Abstract
LC3 is a marker protein that is involved in the formation of autophagosomes and autolysosomes, which are usually characterized and monitored by fluorescence microscopy using fluorescent protein-tagged LC3 probes (FP-LC3). FP-LC3 and even endogenous LC3 can also be incorporated into intracellular protein aggregates in an autophagy-independent manner. However, the dynamic process of LC3 associated with autophagosomes and autolysosomes or protein aggregates in living cells remains unclear. Here, we explored the dynamic properties of the two types of FP-LC3-containing puncta using fluorescence microscopy techniques, including fluorescence recovery after photobleaching (FRAP) and fluorescence resonance energy transfer (FRET). The FRAP data revealed that the fluorescent signals of FP-LC3 attached to phagophores or in mature autolysosomes showed either minimal or no recovery after photobleaching, indicating that the dissociation of LC3 from the autophagosome membranes may be very slow. In contrast, FP-LC3 in the protein aggregates exhibited nearly complete recovery (more than 80%) and rapid kinetics of association and dissociation (half-time < 1 sec), indicating a rapid exchange occurs between the aggregates and cytoplasmic pool, which is mainly due to the transient interaction of LC3 and SQSTM1/p62. Based on the distinct dynamic properties of FP-LC3 in the two types of punctate structures, we provide a convenient and useful FRAP approach to distinguish autophagosomes from LC3-involved protein aggregates in living cells. Using this approach, we find the FP-LC3 puncta that adjacently localized to the phagophore marker ATG16L1 were protein aggregate-associated LC3 puncta, which exhibited different kinetics compared with that of autophagic structures.
Introduction
Macroautophagy, hereafter referred to as autophagy, is an intracellar catabolic process in which cytoplasmic molecules and organelles are engulfed by double-membraned autophagosomes and transported to the lysosomes for degradation.Citation1 In mammalian cells, autophagosomes are thought to derive from precursor membranes called phagophores through membrane expansion. Approximately 20 autophagy-related (ATG) proteins are implicated in autophagy regulation.Citation2 The majority of the ATG proteins reside on the phagophores (e.g., ATG14, WIPI1, ATG16L1, ATG5), not on the enclosed autophagosomes.Citation3,Citation4 Until now, only the microtubule-associated protein light chain 3 (LC3) has been observed on all autophagic structures (i.e., phagophores, autophagosomes, and autolysosomes).Citation5 Immediately after synthesis, LC3 is cleaved by ATG4 (a cysteine protease) to produce LC3-I.Citation6 Next, ATG7 and ATG3 catalyze the conjugation of LC3-I and phosphatidylethanolamine (PE) to generate LC3-II.Citation5 ATG16L1 recruits the ATG12–ATG5 conjugate to form the multimeric ATG12–ATG5-ATG16L1 complex, which specifies the site and facilitates the process of LC3 lipidation.Citation7 The ATG16L1 complex associates with the phagophore and the outer surface of the nascent autophagosome and then leaves immediately before or following membrane closure.Citation8 In contrast to the ATG16L1 complex, LC3 attaches to both the inner and outer sides of the nascent autophagosomes.Citation5 The LC3 bound to the outer side of the autophagosome may be cleaved by ATG4 and may detach from the autophagosomes before or after fusion with the lysosomes,Citation9 which denoted as autolysosomes (, left panel). Thus, LC3 appears to be a bona fide autophagy marker, and fluorescent protein-tagged LC3 (FP-LC3), such as GFP-LC3 or mRFP-GFP-LC3, is widely used to label autophagosomal structures.Citation9,Citation10 Although the diffusional properties of nuclear/cytoplasmic FP-LC3 pool in living cells has been explored in some detail,Citation11,Citation12 the dynamic properties of LC3 associated with the autophagic structures remain largely unknown.Citation13
Figure 1. The two types of mRFP-GFP-LC3 puncta have different dynamics in living cells. (A) Schematic diagrams of LC3 associated with the autophagosomal structures and protein aggregates. Left panel: LC3 is bound to both sides of the autophagosomes. Immediately before and after fusion with the lysosomes, the outer membrane-bound LC3 is released from the autophagosomes, and the inner membrane-bound LC3 is degraded in the autolysosomes. Right panel: During the formation of LC3-involved aggregate, the ubiquitinated proteins first interact with SQSTM1, then become protein aggregates through the oligomerization of SQSTM1. LC3/LC3G120A are recruited to the aggregates through their interaction with the LC3 interacting region (LIR) of SQSTM1. (B) FRAP analysis of the mRFP-GFP-LC3 puncta (arrows) in HBSS-starved HeLa cells. Scale bar: 5 μm.

Recently, it has been reported that LC3 can be incorporated into protein aggregates without being conjugated to phosphatidylethanolamine (PE).Citation14-Citation16 Protein aggregates are poorly soluble oligomers of non-native polypeptide chains that tend to accumulate in inclusion bodies (IBs), which can be visible by microscopy.Citation17,Citation18 The intracellular and/or extracellular deposition of abnormal proteins is observed under stress conditions and is also a common feature of many neurodegenerative disorders, such as Alzheimer, Parkinson, and Huntington diseases.Citation19 It has been reported that IBs frequently contain polyubiquitin and the polyubiquitin-binding protein SQSTM1/p62.Citation16,Citation20 LC3 is also a ubiquitin-like protein. The autophagy-independent association of LC3 with the protein aggregates is dependent on its interaction with SQSTM1 (, right panel).Citation21 These findings highlight the diverse localization and function of LC3. However, caution should be exercised when interpreting the results of GFP-LC3 (or endogenous LC3) puncta formation assay because the LC3-involved protein aggregates may not be easily distinguished from the LC3-containing autophagosomes/autolysosomes in living cells using fluorescence microscopy.Citation8,Citation14,Citation22 Thus, it is necessary to develop an effective method to direct identify whether a FP-LC3 punctum in living cells is a true autophagosomal structure or not.
In this study, we generated FP-tagged LC3, the lipidation-defective mutant LC3G120A, and additional marker proteins. The dynamic properties of LC3 localized to the two types of punctate structures and the biophysical properties of LC3-involved aggregates were imaged and analyzed using dynamic fluorescence microscopy techniques, including fluorescence recovery after photobleaching (FRAP),Citation11,Citation23-Citation25 fluorescence redistribution after photoactivation (FRAPa),Citation26,Citation27 and fluorescence resonance energy transfer (FRET).
Results
Two distinct types of GFP-LC3 puncta were revealed by FRAP
LC3 plays an important role in autophagosomal membrane expansion and closure. Because the assembly and disassembly of many biofunctional protein complexes are highly controlled and regulated, we examined the dynamic properties of LC3 using FRAP, which is a technique that measures the mobility of fluorescent molecules in living cells. HeLa cells transiently expressing mRFP-GFP-LC3 were starved in HBSS for 1 h. The mRFP-GFP-LC3 puncta (arrows in ) in living cells were photobleached using a 488 nm laser line, and the recovery of the mRFP and GFP signals were monitored using confocal microscopy. We found that most of the mRFP-GFP-LC3B puncta showed no fluorescence recovery, and only a few puncta (8 out of 127 dots examined) showed a rapid and nearly complete fluorescence recovery (half-time < 1 sec) (). We found the distinct dynamic properties of LC3 in living cells surprising. Notably, a few of the LC3 puncta displayed fast fluorescence recovery, indicating that LC3 underwent a rapid exchange between the punctate structure and cytoplasmic pool. As the schematic diagrams show in , the LC3 in the phagophore assembly sites or the protein aggregates may access to the cytoplasm pool, and LC3 undergoing rapid exchange may associate with these structures. To identify the types of LC3 fluorescent puncta and analyze their dynamic properties, LC3 and its mutant LC3G120A and some marker proteins were tagged with different fluorescent proteins. The cells expressing these FP-labeled proteins were subjected to dynamic imaging analysis.
