11.038; PMID: 15607973
- Collins CA, De Mazière A, van Dijk S, Carlsson F, Klumperman J, Brown EJ. Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog 2009; 5:e1000430; http://dx.doi.org/10.1371/journal.ppat.1000430; PMID: 19436699
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus.. Science 2004; 306:1037 - 40; http://dx.doi.org/10.1126/science.1103966; PMID: 15528445
- Starr T, Child R, Wehrly TD, Hansen B, Hwang S, López-Otin C, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012; 11:33 - 45; http://dx.doi.org/10.1016/j.chom.2011.12.002; PMID: 22264511
- Gutierrez MG, Vázquez CL, Munafó DB, Zoppino FC, Berón W, Rabinovitch M, et al. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol 2005; 7:981 - 93; http://dx.doi.org/10.1111/j.1462-5822.2005.00527.x; PMID: 15953030
- Sturgill-Koszycki S, Swanson MS. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J Exp Med 2000; 192:1261 - 72; http://dx.doi.org/10.1084/jem.192.9.1261; PMID: 11067875
- Moreau K, Lacas-Gervais S, Fujita N, Sebbane F, Yoshimori T, Simonet M, et al. Autophagosomes can support Yersinia pseudotuberculosis replication in macrophages. Cell Microbiol 2010; 12:1108 - 23; http://dx.doi.org/10.1111/j.1462-5822.2010.01456.x; PMID: 20180800