The fluorescent signals of mRFP-LC3 in both the early autophagic structures and mature autolysosomes only showed either minimal or no recovery after photobleaching
First, we examined whether the dynamic exchange phenomena of LC3 was present in the early autophagic structures and mature autolysosomes. During autophagosome formation, phosphatidylinositol 3-phosphate (PtdIns3P) was locally produced by the phosphatidylinositol 3-kinase (PtdIns3K) complex and decorated the autophagic structures. WIPI1 (the human WD repeat domain phosphoinositide interacting 1 protein), a downstream effector of PtdIns3P recruited to the phagophore,Citation3,Citation28 was used to identify the early autophagic structures in our study. The GFP-WIPI1+/mRFP-LC3+ puncta in HBSS-starved HeLa cells were subjected to two-color FRAP analysis (). In our experimental setup, the fluorescent molecules in a 2.691 μm × 2.691 μm square box were bleached using a 400 ms pulse of 488 nm and 543 nm light at 100% power. During the imaging period, less than 3% of GFP signal bled through the RFP channel, and there was no leakage of the mRFP/mCherry signal into the GFP channel (Fig. S1). As shown in , although GFP-WIPI1 exhibited a rapid and substantial recovery, there was minimal fluorescence recovery for mRFP-LC3 in the GFP-WIPI1+/mRFP-LC3+ structures, indicating that the rapid exchange between the puncta and cytoplasmic pool did not occur in the starvation-induced early autophagosomes. We also used another phagophore marker ZFYVE1/DFCP1 (zinc finger, FYVE domain containing 1) to identify the early autophagic structures. ZFYVE1 translocates to an ER-derived punctate compartment called the omegasome, which is in very close proximity to the LC3 puncta in autophagosome biogenesis.Citation3,Citation29 The GFP-ZFYVE1+/mRFP-LC3+ puncta in HBSS-starved HeLa cells were subjected to two-color FRAP analysis (Fig. S2A and S2B). mRFP-LC3 showed little fluorescence recovery in the GFP-ZFYVE1+/mRFP-LC3+ structures after photobleaching. In addition, we noted that GFP-ZFYVE1 exhibited little amount of recovery compared with GFP-WIPI1 (Fig. S2C and S2D). These results suggest that the dissociation of LC3 from the outer and/or inner autophagosome membranes may be very slow and not constitutive.
Figure 2. FRAP analysis of the dynamic exchange of LC3 in early autophagic structures and mature autolysosomes. (A and B) HeLa cells coexpressing GFP-WIPI1 and mRFP-LC3 were starved in HBSS for 2 h. The GFP-WIPI1+/mRFP-LC3+ vesicles (arrows) were subjected to two-color FRAP analysis. The recovery kinetics of GFP-WIPI1 and mRFP-LC3 in the autophagosomes are shown in the graph (B). Error bars: the standard error of the mean (s.e.m.) with n = 18. (C and D) HeLa cells expressing mRFP-LC3 were starved for 1.5 h and then stained with 100 nM LysoTracker Green for 30 min. mRFP-LC3 puncta positive for the LysoTracker Green signal were subjected to FRAP analysis. The recovery kinetics of mRFP-LC3 in autolysosomes are shown in graph (D). Error bars: s.e.m. with n = 15. (E and F) HeLa cells were cotransfected with GFP-LC3 and mRFP-LC3 for 24 h and then treated with 50 mM NH4Cl for 1.5 h. The mRFP-LC3 puncta were subjected to FRAP analysis. The recovery kinetics of mRFP signal in autolysosomes are shown in graph (F). Error bars: s.e.m. with n = 8. The inset images in (B–F) are the representative FRAP images. For all FRAP analysis, the puncta were outlined (white circle) and the intensities were extracted. Scale bars: 5 μm. Photobleaching box size: 2.691 μm × 2.691 μm.
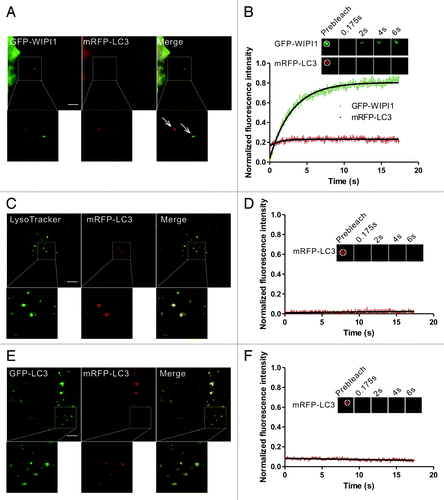
The outer membrane of enclosed autophagosomes fuses with lysosomes to become autolysosomes, which are acidic structures.Citation9 We observed no fluorescence recovery in the mRFP-LC3 puncta that colocalized with LysoTracker Green, a marker for the late endosomes/lysosomes (). This result is consistent with our hypothesis that LC3 is trapped in the membrane structures of autolysosomes. NH4Cl, a VMA21-independent neutralyzer of lysosomal pH, was also used to enrich the PE-linked forms of LC3.Citation30 FRAP analysis of these structures demonstrated that no obvious recovery for mRFP-LC3 in NH4Cl−treated cells (). Taken together, our data confirmed the lack of rapid LC3 exchange in both the early autophagic structures and mature autolysosomes.
GFP-LC3 and GFP-LC3G120A in the protein aggregates displayed rapid dynamic exchange between the fluorescent puncta and cytoplasmic pool
Because the LC3 protein associated with the autophagosomal structures did not show rapid dynamic properties, we suspected that the rapid exchange of LC3 may occur in the protein aggregates, in which LC3 also was recruited. To establish a simple method to identify the aggregate-associated LC3 puncta in living cells, LC3G120A, an LC3 mutant that cannot be recruited to the autophagosomal membrane,Citation5,Citation9 was used as a marker for the LC3-involved aggregates. Puromycin, an inhibitor of protein synthesis, was used to induce the formation of LC3 puncta in an autophagy-independent manner.Citation16,Citation31,Citation32 Under standard cell culture conditions, only around 8% of the mCherry-LC3G120A-expressing HeLa cells showed punctate structures, and treatment with puromycin for 2.5 h resulted in the accumulation of LC3G120A puncta in nearly 45% of the cells (Fig. S3A). In the negative control, HeLa cells expressing the mCherry tag alone rarely showed such punctate structures (Fig. S3B). When HeLa cells coexpressing GFP-LC3 and mCherry-LC3G120A were treated with puromycin for 2.5 h, confocal imaging showed that both LC3 and LC3G120A were recruited and localized in the same protein aggregates (). As a negative control, GFP alone was not colocalized with the mCherry-LC3G120A puncta (Fig. S3C).
Figure 3. FRAP and FRAPa analysis of the LC3/LC3G120A proteins in the protein aggregates. (A) Confocal imaging of puromycin-treated HeLa cells coexpressing GFP-LC3 and mCherry-LC3G120A. The arrows indicate the colocalized GFP-LC3 and mCherry-LC3G120A puncta. Scale bar: 10 μm. (B) FRAP analysis of the GFP-LC3 puncta positive for mCherry signal in (A). The quantitative and normalized fluorescence recovery kinetics of GFP-LC3 after photobleaching are shown in the graph. Error bars: s.e.m. with n = 9. Inset: representative FRAP images. (C) The GFP-LC3G120A expressing HeLa cells were treated with puromycin for 2.5 h. The quantitative and normalized fluorescence recovery kinetics of GFP-LC3G120A are shown in the graph. Error bars: s.e.m. with n = 12. Inset: representative FRAP images. (D) The mobile fraction for aggregate-associated GFP-LC3/LC3G120A and mRFP-LC3 puncta associated with the autophagic structures. Data were collected from previous figures and showed as the mean ± SD. (E) Two-color FRAP analysis showed fast recovery kinetics of LC3/LC3G120A and slow recovery kinetics of ubiquitin in IBs. Left panel: Representative two-color FRAP images. Right panel: the normalized fluorescence recovery kinetics of mRFP-UBC and GFP-LC3/LC3G120A in the colocalized aggregates. Error bars: s.e.m. with n = 9 for LC3+/UBC+ puncta, n = 7 for LC3G120A+/UBC+ puncta. Scale bar: 5 μm. (F) FRAPa analysis of Dendra2-LC3 in the protein aggregates. The red fluorescence intensity of Dendra2-LC3 puncta was monitored immediately after photoconversion by irradiation at 405 nm in live cells (dots) or fixed cells (squares). The normalized diffusion kinetics of red fluorescence are shown in the graph. Error bars: s.e.m. with n = 8 (live cells) and n = 3 (fixed cells). Inset: representative two-color FRAPa images. For all FRAP and FRAPa analysis, the puncta were outlined (white circle) and the intensities were extracted. The box size used in all of the photobleaching and photoactivation experiments was 2.691 μm × 2.691 μm.
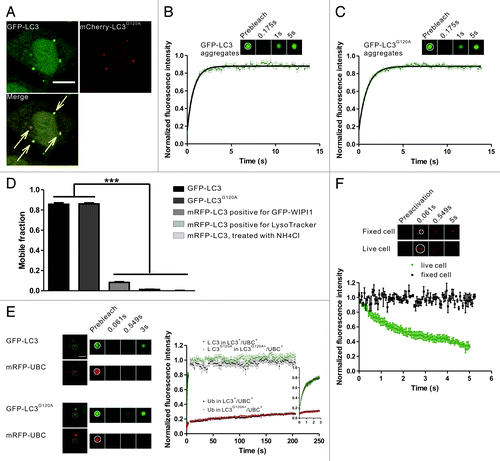
To monitor the dynamics of LC3 in the IBs, HeLa cells coexpressing GFP-LC3 and mCherry-LC3G120A were treated with puromycin, in which mCherry-LC3G120A was used as a marker to identify the protein aggregates. Next, the GFP-LC3 fluorescent puncta were subjected to FRAP analysis (). After photobleaching using a 488 nm laser line, nearly all of the examined foci showed a fast recovery with a half-time about 500 ms (). GFP-LC3G120A exhibited similar kinetics of fluorescence recovery to that of GFP-LC3 (). GFP-LC3G120A and GFP-LC3+/mCherry-LC3G120A+ puncta spontaneously formed in cells without puromycin stimulation also showed rapid dynamic exchange (data not shown). In contrast, all of the puncta with only the GFP-LC3 signal, but not the mCherry-LC3G120A signal, did not exhibit fluorescence recovery (data not shown), suggesting these puncta may mark the autophagic structures.
In FRAP analysis, mobile fraction (Mf) is a measure of fraction of fluorescent molecule that can exchange between the region of interest (ROI) and the surrounding area. As shown in , the mobile fraction were more than 80% for GFP-LC3 and GFP-LC3G120A, while less than 10% for LC3 associated with the autophagic structures (this is not due to the different tags used, because similar mobility were observed for the GFP or mRFP tags).
To verify that the LC3/LC3G120A fluorescent puncta that exhibited rapid exchange was protein aggregates, mRFP-UBC (ubiquitin C) was used as a marker for protein aggregates. In puromycin-treated HeLa cells that coexpressed mRFP-UBC and GFP-LC3 or GFP-LC3G120A, nearly all of the GFP-LC3/LC3G120A puncta colocalized with mRFP-UBC (Fig. S4). FRAP analysis of the GFP+/mRFP+ puncta demonstrated that GFP-LC3/LC3G120A exhibited a rapid fluorescence recovery, while mRFP-UBC showed very slow recovery after laser irradiation (), which suggests that the polyubiquitinated protein was largely sequestered in the protein aggregates.
To test directly whether LC3 is rapidly released from the IBs, we used a complementary technique to FRAP, fluorescence redistribution after photoactivation (FRAPa), which measures the mobility of selectively photoconverted fluorescent molecules. We utilized the photochemical properties of Dendra2, a fluorescent protein that irreversibly converts from a green fluorescent form to a red fluorescent form upon irradiation at 405 nm.Citation33,Citation34 HeLa cells were transfected with Dendra2-LC3 for 24 h and then treated with puromycin for 2.5 h. The red fluorescence intensity of Dendra2-LC3 was monitored immediately after photoactivation. A substantial fluorescence loss occurred rapidly in live cells (half-time ~1 sec) compared with fixed cells ().
Aggregate-associated LC3 puncta induced by other conditions also exhibited rapid dynamic exchange
Previous reports have shown that some autophagy inhibitors (e.g., the PtdIns3K inhibitors LY294002 and wortmannin) and proteasomal inhibitors (e.g., MG-132) efficiently induce the aggregation and autophagy-independent incorporation of LC3 into protein aggregates.Citation21,Citation32,Citation35 To further confirm that the dynamic exchange of LC3 in membrane-free protein aggregates was a common phenomenon, we next used these pharmacological agents to induce the formation of aggregate-associated LC3 puncta. FP-LC3G120A puncta occurred in 50% to 60% of cells treated with LY294002, wortmannin, or MG-132. FRAP analysis of these puncta also showed the rapid fluorescence recovery of GFP-LC3/LC3G120A (; Fig. S5). In contrast, the well-known autophagy inducer rapamycin and lysosomotropic reagent chloroquine (CQ) both induced GFP-LC3 puncta but not mCherry-LC3G120A puncta. These GFP-LC3 puncta showed no fluorescence recovery after photobleaching (Fig. S6).
Figure 4. Rapid dynamic exchange of LC3 occurs in protein aggregates induced by other conditions. (A–C) HeLa cells that were transiently cotransfected with GFP-LC3 and mCherry-LC3G120A for 24 h were treated with 50 μM LY294002 for 6 h (A), 200 nM wortmannin for 6 h (B) or 10 μM MG-132 for 9 h (C), and representative images are shown. The GFP-LC3 puncta that were positive for the mCherry signal were then subjected to FRAP analysis. The quantitative and normalized fluorescence recovery kinetics of GFP-LC3 after photobleaching are shown in the right graph. Error bars: s.e.m. with n = 8 (A), 8 (B), 22 (C). Inset: representative FRAP images. (D) atg5−/− MEFs that were transiently cotransfected with GFP-LC3 and mCherry-LC3G120A for 24 h. The GFP-LC3 puncta that were positive for the mCherry signal were then subjected to FRAP analysis. The quantitative and normalized fluorescence recovery kinetics of GFP-LC3 after photobleaching are shown in the right graph. Error bars: s.e.m. with n = 15. Inset: representative FRAP images. R-LC3G120A, mCherry-LC3G120A. Scale bars: 5 μm. Photobleaching box size: 2.691 μm × 2.691 μm.
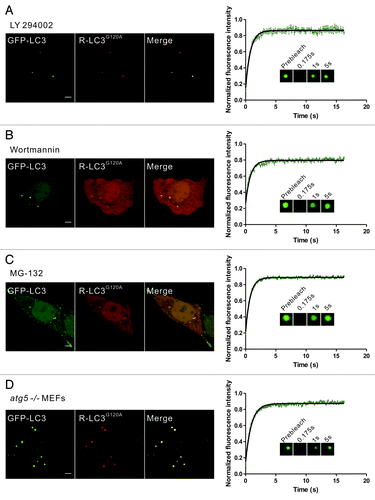
Moreover, GFP-LC3 transiently expressed in autophagy-deficient atg5−/− mouse embryonic fibroblasts (MEFs) also forms non-autophagic punctate structures.Citation14,Citation36 In atg5−/− MEFs transiently transfected with GFP-LC3/mCherry-LC3G120A or GFP-LC3G120A for 24 h, almost all of the examined GFP-LC3/LC3G120A foci showed a fast recovery after photobleaching (; Fig. S7A). However, we still found very few GFP-LC3G120A or mRFP-LC3/mCherry-LC3G120A puncta colocalized with either LysoTracker Red marker or GFP-RAB7A and did not show recovery after photobleaching in atg5−/− MEFs at 24 h after transfection (Fig. S7B–D). This finding was further confirmed by the observation that all of the dots that were negative for late endosome/lysosome markers displayed fast fluorescence recovery (~200 dots examined) (Fig. S7B–D). In fact, as shown in Fig. S7E–F, at 48 h after transfection, nearly all of the cells displayed many LC3/LC3G120A dots that were positive for RAB7A, suggesting that the LC3/LC3G120A was transported to the lysosomes by an ATG5-independent process.
LC3/LC3G120A interacted with SQSTM1 transiently in the protein aggregates
It has been reported that SQSTM1 is a general mediator for IB formation,Citation37 and that the incorporation of LC3/LC3G120A into IBs is facilitated by their interaction with SQSTM1.Citation21,Citation32,Citation38 Because LC3/LC3G120A exhibited a rapid exchange between the protein aggregates and cytoplasm, we were interested in the biophysical properties of the interaction between LC3/LC3G120A and SQSTM1 in the protein aggregates. HeLa cells were cotransfected with the mRFP-SQSTM1 and GFP-LC3/LC3G120A plasmids. Confocal fluorescence microscopy demonstrated that nearly all of the GFP-LC3/LC3G120A puncta contained mRFP-SQSTM1 and that the larger and brighter SQSTM1 bodies were likely membrane-free protein aggregates. Two-color FRAP analysis of the GFP+/mRFP+ bodies showed that the slow recovery of SQSTM1 was similar to that of ubiquitin, in contrast with LC3/LC3G120A (). To study the biophysical interaction of LC3 and SQSTM1 in living cells, we performed fluorescence resonance energy transfer (FRET) microscopy using mCerulean (mCer) and mCitrine (mCit) as the FRET donor and acceptor, respectively. Taking advantage of the relatively slow fluorescence recovery of SQSTM1 compared with LC3, we could detect the FRET signal in living cells using the acceptor photobleaching method, which was usually applied to fixed samples. Here, HeLa cells transfected with the plasmid encoding mCerulean and mCitrine linked by 12 amino acids served as a positive control, and HeLa cells cotransfected with the separate mCerulean and mCitrine plasmids served as a negative control. However, when we examined the cells that coexpressed mCerulean-LC3 and mCitrine-SQSTM1, a minimal FRET signal was detected (). This result may be due to the unfavorable orientation of mCitrine and mCerulean or the large distance between the N-terminus and the LC3-interacting region (LIR) of SQSTM1. We then fused the mCitrine moiety at the C-terminus of SQSTM1 (SQSTM1-mCitrine) or adjacent to the LIR region of SQSTM1 (s-SQSTM1-mCitrine, a functional construct that colocalized with mRFP-SQSTM1) (), and the resulting FRET signals were approximately 4 to 6 times higher compared with mCerulean-LC3/mCitrine-SQSTM1 (). Taken together, our FRAP and FRET results suggest that LC3/LC3G120A interacts with the C-terminal LIR of SQSTM1 and that their association is transient in living cells.
Figure 5. FRAP and FRET analysis of the dynamics and interaction of LC3/LC3G120A and SQSTM1 in the protein aggregates. (A) Two-color FRAP analysis of mRFP-SQSTM1 and GFP-LC3/LC3G120A colocalized puncta in living cells. Left panel: representative confocal images. Right panel: the normalized recovery kinetics of mRFP-SQSTM1 and GFP-LC3/LC3G120A. Error bars: s.e.m. with n = 8. Scale bars: 5 μm. (B) The FRET signal between mCerulean-LC3 and mCitrine-tagged SQSTM1 in HeLa cells. The mCerulean+/mCitrine+ aggregates in live cells were subjected to acceptor photobleaching FRET analysis. The FRET efficiencies of the positive and negative controls were determined in the fixed cells. All FRET data are shown in the graph. *p < 0.05 and ***p < 0.001. Error bars: s.e.m. with n = 15. Inset: representative confocal images of acceptor photobleaching FRET analysis. (C) The schematic diagrams of s-SQSTM1-mCitrine and confocal imaging of HeLa cells cotransfected with s-SQSTM1-mCitrine and mRFP-SQSTM1. Scale bar: 10 μm. (D) Acceptor photobleaching FRET analysis of the interaction of SQSTM1-SQSTM1 (mCerulean-SQSTM1/mCitrine-SQSTM1, mCerulean-SQSTM1/SQSTM1-mCitrine, and mCitrine-SQSTM1/SQSTM1-mCerulean) in HeLa cells. The quantitative FRET efficiencies are shown in the graph. ***p < 0.001. Error bars: s.e.m. with n = 15. Inset: representative confocal images of acceptor photobleaching FRET analysis. (E) Schematic diagram of the arrangement of SQSTM1 in the protein aggregates. For all FRAP analysis, the puncta were outlined (white circle) and the intensities were extracted. Photobleaching box size: 2.691 μm × 2.691 μm.
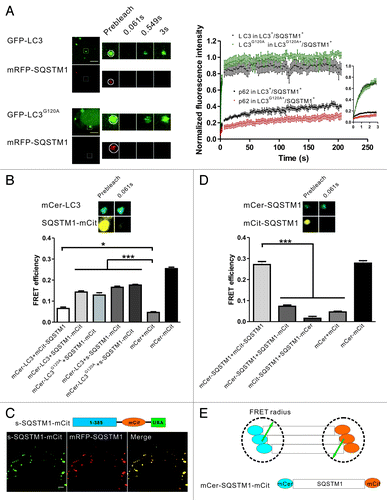
We next examined whether the arrangement of SQSTM1 in aggregates occurred in a regular pattern. FRET analysis showed that mCerulean-SQSTM1/SQSTM1-mCitrine or mCitrine-SQSTM1/SQSTM1-mCerulean in the protein aggregates displayed either very weak or no FRET signals (). However, the FRET signal of mCerulean-SQSTM1/mCitrine-SQSTM1 was strong and comparable to the positive control (). These results suggest that the oligomerization of SQSTM1 orients the N-terminus in close proximity and that the fluorescent proteins tagged at N-terminus of SQSTM1 are within the effective FRET radius. However, the distance between the N-terminus and C-terminus of SQSTM1 was likely outside the of FRET distance limit ().
ATG16L1 was recruited proximal to the aggregate-associated LC3 puncta
Since ATG16L1 is a phagophore marker, we also used GFP-ATG16L1 to label the early autophagic structures.Citation8,Citation39-Citation41 Under starvation conditions, there were fewer mRFP-LC3 puncta in HeLa cells coexpressing GFP-ATG16L1 and mRFP-LC3 (). This finding was consistent with another report that the overexpression of ATG16L1 inhibited autophagosome formation.Citation7 However, we observed that a fraction of ATG16L1 puncta localized adjacently to the LC3 puncta (, lower panel). Time-lapse imaging showed that the GFP-ATG16L1 signal did not disappear after 20 min (data not shown), which is much longer than the lifetime of the GFP-ATG5 labeled early autophagic structures (usually < 10 min).Citation8,Citation42,Citation43 We hypothesized that the LC3 puncta that localized adjacent to the ATG16L1 puncta were aggregate-associated. FRAP analysis showed that nearly all of these mRFP-LC3 puncta demonstrated a rapid exchange, which was similar to the aggregate-associated LC3 puncta (). We also observed this phenomenon in wild-type MEFs and atg5−/− MEFs (Fig. S8), indicating that the mRFP-LC3 puncta that localized adjacent to GFP-ATG16L1 were IBs, but not true autophagic structures.
Figure 6. GFP-ATG16L1 localized adjacent to the aggregate-associated LC3 puncta. (A and B) HeLa cells transiently coexpressing either GFP/mRFP-LC3 or GFP-ATG16L1/mRFP-LC3 were starved in HBSS for 1.5 h. The right graph shows quantification results of the dot-positive cells (containing more than 15 dots). The mRFP-LC3 puncta that localized adjacent to the GFP-ATG16L1 puncta are indicated by arrows. (C) HeLa cells were transiently cotransfected with GFP-ATG16L1 and mRFP-LC3 for 24 h. The adjacent puncta were then subjected to two-color FRAP analysis. The recovery kinetics of mRFP-LC3 are shown, as indicated. Error bars: s.e.m. with n = 25. The percentage of GFP-ATG16L1 colocalized adjacent to mRFP-LC3 puncta with fast recovery is shown in the right graph (55 dots examined). (D) Confocal imaging of HeLa cells coexpressing mCerulean-LC3, mCitrine-16L1 and mCherry-LC3G120A. The arrows indicate LC3 puncta localized adjacent to ATG16L1, which colocalized with mCherry-LC3G120A. The percentage of ATG16L1 adjacently colocalized LC3 puncta positive for LC3G120A is shown in the right graph (92 dots observed). Scale bars: 5 μm.
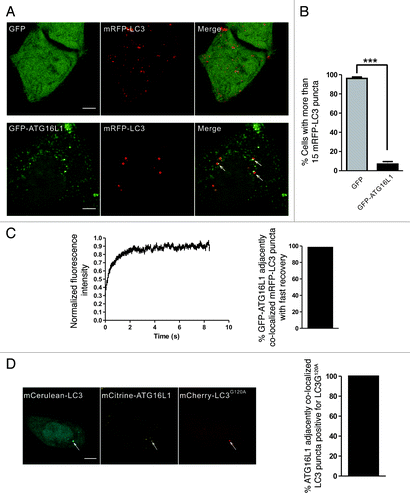
To further confirm the previous finding, we cotransfected HeLa cells with mCitrine-ATG16L1, mCerulean-LC3 and mCherry-LC3G120A. A localization analysis demonstrated that the LC3 puncta that were localized adjacent to the ATG16L1 were always positive for LC3G120A, a marker of IBs (). Taken together, these results show that ATG16L1 is recruited proximally to the aggregate-associated LC3 puncta in an ATG5-independent manner.
Discussion
In this study, we investigated the biophysical properties of LC3 associated with autophagosomes/autolysosomes and protein aggregates. First, we examined the dynamic properties of LC3 in the early autophagic structures and mature autolysosomes. The initial recruitment of LC3 to the phagophore assembly sites is a relatively slow process that requires hundreds of seconds.Citation29,Citation43 Once formed, our results demonstrated that the LC3 associated with the outer membrane of autophagosomes does not exchange with the cytoplasmic pool in a constitutive and rapid manner. Further studies are required to clarify the exact dynamics of LC3 dissociation from the outer membranes of nascent autophagosomes. Upon maturation of the autolysosomes, LC3 was trapped in the membrane structures and exhibited no exchange with cytoplasmic pool in FRAP analysis. We also found that in the early autophagic structures, the PtdIns3P-binding protein ZFYVE1 showed little fluorescence recovery post-bleaching, while another PtdIns3P effector, WIPI1, displayed rapid and substantial recovery. These differences may be due to their distinct biochemical properties, different membrane locations,Citation3,Citation28,Citation29 or other potential binding partners. These results also implied that although many ATG proteins are recruited to the autophagosome formation site, their dynamic properties may vary. Further biophysical and biochemical studies will provide more information regarding the dynamic properties of the ATG proteins and elucidate how they function.
Previous reports identified SQSTM1 as a common component of protein aggregates and showed that it is required for the formation of IBs.Citation21,Citation31,Citation32,Citation44 It has been suggested that SQSTM1 recognizes ubiquitinated proteins through its C-terminal ubiquitin-associated (UBA) domain and self-oligomerizes through its N-terminal Phox and Bem1 (PB1) domain. Both processes are important for SQSTM1 aggregation.Citation37,Citation45 LC3 can be incorporated into the IBs through its interaction with the LIR of SQSTM1.Citation21,Citation46 However, before these ubiquitin- and SQSTM1-positive aggregates become large enough to be detected by light microscopy, they likely have been targeted to the autophagosome through the LC3-SQSTM1 interaction.Citation32,Citation45 In this study we found that LC3 associated and dissociated rapidly with IBs in an autophagy-independent manner. Although ubiquitin and SQSTM1 dynamically associate with IBs, the rates of exchange are much lower than that of LC3. It is also noted that both our and others’ work demonstrated the dynamic property of SQSTM1 and ubiquitin involved in IBs,Citation47-Citation49 however, the exchange rates measured are slightly varied, which may be due to the different expression levels of FP-tagged proteins or different experiment methods. When mRFP-SQSTM1 expressed in HeLa cells alone, a much higher exchange rate was observed (Fig. S9).
Our results also showed that the composite organization of these IBs involved ordered molecular interactions. This was mainly due to the structure of the proteins involved. Together, our results indicated that the oligomerization of SQSTM1 and the interaction between SQSTM1 and mono- and poly-ubiquitin were more stable than the LC3-SQSTM1 interaction. LC3, similar to HSP70 (heat shock 70kDa protein),Citation24 a molecular chaperone, associated transiently with IBs and was not sequestered irreversibly, suggesting that IBs may not disrupt the normal cellular function of LC3 by sequestering it. The ubiquitin-SQSTM1-LC3 interaction mode may also be applied to the SQSTM1-mediated process that incorporates cargo into the autophagosomes.
The LC3 mutant that is defective in lipidation (LC3∆G) was used as a negative control to distinguish between the LC3-involved aggregates and autophagosomes.Citation50 In our study, nearly all of the LC3G120A puncta examined exhibited rapid exchange. However, similar to the results obtained with atg5−/− MEFs, in puromycin-treated HeLa cells expressing mCherry-LC3G120A alone, puncta showing no fluorescence recovery after photobleaching were occasionally observed. We suspected that these puncta may have been trapped in membrane-enclosed structures such as autolysosomes. After 24 h of mCherry-LC3G120A expression in HeLa cells, LysoTracker Green staining demonstrated that puromycin induced the vast majority of the mCherry-LC3G120A puncta that were negative for the green fluorescent signal and very few of the mCherry-LC3G120A puncta that were positive for the LysoTracker Green signal (Fig. S10A). Similarly, very few GFP-LC3G120A puncta were positive for LysoTracker Red signal (Fig. S10B), which is consistent with previous reports and may be due to the incomplete quenching of the GFP fluorescent signal in acidic structures.Citation51,Citation52 We also noted that after 48 h of mCherry-LC3G120A expression in HeLa cells, a greater number of mCherry-LC3G120A puncta were found in acidic structures (Fig. S10C). These results indicate that mCherry-LC3G120A can form two types of bodies: the first is a type of inclusion body or a membrane-free protein aggregate, and the other is a membrane-confined lysosomal structure that is likely formed through autophagy. In contrast, nearly no GFP-LC3G120A puncta formed or were positive for LysoTracker Red in the transfected cells cultured for 48 h in regular medium (Fig. S10D; Fig. S11A, left panel). However, 24 h of NH4Cl (a lysosome inhibitor) treatment induced many fluorescent puncta in the GFP-LC3G120A-transfected HeLa cells with 48 h culture (Fig. S11A). As a control, the same treatment did not induced fluorescent puncta in GFP-transfected cells (Fig. S11B). As shown in Figure S11C and S11D, under 24 h NH4Cl treatment, either the GFP-LC3G120A puncta colocalized with mRFP-LC3 or the mCitrine-LC3G120A puncta (similar to GFP-LC3G120A, data not shown) colocalized with mCerulean-RAB7A, suggesting that these GFP/mCitrine-LC3G120A puncta were in autophagosomal and lysosomal structures. This type of GFP-LC3G120A puncta also formed under 24 h of CQ treatment (data not shown). Thus, under certain conditions, it is important to rule out these artifacts when using LC3G120A as a negative control for autophagy.
In summary, using dynamic fluorescence microscopy techniques (e.g., FRAP, FRAPa and FRET), we found that LC3 exhibited rapid kinetics of association and dissociation with the protein aggregates while tightly attached to the autophagosomal membrane. Taking advantage of the distinct biophysical properties of the two types of LC3 puncta, we could distinguish whether a specific FP-LC3 punctum was an autophagosome or a LC3-involved protein aggregate in living cells by qualitative FRAP analysis.
Materials and Methods
Chemicals
Chloroquine (C6628), ammonium chloride (A9434), LY294002 (L9908), wortmannin (W1628) and puromycin (P8833) were purchased from Sigma-Aldrich. LysoTracker Green (L7526) was purchased from Invitrogen. Rapamycin (S1842), MG-132 (S1748) and LysoTracker Red (C1046) were purchased from Beyotime Institute of Biotechnology.
Plasmids
GFP-ZFYVE1 and mRFP-LC3 were gifts of Nicholas T. Ktistakis.Citation29 The MAP1LC3B/LC3B, WIPI1, ATG16L1 and RAB7A cDNAs were obtained by PCR from total cDNA from HeLa cells and then cloned into the EGFP-C1 vector (Clontech, USA, 6084–1). LC3B was also cloned into the mCerulean-C1 vector to generate mCerulean-LC3B. ATG16L1 was cloned into the mCitrine-C1 vector to generate mCitrine-ATG16L1. RAB7A was also cloned into the mCerulean-C1 vector. LC3G120A was amplified and inserted into pcDNA3.1(+)-mCherry. LC3G120A was also amplified and then cloned into the EGFP-C1, mCerulean-C1 and mCitrine-C1 vectors. The EGFP in EGFP-LC3B was replaced by Dendra2 to generate Dendra2-LC3B. The cDNAs for UBC (ubiquitin C) and SQSTM1 were obtained by PCR from total cDNA from HeLa cells. The amplified products were cloned into pcDNA3.1(+)-mRFP and pcDNA3.1(+)-mRFP/mCerulean/mCitrine. The SQSTM1 DNA was also amplified and inserted into the mCerulean-N1 and mCitrine-N1 vectors. The DNA coding for the UBA domain of SQSTM1 was amplified and cloned into mCitrine-C1 to generate mCitrine-UBA. The DNA coding for the N-terminal 385 amino acids of SQSTM1 was inserted into mCitrine-UBA to generate s-SQSTM1-mCitrine. A linker of 12 amino acids was included between the two fluorescent tags in mCerulean-mCitrine. The primers are listed in Table S1.
Cell culture, transfection and treatment
HeLa cells (obtained from ATCC, CCL-2), wild-type MEFs and atg5−/− MEFs (a generous gift from Dr. Noboru Mizushima, Tokyo Metropolitan Institute of Medical Science) were maintained in DMEM medium containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in 5% CO2. The cells were transfected with the indicated plasmids using Lipofectamine™ 2000 (Invitrogen Co., 11668–019) 20 to 24 h before imaging analysis. To observe the autophagic structures, the cells were treated with HBSS, rapamycin (1 μM), CQ (50 μM) or NH4Cl (50 mM) for the indicated period of time. To induce protein aggregates, the cells were treated with puromycin (5 μg/ml), wortmannin (200 nM), LY294002 (50 μM), or MG-132 (10μM) for the indicated period of time.
Confocal imaging
The cells were grown on 35-mm glass-bottomed dishes (LabTek) and mounted on the stage of a commercial FV1000 confocal microscope (Olympus). The images were captured using a 60 × 1.4 NA objective. The GFP and RFP (mCherry or mRFP) signals were imaged using a line sequential scan setting with excitation laser lines at 488 and 543 nm, respectively. The emission signals were collected at 495 to 530 nm (GFP, channel 1) and 590 to 650 nm (RFP, channel 2).
Qualitative and quantitative FRAP analysis
For qualitative FRAP analysis of mRFP-GFP-LC3, the 488 and 543 nm laser lines were used to excite GFP and mRFP, and the emission data were collected at 495 to 530 nm and 590 to 650 nm, respectively. Next, a 3 sec pulse of the 488 and 543 nm laser lines at 100% power was used to bleach the GFP/mRFP puncta, and then the fluorescent signals were acquired every 5 sec.
For quantitative two-color (or one-color, that is, GFP or RFP signal only) FRAP analysis, the 488 and 543 nm laser lines were used to excite GFP and RFP simultaneously, and the emission data were collected on two channels, respectively. There was minimal crosstalk between the two channels. After irradiation with three iterations of the 488 and 543 nm laser lines at 100% power, the GFP and RFP signals in the region of interest (ROI) were nearly completely photobleached. The fluorescence emissions were collected using the same settings as pre-photobleaching every 175 ms. For mRFP-LC3 colocalized with GFP-ZFYVE1, eight iterations of the 488 and 543 nm laser lines at 100% power were used to bleach the mRFP tag and followed by imaging every 61 ms. The recovery of GFP and RFP signals associated with the punctate structures (per-pixel intensity) was measured using the SpotTracker plug-in of ImageJ (National Institutes of Health) and the puncta with obvious Z axis-drift were discarded. The cytoplasmic per-pixel fluorescent intensity in a fixed region of interest (ROI) adjacent to the puncta were measured and subtracted from the puncta signal to correct for the cytoplasmic diffusion component and background.Citation53 The resulting data were normalized to the pre-bleached images and then plotted using Prism 5.0 (GraphPad Software). The average of the corrected FRAP curves were fitted using the Sigmaplot software (Systat Software) and the equation F(t) = Fmin + (Fmax − Fmin)(1 − exp−kt),Citation25 where F(t) is the intensity of fluorescence at time t, Fmin is the intensity of fluorescence immediately post-bleaching, Fmax is the intensity of fluorescence following complete recovery, and k is the rate constant of the exponential recovery. The recovery half-time (t1/2 = In2/k) and mobile fraction (Mf) were calculated from the fitted FRAP curve. Mobile Fraction (Mf) was calculated as the following formula: Mf = (F∞ − F0) / (Fpre − F0), where F∞ is the stable fluorescent intensity of the puncta after sufficient recovery, F0 is the fluorescent intensity immediately after bleaching, and Fpre is the fluorescent intensity before bleaching.Citation54
For two-color (or one-color) FRAP analysis of LC3/UBC, LC3G120A/UBC, LC3/SQSTM1, LC3G120A/SQSTM1, and SQSTM1, following prebleach imaging, a 480 ms pulse of the 488 and 543 nm laser lines at 100% power was used to bleach GFP and RFP both. Then the fluorescence emissions were collected every 61 ms for the first 46 post-bleaching images and every 2 sec for the next 100 post-bleaching images. For mRFP-LC3 colocalized with GFP-ATG16L1, 100 post-bleaching images were acquired every 61 ms. The recovery of GFP and RFP signals associated with the punctate structures was measured using the SpotTracker plugin of ImageJ and background corrected, and the resulting data were normalized to the pre-bleached images and plotted using Prism 5.0.
FRAPa analysis
For FRAPa analysis, a 480 ms pulse of the 405 nm laser was used to photoactivate the Dendra2-LC3B puncta, and the fluorescence emissions at 590 to 650 nm were collected using the same settings as pre-photoactivation every 61 ms. The extracted data were normalized to the immediate post-activation images and plotted using Prism 5.0.
Acceptor photobleaching FRET in live cells
The 458 nm and 515 nm laser lines were used to excite mCerulean and mCitrine simultaneously, and their emission data were collected at 465 to 490 nm and 525 to 560 nm, respectively. There was no leakage of the mCitrine signal into the mCerulean channel. Next, a 480 ms pulse of the 515 nm laser at 100% power was used to bleach mCitrine, which was followed by image acquisition using the same settings as pre-bleaching with no delay. The intensities of mCerulean pre- and post-bleaching were extracted, and the background was subtracted using ImageJ. The FRET efficiency was measured using the following formula: EFRET = (Idonor, post − Idonor, pre) / Idonor, post . EFRET: FRET efficiency, Idonor, post: donor (mCerulean) fluorescence intensity before mCitrine bleaching, Idonor, post: donor (mCerulean) fluorescence intensity immediately post-mCitrine bleaching.Citation55
For the positive and negative FRET controls, the samples were washed with PBS and fixed in 3.75% paraformaldehyde in PBS.
Statistical analysis
The statistical significance between groups was determined using the unpaired t-test in Prism 5.0 (GraphPad Software). A p value < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01 and ***p < 0.001.
| Abbreviations: | ||
| ATG16L1 | = | autophagy-related 16-like 1 |
| ATG5 | = | autophagy-related 5 |
| ATG4 | = | autophagy-related 4 |
| ATG7 | = | autophagy-related 7 |
| ATG3 | = | autophagy-related 3 |
| ATG12 | = | autophagy-related 12 |
| PE | = | phosphatidylethanolamine |
| IBs | = | inclusion bodies |
| SQSTM1/p62 | = | sequestosome 1 |
| PtdIns3P | = | phosphatidylinositol 3-phosphate |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| WIPI1 | = | WD repeat domain, phosphoinositide interacting 1 |
| ZFYVE1/DFCP1 | = | zinc finger, FYVE domain containing 1 |
| VMA21 | = | VMA21 vacuolar H+-ATPase homolog |
| NH4Cl | = | Ammonium chloride |
| ROI | = | region of interest |
| UBC | = | ubiquitin C |
| CQ | = | chloroquine |
| MEFs | = | mouse embryonic fibroblasts |
| LIR | = | LC3-interacting region |
| UBA | = | ubiquitin-associated |
| MAP1LC3/LC3 | = | microtubule-associated protein light chain 3 |
| FRAP | = | fluorescence recovery after photobleaching |
| FRET | = | fluorescence resonance energy transfer |
| mCer | = | mCerulean |
| mCit | = | mCitrine |
Additional material
Download Zip (21.5 MB)Acknowledgments
We thank Dr. Noboru Mizushima (Tokyo Metropolitan Institute of Medical Science) for kindly providing the wild-type and atg5−/− MEF cells, Dr. Nicholas T. Ktistakis (the Babraham Institute, UK) for providing mRFP-LC3 and GFP-ZFYVE1, Dr. Roger Tsien (University of California, San Diego) for pRSETB-mRFP1 and pRSETB-mCherry, Dr. David Piston (Vanderbilt University School of Medicine) for pmCerulean-C1, Dr. Joel Swanson (University of Michigan Medical School) for pmCitrine-C1, Dr. Konstantin A. Lukyanov (Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Moscow) for Dendra2. We also thank the Analytical and Testing Center of Huazhong University of Science and Technology for spectral measurements. This work was supported by the National Basic Research Program of China (Grant No. 2011CB910401), Science Fund for Creative Research Group of China (Grant No.61121004), National Natural Science Foundation of China (Grant No. 81172153), and Specific International Scientific Cooperation of China (Grant No.2010DFR30820) .
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/23814
References
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004 - 10; http://dx.doi.org/10.1038/nrm2529; PMID: 18971948
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010; 12:831 - 5; http://dx.doi.org/10.1038/ncb0910-831; PMID: 20811355
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010; 6:764 - 76; http://dx.doi.org/10.4161/auto.6.6.12709; PMID: 20639694
- Longatti A, Tooze SA. Vesicular trafficking and autophagosome formation. Cell Death Differ 2009; 16:956 - 65; http://dx.doi.org/10.1038/cdd.2009.39; PMID: 19373247
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/10.1093/emboj/19.21.5720; PMID: 11060023
- Fass E, Amar N, Elazar Z. Identification of essential residues for the C-terminal cleavage of the mammalian LC3: a lesson from yeast Atg8. Autophagy 2007; 3:48 - 50; PMID: 17102583
- Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092 - 100; http://dx.doi.org/10.1091/mbc.E07-12-1257; PMID: 18321988
- Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol 2001; 152:657 - 68; http://dx.doi.org/10.1083/jcb.152.4.657; PMID: 11266458
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 60; PMID: 17534139
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101 - 11; http://dx.doi.org/10.1091/mbc.E03-09-0704; PMID: 14699058
- Drake KR, Kang M, Kenworthy AK. Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. PLoS One 2010; 5:e9806; http://dx.doi.org/10.1371/journal.pone.0009806; PMID: 20352102
- Kraft LJ, Kenworthy AK. Imaging protein complex formation in the autophagy pathway: analysis of the interaction of LC3 and Atg4BC⁷⁴A in live cells using Förster resonance energy transfer and fluorescence recovery after photobleaching. J Biomed Opt 2012; 17:011008; http://dx.doi.org/10.1117/1.JBO.17.1.011008; PMID: 22352642
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 2011; 18:1042 - 52; http://dx.doi.org/10.1016/j.chembiol.2011.05.013; PMID: 21867919
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323 - 8; PMID: 17387262
- Ciechomska IA, Tolkovsky AM. Non-autophagic GFP-LC3 puncta induced by saponin and other detergents. Autophagy 2007; 3:586 - 90; PMID: 17786021
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006; 2:189 - 99; PMID: 16874109
- Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 2000; 10:524 - 30; http://dx.doi.org/10.1016/S0962-8924(00)01852-3; PMID: 11121744
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science 2002; 296:1991 - 5; http://dx.doi.org/10.1126/science.1067122; PMID: 12065827
- Bjørkøy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy 2006; 2:138 - 9; PMID: 16874037
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/10.1083/jcb.200507002; PMID: 16286508
- Shvets E, Elazar Z. Autophagy-independent incorporation of GFP-LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 2008; 4:1054 - 6; PMID: 18776740
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313 - 26; http://dx.doi.org/10.1016/j.cell.2010.01.028; PMID: 20144757
- Reits EA, Neefjes JJ. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat Cell Biol 2001; 3:E145 - 7; http://dx.doi.org/10.1038/35078615; PMID: 11389456
- Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI. Polyglutamine protein aggregates are dynamic. Nat Cell Biol 2002; 4:826 - 31; http://dx.doi.org/10.1038/ncb863; PMID: 12360295
- Weisswange I, Newsome TP, Schleich S, Way M. The rate of N-WASP exchange limits the extent of ARP2/3-complex-dependent actin-based motility. Nature 2009; 458:87 - 91; http://dx.doi.org/10.1038/nature07773; PMID: 19262673
- Calvert PD, Peet JA, Bragin A, Schiesser WE, Pugh EN Jr.. Fluorescence relaxation in 3D from diffraction-limited sources of PAGFP or sinks of EGFP created by multiphoton photoconversion. J Microsc 2007; 225:49 - 71; http://dx.doi.org/10.1111/j.1365-2818.2007.01715.x; PMID: 17286695
- Mazza D, Braeckmans K, Cella F, Testa I, Vercauteren D, Demeester J, et al. A new FRAP/FRAPa method for three-dimensional diffusion measurements based on multiphoton excitation microscopy. Biophys J 2008; 95:3457 - 69; http://dx.doi.org/10.1529/biophysj.108.133637; PMID: 18621824
- Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27:107 - 32; http://dx.doi.org/10.1146/annurev-cellbio-092910-154005; PMID: 21801009
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 2008; 182:685 - 701; http://dx.doi.org/10.1083/jcb.200803137; PMID: 18725538
- Chakrama FZ, Seguin-Py S, Le Grand JN, Fraichard A, Delage-Mourroux R, Despouy G, et al. GABARAPL1 (GEC1) associates with autophagic vesicles. Autophagy 2010; 6: 495 - 505; http://dx.doi.org/10.4161/auto.6.4.11819; PMID: 20404487
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010; 6:330 - 44; http://dx.doi.org/10.4161/auto.6.3.11226; PMID: 20168092
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131 - 45; http://dx.doi.org/10.1074/jbc.M702824200; PMID: 17580304
- Chudakov DM, Lukyanov S, Lukyanov KA. Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc 2007; 2:2024 - 32; http://dx.doi.org/10.1038/nprot.2007.291; PMID: 17703215
- Zhu M-Q, Zhang G-F, Li C, Li Y-J, Aldred MP, Li ADQ. Photoswitchable Nanofluorophores for Innovative Bioimaging. J Innovative Opt Health Sci 2011; 04:395; http://dx.doi.org/10.1142/S1793545811001423
- Kuusisto E, Suuronen T, Salminen A. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun 2001; 280:223 - 8; http://dx.doi.org/10.1006/bbrc.2000.4107; PMID: 11162503
- Hou H, Zhang Y, Huang Y, Yi Q, Lv L, Zhang T, et al. Inhibitors of phosphatidylinositol 3′-kinases promote mitotic cell death in HeLa cells. PLoS One 2012; 7:e35665; http://dx.doi.org/10.1371/journal.pone.0035665; PMID: 22545128
- Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 2008; 4:1063 - 6; PMID: 18776737
- Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci 2008; 121:2685 - 95; http://dx.doi.org/10.1242/jcs.026005; PMID: 18653543
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679 - 88; http://dx.doi.org/10.1242/jcs.00381; PMID: 12665549
- Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC. Autophagosome precursor maturation requires homotypic fusion. Cell 2011; 146:303 - 17; http://dx.doi.org/10.1016/j.cell.2011.06.023; PMID: 21784250
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 2004; 36:2491 - 502; http://dx.doi.org/10.1016/j.biocel.2004.02.005; PMID: 15325587
- Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin?. Cell Death Differ 2009; 16:984 - 90; http://dx.doi.org/10.1038/cdd.2009.54; PMID: 19424283
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656 - 67; http://dx.doi.org/10.1016/j.cell.2010.04.009; PMID: 20478256
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149 - 63; http://dx.doi.org/10.1016/j.cell.2007.10.035; PMID: 18083104
- Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283:22847 - 57; http://dx.doi.org/10.1074/jbc.M802182200; PMID: 18524774
- Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol 2011; 192:17 - 27; http://dx.doi.org/10.1083/jcb.201009067; PMID: 21220506
- Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, et al. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 2004; 164:667 - 75; http://dx.doi.org/10.1083/jcb.200312073; PMID: 14981091
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44:279 - 89; http://dx.doi.org/10.1016/j.molcel.2011.07.039; PMID: 22017874
- Dumit VI, Dengjel J. Autophagosomal protein dynamics and influenza virus infection. Front Immunol 2012; 3:43; http://dx.doi.org/10.3389/fimmu.2012.00043; PMID: 22566925
- Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K. Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 2008; 4:131 - 4; PMID: 18000393
- Zhou C, Zhong W, Zhou J, Sheng F, Fang Z, Wei Y, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012; 8:1215 - 26; http://dx.doi.org/10.4161/auto.20284; PMID: 22647982
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy 2005; 1:23 - 36; http://dx.doi.org/10.4161/auto.1.1.1495; PMID: 16874023
- Loerke D, Wienisch M, Kochubey O, Klingauf J. Differential control of clathrin subunit dynamics measured with EW-FRAP microscopy. Traffic 2005; 6:918 - 29; http://dx.doi.org/10.1111/j.1600-0854.2005.00329.x; PMID: 16138905
- Snapp EL, Altan N, Lippincott-Schwartz J. Measuring protein mobility by photobleaching GFP chimeras in living cells. Curr Protoc Cell Biol 2003; Chapter 21:Unit 21 1.
- Wang X-P, Yu H-N, Chen T-S. Quantitative Fret Measurement Based on Confocal Microscopy Imaging and Partial Acceptor Photobleaching. J Innovative Opt Health Sci 2012; 05:1250015; http://dx.doi.org/10.1142/S1793545812500150